Back to Journals » OncoTargets and Therapy » Volume 10
Prognostic immune-related gene models for breast cancer: a pooled analysis
Authors Zhao J, Wang Y, Lao Z, Liang S, Hou J, Yu Y, Yao H, You N, Chen K
Received 14 June 2017
Accepted for publication 3 August 2017
Published 11 September 2017 Volume 2017:10 Pages 4423—4433
DOI https://doi.org/10.2147/OTT.S144015
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Yao Dai
Jianli Zhao,1,2,* Ying Wang,1,2,* Zengding Lao,3,* Siting Liang,3 Jingyi Hou,4 Yunfang Yu,1,2 Herui Yao,1,2 Na You,3 Kai Chen1,2
1Breast Tumor Center, Sun Yat-sen Memorial Hospital, Sun Yat-sen University, Guangzhou, China; 2Guangdong Provincial Key Laboratory of Malignant Tumor Epigenetics and Gene Regulation, Sun Yat-Sen Memorial Hospital, Sun Yat-Sen University, Guangzhou, China; 3School of Mathematics, Sun Yat-Sen University, Guangzhou, China; 4Department of Orthopedics, Sun Yat-Sen Memorial Hospital, Sun Yat-Sen University, Guangzhou, China
*These authors contributed equally to this work
Abstract: Breast cancer, the most common cancer among women, is a clinically and biologically heterogeneous disease. Numerous prognostic tools have been proposed, including gene signatures. Unlike proliferation-related prognostic gene signatures, many immune-related gene signatures have emerged as principal biology-driven predictors of breast cancer. Diverse statistical methods and data sets were used for building these immune-related prognostic models, making it difficult to compare or use them in clinically meaningful ways. This study evaluated successfully published immune-related prognostic gene signatures through systematic validations of publicly available data sets. Eight prognostic models that were built upon immune-related gene signatures were evaluated. The performances of these models were compared and ranked in ten publicly available data sets, comprising a total of 2,449 breast cancer cases. Predictive accuracies were measured as concordance indices (C-indices). All tests of statistical significance were two-sided. Immune-related gene models performed better in estrogen receptor-negative (ER-) and lymph node-positive (LN+) breast cancer subtypes. The three top-ranked ER- breast cancer models achieved overall C-indices of 0.62–0.63. Two models predicted better than chance for ER+ breast cancer, with C-indices of 0.53 and 0.59, respectively. For LN+ breast cancer, four models showed predictive advantage, with C-indices between 0.56 and 0.61. Predicted prognostic values were positively correlated with ER status when evaluated using univariate analyses in most of the models under investigation. Multivariate analyses indicated that prognostic values of the three models were independent of known clinical prognostic factors. Collectively, these analyses provided a comprehensive evaluation of immune-related prognostic gene signatures. By synthesizing C-indices in multiple independent data sets, immune-related gene signatures were ranked for ER+, ER-, LN+, and LN- breast cancer subtypes. Taken together, these data showed that immune-related gene signatures have good prognostic values in breast cancer, especially for ER- and LN+ tumors.
Keywords: breast cancer, prognostic models, immune-related gene
Introduction
Breast cancer is the most common cancer among women. As a clinically and biologically heterogeneous disease, accurate prognostic predictions are very important for breast cancer treatment planning. In the past, the pathological Tumor-Node-Metastasis Classification of Malignant Tumors staging system of the American Joint Committee on Cancer was the predominant prognostic prediction tool. In recent decades, molecular breast cancer subtypes have been identified based on pathological markers including estrogen receptor (ER), progesterone receptor, human epidermal growth factor 2 (HER2), and Ki67. Measured expression levels of these biomarkers are routinely used in clinical practices to stratify patients for prognostic predictions and to select treatments.1–4 In particular, four distinct subtypes of breast cancers have been defined: luminal A-like, luminal B-like, HER2-positive, and triple negative. These subtypes were first adopted by the St Gallen International Expert Consensus panel in 2013 as a basis for selecting adjuvant systemic therapy regimens (ie, endocrine therapy, chemotherapy, or anti-HER2 therapy).2 Nevertheless, diagnosis and treatment decision-making for breast cancer still relies on classical histopathological and immunohistochemical techniques. More quantitative diagnostic approaches and individualized treatment plans remain unmet needs. To meet these needs, multigene prognostic models have emerged, including 21-gene recurrence score,5–7 PAM50,8–13 Mammaprint®,14–16 and Breast Cancer Index™.17–20
Historically, prognostic multigene predictors strictly consisted of proliferation-associated genes.21,22 The tests were highly effective at classifying recurrence risks for ER-positive (ER+) and lymph node–negative (LN-) breast cancer subtypes; prognoses for these subtypes were well predicted by Ki67 indices or levels of DNA synthesis or cell cycle regulatory genes. Although most ER-negative (ER-) or LN-positive (LN+) breast cancers are highly proliferative, multigene predictors do not have discriminative power for prognoses within either subtype. Therefore, alternative prognostic signatures must be proposed for these subtypes that are not limited to proliferation-related genes.23,24 Recent preclinical studies have underscored inflammation and the stromal immune landscape as important drivers of breast cancer. There has been growing interest in establishing prognostic multigene signatures focused on immune-related genes, such as the STAT-1 gene, interferons, and immune response genes.25–32
Currently, no immune-related signature has been widely accepted in clinical practice; it is unknown whether one test is the most robust. In this study, all published immune-related prognostic gene models were systematically reviewed to evaluate performances across independent data sets. The aims of this study were two-fold: first, to investigate the prognostic effects of immune-related gene signatures and, second, to identify the most robust signature for the prognostication of nonmetastatic breast cancer.
Methods
The design of this study had two major sections. First, published literature describing prognostic immune-related gene signatures and publicly available genomic data sets were systematically reviewed. Second, a multistudy evaluation of immune-related prognostic models that met specific inclusion criteria was performed. The literature search strategies as well as inclusion criteria of prognostic models and data sets are detailed in the following subsections.
Literature search strategies
Systematic literature searches of articles published between July 2006 and March 2017 were performed independently by two authors (KC and JZ). In both cases, Internet-based searches of the PubMed and Web of Science platforms were performed without language or regional restrictions, using combinations of the following keywords: “breast cancer,” “breast neoplasm,” “breast carcinoma,” “immune-related gene,” “immune response gene,” “prognostic model,” and “immune-related predictor.” Reference lists of retrieved articles were manually screened to identify relevant articles. If a single study generated multiple publications, then either the highest quality publication or the most recent publication was chosen for subsequent analyses.
Inclusion criteria of prognostic models
Published prognostic models of immune-related gene signatures were evaluated for inclusion in the current study. Eligible studies included the following: 1) a claim of providing prognostic value for nonmetastatic breast cancer cases; 2) a model that was based on mRNA expression levels, as quantified by microarray technologies; 3) a model that was developed using a training data set and then validated with a test data set; 4) a training data set of at least 30 subjects; 5) defined outcome measurements of either overall survival, disease-free survival (DFS), or recurrence-free survival (RFS); 6) a model that was fully specified or could be rederived from the specified data set; and 7) a model for which ≥80% of genes could also be identified in the validation data set.
Inclusion criteria of data sets and samples
Publicly available microarray data sets were selected as validation data sets for this study. Eligible data sets included the following: 1) gene expression data gathered from human nonmetastatic breast cancer sources; 2) continuous time-to-event survival data; 3) at least 40 samples with 15 deaths; 4) measurements of gene expression profiles using U133A GeneChip technologies (Affymetrix/Thermo Fisher Scientific, Santa Clara, CA, USA); and 5) clinical characteristics of the enrolled subjects, including ER levels and LN status.
Evaluation of published prognostic models on independent data
Immune-related gene prognostic models that met the inclusion criteria were evaluated on the included data sets. CEL files containing pixel intensity calculations of the validation data sets were downloaded from public databases (such as the Gene Expression Omnibus). Gene expression levels were normalized based upon background corrections. For each prognostic model, the risk was calculated for each sample in the validation data set. The risk score calculation was defined as follows: score =Σwixi/Σ|wi|, where xi was the expression level of the involved gene in the model, and wi was either +1 or −1, depending on the correlation between the gene and the prognosis of the paired subject described in the original publication.23,24,33
Concordance indices (C-indices) were calculated as previously described.34 For each model tested on each validation data set, the C-index was defined as the probability that a patient was predicted to have a lower risk score when compared to another patient who survived longer than the first patient. A C-index of 0.5 indicated that the model did not predict risk better than random guess. A score of 1 indicated perfect prediction of risk. C-indices generated across all of the validation data sets were synthesized into a summary value using random effects modeling. All computations were completed using R (version 3.3.2).
Stratification by ER status and LN status
The discriminative power of each immune-related prognostic model for predicting breast cancer prognosis was evaluated in patients depending on ER and LN status. Patients were stratified into four subgroups (ER+ versus ER- and LN+ versus LN-). C-indices were computed in each of these four subgroups for every data set and then synthesized into a final assessment.
Statistical analyses
All outcomes were reported with 95% CIs. Statistical heterogeneities between data sets were assessed using χ2 tests, with significance levels set to P<0.05. Statistical tests were two-sided, and P-values <0.05 were considered statistically significant. Random effects modeling was used if there was significant heterogeneity; otherwise, fixed effects modeling was reported.
Results
A systematic review of prognostic immune-related gene signatures for breast cancer was performed. After excluding redundant literature, a total of 393 papers were investigated. From these papers, eight models emerged as candidates that passed the study inclusion criteria and were selected for further analyses (Figure 1). Table 1 summarizes the features of the included models. The performance of each model was evaluated in each of ten data sets that were selected based on above-stated inclusion criteria (Table 2).
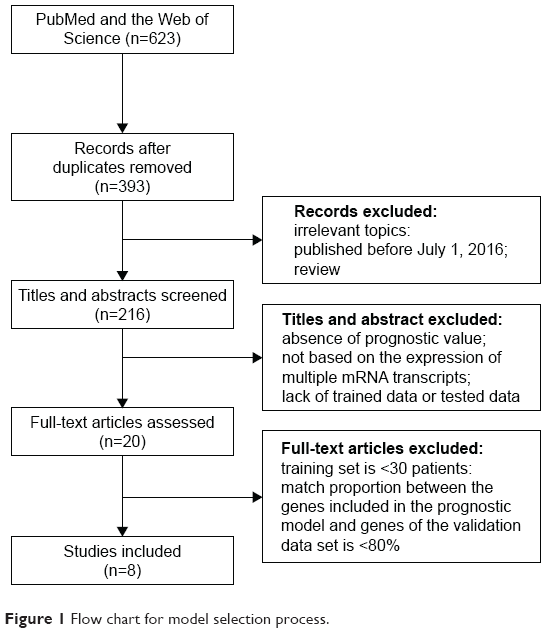 | Figure 1 Flow chart for model selection process. |
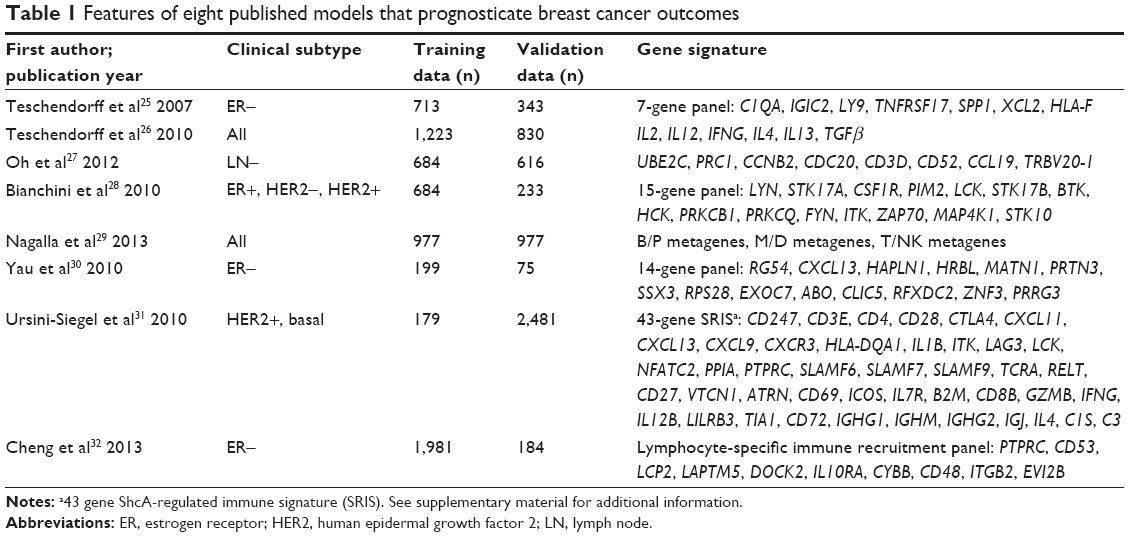 | Table 1 Features of eight published models that prognosticate breast cancer outcomes |
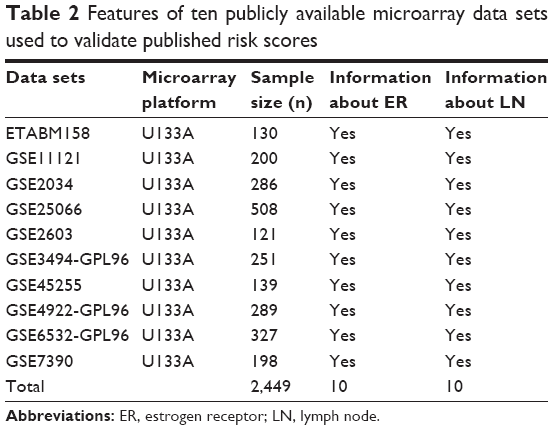 | Table 2 Features of ten publicly available microarray data sets used to validate published risk scores |
Heterogeneity analyses
Heterogeneities across data sets were assessed using each immune-related gene model. Based on the results, C-indices of summary values were synthesized using either random effects or fixed effects models. Both methodologies produced similar summary values, resulting in similar ranking patterns for the immune-related gene models. Given that significant heterogeneity was identified in several models (Table 3), the random effects modeling methodology was chosen to synthesize C-indices across the prognostic models.
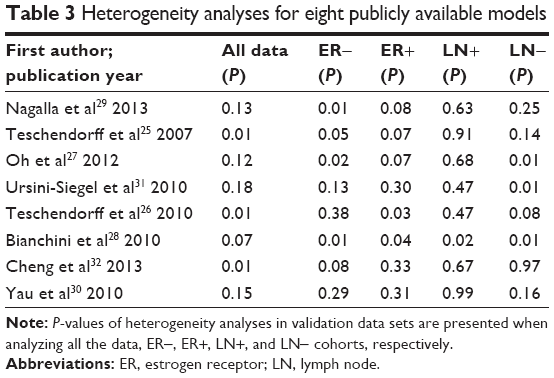 | Table 3 Heterogeneity analyses for eight publicly available models |
Evaluation of prognostic models on independent data sets
Prognostic accuracy of each model was evaluated in every data set, with the exception of the training data set used by the original authors. Evaluations were quantified by C-indices (Figure 2A), and the models were ranked by summary values, from high to low. The top-ranked model was authored by Oh et al,27 with a summary C-index of 0.61. Two other models had better than chance predictive values, with summary C-indices of 0.54 and 0.53, respectively.25,32
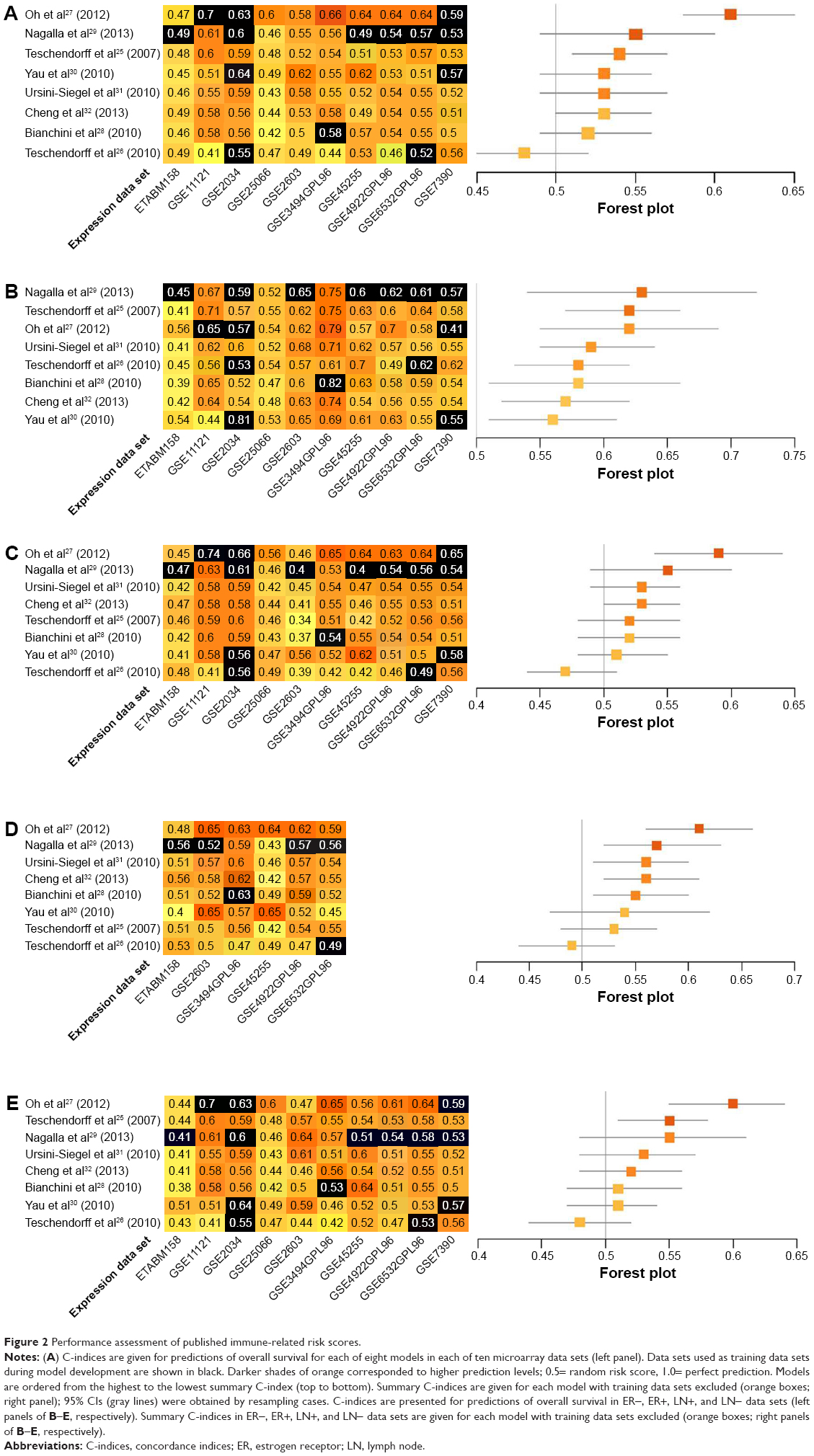 | Figure 2 Performance assessment of published immune-related risk scores. |
Stratification by known prognostic factors
Further analyses were performed within subgroups that were stratified by ER status (ER+ versus ER-), or LN status (LN+ versus LN-). Only a subset of data sets, those in which these factors were recorded, could be assessed. Most immune-related gene models performed better in ER- data sets. The top-ranked three models for ER- breast cancer were authored by Nagalla et al,29 Teschendorff et al,25 and Oh et al.27 These models achieved summary C-indices between 0.62 and 0.63. The remaining five models26,28,30–32 also provided predictions that were better than chance, with summary C-indices varying from 0.56 to 0.59 (Figure 2B). Interestingly, summary C-indices in data from ER+ subjects were lower than in data from ER- subjects. The two top-ranking models for ER+ subjects were those authored by Oh et al27 and Cheng et al32 with summary C-indices of 0.59 and 0.53, respectively. The other six models showed no predictive advantage above chance in ER+ subjects (Figure 2C).
Data were also stratified by the status of invasion to the LNs. The prognostic models performed better in LN+ data sets. The four top-ranking models for LN+ were designed by Oh et al,27 Nagalla et al,29 Ursini-Siegel et al,31 and Cheng et al.32 Summary C-indices in these models ranged from 0.56 to 0.61. In the remaining four models,25,26,28,30 no predictive advantages above chance were observed (Figure 2D). Synthesized C-indices for LN- subjects were lower than those calculated for LN+ subjects. Only two models, originally published by Oh et al27 and Teschendorff et al,25 predicted outcomes better than chance (Figure 2E).
Univariate and multivariate analyses
Univariate analyses were used to investigate correlations between risk scores and the presence of ERs or LN invasion. Risk scores from the prognostic models designed by Nagalla et al,29 Teschendorff et al,25 Cheng et al,32 Bianchini et al,28 Yau et al,30 and Ursini-Siegel et al31 significantly correlated with ER status (range of odds ratios: 1.01–1.06; P<0.05). Risk scores from the models of Oh et al27 and Yau et al30 significantly correlated with LN status (odds ratios: 1.02 and 1.22, respectively; P<0.05). In multivariate analyses that controlled for the status of ER, LN, HER2, and age, DFS was associated with risk scores in the models of Oh et al27 (P=0.0443), Cheng et al32 (P=0.0391), and Bianchini et al28 (P=0.0343). These findings demonstrated that modeling immune-related gene signatures may provide unique information to prognosticate breast cancer cases.
Discussion
Gene expression profile studies on breast cancer have uncovered new insights into the genetic pathways that contribute to tumorigenesis. This research has inspired the development of gene expression signatures to predict patient outcomes. It is generally understood that proliferation-associated genes account for the majority of predictive power of many previously reported prognostic genetic tools, such as the 21-gene Oncotype DX test (Genomic Health, Inc., Redwood City, CA, USA).5,7 However, these prognostic models are only applicable for low proliferation tumors, as found in ER+ and LN- breast cancer subtypes. Therefore, developing new prognostic signatures, independent of proliferation phenotypes, is a challenging unmet need.23,24
In recent years, research revealed that immune-related gene signatures were associated with both clinical outcomes and pathological complete response rates in breast cancer.25,35–37 A whole-transcriptome analysis of the NCCTG-N9831 adjuvant trastuzumab trial in early-stage HER2+ breast cancer revealed that tumors enriched for immune-functioning genes had increased RFS rates. Conversely, trastuzumab did not increase RFS in patients whose tumors lacked elevated immune-functioning gene expression scores. These data suggested that the increased expression of a subset of immune-functioning genes predicted benefit from adjuvant trastuzumab.38 Additionally, the Finland Herceptin trial reported that higher levels of tumor-infiltrating lymphocytes at diagnosis had a significant inverse association with distant recurrence rates in primary ER-/HER2- breast cancers.39
Previously, investigators pooled genomic data to identify predictors of clinical outcomes in breast cancer.25–32 The current study is the largest pooled analysis of immune-related prognostic gene models for breast cancer; all published immune-related prognostic gene models were evaluated. For ER- breast cancer, the top-ranking risk score was achieved by the model of Nagalla et al.29 This model comprised three distinct immunity-related metagenes indicative of tumor–immune cell interactions. The first metagene, the B/P cluster, consisted primarily of immunoglobulin-encoding genes that were expressed in B-cell and plasma cell populations. The second metagene was the T/NK cluster, which contained components of the T-cell receptor–cluster of differentiation 3 complex and granzymes. This was largely restricted to T cells and natural killer cells. The third metagene was the M/D cluster, which included the major histocompatibility complex class II and myeloid-specific markers. These were expressed at the highest levels in monocytes and dendritic cells. This prognostic model covered the most comprehensive array of immune-related genes in the cohort studied here. This may explain why the model of Nagalla et al had the highest prognostic value.
In the current study, seven of ten validation data sets were training data sets used to develop the model of Nagalla et al.29 This reduced the statistical power for the validation of this model. Therefore, the model of Teschendorff et al,25 the second best prognostic model for ER- breast cancer, may have the most clinical value. This model employed a methodology called profile analysis using clustering and kurtosis (PACK). PACK was a semi-supervised algorithm, consisting of two main steps. First, a feature selection criterion was performed. Second, a supervised step correlated the selected features with specific phenotypes.40 Despite being a conservative procedure that removed true positives, PACK efficiently removed false positives, providing more reliable identifications of prognostic markers. Another advantage of PACK was that it tested prognostic genes by applying two statistical tools: singular value decomposition41 and the shrunken centroids classifier.42 Two of the potential prognostic genes identified by the model of Teschendorff et al25 were associated with breast cancer clinical outcomes. Specifically, C1QA, a gene involved in the classical complement pathway, harbored a single nucleotide polymorphism that correlated with distant breast cancer metastases.43 Two other studies also found prognostic value for OPN in metastatic breast cancer.44,45
In the current study, the prognostic model by Oh et al27 ranked first in ER+ breast cancer and third in ER- breast cancer. This was the only model that integrated proliferation- and immunity-related genes into a single prognostic model for nonmetastatic breast cancer patients. Additionally, this model used a large data set, which reduced size-based sampling problems. Prognostic genes were selected based upon both statistical significance and biological relevance. Together, these factors explain the success of the model of Oh et al in prognosticating breast cancer outcomes.
The current study also revealed that immune-related gene models performed best in ER- and LN+ breast cancer subtypes. This finding directly opposed current proliferation-related gene models. ER- and LN+ breast cancers tend to present as high-grade diseases that frequently harbor p53 mutations, leading to worsen prognoses. These molecular and pathological tendencies may explain why differences in clinical outcome within the ER- subgroup cannot be explained by the differential activation of cell cycle pathways. Thus, proliferation-related gene models do not have prognostic power in this breast cancer subtype. Unlike proliferation-related functional gene signatures, immune-related gene expression profiles have more individualized properties. Immune-related models have achieved statistical significance in both high and low proliferation breast neoplasms.29 Therefore, immune-related gene signature–based tools may be advantageous for the prognostication of ER- and LN+ breast cancers. Future use of these tools may complement more established proliferation-based gene expression panels.
It is of high clinical relevance to identify patients who will experience poor outcomes prior to the initiation of therapy. Patients with lower immune-related gene expression scores may require increased follow-up frequency and systemic management after traditional adjuvant therapy (eg, metronomic chemotherapy for triple-negative breast cancer or ovary functional suppression for ER+ breast cancer). Alternatively, these patients might be candidates for future clinical trials evaluating therapeutic approaches to enhance immune activity within breast tumors, such as by targeting checkpoint proteins.46
The coverage of immune-related gene signatures included many critical and well-studied immunomodulatory genes, such as TGFβ, CD28, and CTLA4. Members of the immunomodulatory Siglec gene family were not included in these models. Most of the Siglec family members contain immunoreceptor tyrosine-based inhibitory motifs in regions exposed to cytosolic compartments. Siglec proteins are inhibitory receptors for both innate and adaptive immune cells and may attenuate reductions in tumor cell immunity. CD169/Siglec-1 is bound to cell surface–associated (MUC1) receptors that were expressed on breast cancer cells.47 High densities of CD169+ macrophages in regional LNs surrounding colorectal tumors, endometrial carcinomas, malignant melanomas, and breast neoplasms were associated with improved clinical prognoses. These findings may be due to increases in cytotoxic immune cell infiltrates in these tumors.48–50 Therefore, expression levels of Siglec immunomodulatory gene family members could affect prognostic breast cancer outcomes. Functions of Siglec gene products have been well studied in oncogenic model systems. Further investigations should concentrate on clinical oncology. Future immune-related prognostic gene models for breast cancer should consider assessing Siglec gene expression levels.
The study reported here had several limitations. First, the selected models included only intracellular immune-related genes. Therefore, it remains unknown whether interstitial genes in the microenvironment, such as CCL-18,51 can prognosticate breast cancer patient outcomes. Second, useful gene expression profiles that could be included in this study were limited by incomplete clinical information. This caveat was an unavoidable inadequacy found in all publicly available data sets. Third, evaluations of these gene models were limited to the breast cancer subtypes discussed above. Few authors have investigated the impact of immune-related prognostic gene models on conventional breast cancer subtypes. Few studies have published validation data sets containing clinical information that specifies patient molecular subtypes. Rather, previous studies published information specifying ER status and regional LN involvement. This allowed, in the current study, for the stratification of summary C-indices by these characteristics. The model by Nagalla et al29 was the only tool in which subgroup analyses were performed in subtypes. In those studies, basal-like breast tumors were positively associated with T/NK, M/D, and B/P metagenes. Similarly, luminal B tumors trended toward association with the T/NK metagene and were significantly associated with the M/D metagene. None of the metagenes were significantly associated with the luminal A, HER2-enriched, or claudin-low tumor subtypes.
Despite these limitations, the current analysis revealed the top-ranked immune-related prognostic gene models for ER+, ER-, LN+, and LN- breast cancer subtypes. Moreover, these data showed that immune-related gene signatures had good prognostic value in breast cancer, especially for ER- and LN+ tumors. Therefore, immune-related gene models may be clinically relevant and complementary to current proliferation-related gene models. The use of immune-related gene models may fulfill an unmet need in breast cancer prognostication.
Acknowledgments
This study was supported by the National Natural Science Foundation of China (grant numbers: 81402201, 81372817, and 11671409), the National Natural Science Foundation of Guangdong Province (grant number: 2014A030310070), and a grant ([2013]163) from the Key Laboratory of Malignant Tumor Molecular Mechanisms and Translational Medicine of the Guangzhou Bureau of Science and Information Technology.
Disclosure
The authors report no conflicts of interest in this work.
References
Perou CM, Sorlie T, Eisen MB, et al. Molecular portraits of human breast tumours. Nature. 2000;406(6797):747–752. | ||
Goldhirsch A, Winer EP, Coates AS, et al. Personalizing the treatment of women with early breast cancer: highlights of the St Gallen International Expert Consensus on the Primary Therapy of Early Breast Cancer 2013. Ann Oncol. 2013;24(9):2206–2223. | ||
Coates AS, Winer EP, Goldhirsch A, et al. Tailoring therapies–improving the management of early breast cancer: St Gallen International Expert Consensus on the Primary Therapy of Early Breast Cancer 2015. Ann Oncol. 2015;26(8):1533–1546. | ||
Sotiriou C, Neo SY, McShane LM, et al. Breast cancer classification and prognosis based on gene expression profiles from a population-based study. Proc Natl Acad Sci U S A. 2003;100(18):10393–10398. | ||
Paik S, Tang G, Shak S, et al. Gene expression and benefit of chemotherapy in women with node-negative, estrogen receptor-positive breast cancer. J Clin Oncol. 2006;24(23):3726–3734. | ||
Albain KS, Barlow WE, Shak S, et al. Prognostic and predictive value of the 21-gene recurrence score assay in postmenopausal women with node-positive, oestrogen-receptor-positive breast cancer on chemotherapy: a retrospective analysis of a randomised trial. Lancet Oncol. 2010;11(1):55–65. | ||
Paik S, Shak S, Tang G, et al. A multigene assay to predict recurrence of tamoxifen-treated, node-negative breast cancer. N Engl J Med. 2004;351(27):2817–2826. | ||
Parker JS, Mullins M, Cheang MC, et al. Supervised risk predictor of breast cancer based on intrinsic subtypes. J Clin Oncol. 2009;27(8):1160–1167. | ||
Prat A, Cheang MC, Martin M, et al. Prognostic significance of progesterone receptor-positive tumor cells within immunohistochemically defined luminal A breast cancer. J Clin Oncol. 2013;31(2):203–209. | ||
Martin M, Prat A, Rodriguez-Lescure A, et al. PAM50 proliferation score as a predictor of weekly paclitaxel benefit in breast cancer. Breast Cancer Res Treat. 2013;138(2):457–466. | ||
Filipits M, Nielsen TO, Rudas M, et al. The PAM50 risk-of-recurrence score predicts risk for late distant recurrence after endocrine therapy in postmenopausal women with endocrine-responsive early breast cancer. Clin Cancer Res. 2014;20(5):1298–1305. | ||
Scott E, Mamawala M, Epstein JI, et al. Intermediate and longer-term outcomes from a prospective active-surveillance program for favorable-risk prostate cancer. Tosoian JJ, Mamawala M, Epstein JI, Landis P, Wolf S, Trock BJ, Carter HB. J Clin Oncol. 20;33(30):3379–3385. [Epub 2015 Aug 31]. doi:10.1200/JCO.2015.62.5764. Urol Oncol. 2017;35(3):121–122. | ||
Sestak I, Dowsett M, Zabaglo L, et al. Factors predicting late recurrence for estrogen receptor-positive breast cancer. J Natl Cancer Inst. 2013;105(19):1504–1511. | ||
Aalders KC, Kuijer A, Straver ME, et al. Characterisation of multifocal breast cancer using the 70-gene signature in clinical low-risk patients enrolled in the EORTC 10041/BIG 03-04 MINDACT trial. Eur J Cancer. 2017;79:98–105. | ||
Cardoso F, van’t Veer LJ, Bogaerts J, et al. 70-Gene signature as an aid to treatment decisions in early-stage breast cancer. N Engl J Med. 2016;375(8):717–729. | ||
Makama M, Drukker CA, Rutgers EJ, et al. An association study of established breast cancer reproductive and lifestyle risk factors with tumour subtype defined by the prognostic 70-gene expression signature (MammaPrint®). Eur J Cancer. 2017;75:5–13. | ||
Sestak I, Zhang Y, Schroeder BE, et al. Cross-stratification and differential risk by breast cancer index and recurrence score in women with hormone receptor-positive lymph node-negative early-stage breast cancer. Clin Cancer Res. 2016;22(20):5043–5048. | ||
Sgroi DC, Chapman JA, Badovinac-Crnjevic T, et al. Assessment of the prognostic and predictive utility of the Breast Cancer Index (BCI): an NCIC CTG MA.14 study. Breast Cancer Res. 2016;18(1):1. | ||
Sgroi DC, Sestak I, Cuzick J, et al. Prediction of late distant recurrence in patients with oestrogen-receptor-positive breast cancer: a prospective comparison of the breast-cancer index (BCI) assay, 21-gene recurrence score, and IHC4 in the TransATAC study population. Lancet Oncol. 2013;14(11):1067–1076. | ||
Mathieu MC, Mazouni C, Kesty NC, et al. Breast Cancer Index predicts pathological complete response and eligibility for breast conserving surgery in breast cancer patients treated with neoadjuvant chemotherapy. Ann Oncol. 2012;23(8):2046–2052. | ||
Pusztai L. Gene expression profiling of breast cancer. Breast Cancer Res. 2009;11(Suppl 3):S11. | ||
Cheang MC, Chia SK, Voduc D, et al. Ki67 index, HER2 status, and prognosis of patients with luminal B breast cancer. J Natl Cancer Inst. 2009;101(10):736–750. | ||
Wirapati P, Sotiriou C, Kunkel S, et al. Meta-analysis of gene expression profiles in breast cancer: toward a unified understanding of breast cancer subtyping and prognosis signatures. Breast Cancer Res. 2008;10(4):R65. | ||
Desmedt C, Haibe-Kains B, Wirapati P, et al. Biological processes associated with breast cancer clinical outcome depend on the molecular subtypes. Clin Cancer Res. 2008;14(16):5158–5165. | ||
Teschendorff AE, Miremadi A, Pinder SE, Ellis IO, Caldas C. An immune response gene expression module identifies a good prognosis subtype in estrogen receptor negative breast cancer. Genome Biol. 2007;8(8):R157. | ||
Teschendorff AE, Gomez S, Arenas A, et al. Improved prognostic classification of breast cancer defined by antagonistic activation patterns of immune response pathway modules. BMC Cancer. 2010;10:604. | ||
Oh E, Choi YL, Park T, Lee S, Nam SJ, Shin YK. A prognostic model for lymph node-negative breast cancer patients based on the integration of proliferation and immunity. Breast Cancer Res Treat. 2012;132(2):499–509. | ||
Bianchini G, Iwamoto T, Qi Y, et al. Prognostic and therapeutic implications of distinct kinase expression patterns in different subtypes of breast cancer. Cancer Res. 2010;70(21):8852–8862. | ||
Nagalla S, Chou JW, Willingham MC, et al. Interactions between immunity, proliferation and molecular subtype in breast cancer prognosis. Genome Biol. 2013;14(4):R34. | ||
Yau C, Esserman L, Moore DH, Waldman F, Sninsky J, Benz CC. A multigene predictor of metastatic outcome in early stage hormone receptor-negative and triple-negative breast cancer. Breast Cancer Res. 2010;12(5):R85. | ||
Ursini-Siegel J, Cory S, Zuo D, et al. Receptor tyrosine kinase signaling favors a protumorigenic state in breast cancer cells by inhibiting the adaptive immune response. Cancer Res. 2010;70(20):7776–7787. | ||
Cheng WY, Ou Yang TH, Anastassiou D. Development of a prognostic model for breast cancer survival in an open challenge environment. Sci Transl Med. 2013;5(181):181ra150. | ||
Ignatiadis M, Singhal SK, Desmedt C, et al. Gene modules and response to neoadjuvant chemotherapy in breast cancer subtypes: a pooled analysis. J Clin Oncol. 2012;30(16):1996–2004. | ||
Uno H, Cai TX, Tian L, Wei LJ. Evaluating prediction rules for t-year survivors with censored regression models. J Am Stat Assoc. 2007;102(478):527–537. | ||
Alexe G, Dalgin GS, Scanfeld D, et al. High expression of lymphocyte-associated genes in node-negative HER2+ breast cancers correlates with lower recurrence rates. Cancer Res. 2007;67(22):10669–10676. | ||
Schmidt M, Bohm D, von Torne C, et al. The humoral immune system has a key prognostic impact in node-negative breast cancer. Cancer Res. 2008;68(13):5405–5413. | ||
Bianchini G, Qi Y, Alvarez RH, et al. Molecular anatomy of breast cancer stroma and its prognostic value in estrogen receptor-positive and −negative cancers. J Clin Oncol. 2010;28(28):4316–4323. | ||
Perez EA, Thompson EA, Ballman KV, et al. Genomic analysis reveals that immune function genes are strongly linked to clinical outcome in the North Central Cancer Treatment Group n9831 Adjuvant Trastuzumab Trial. J Clin Oncol. 2015;33(7):701–708. | ||
Loi S, Michiels S, Salgado R, et al. Tumor infiltrating lymphocytes are prognostic in triple negative breast cancer and predictive for trastuzumab benefit in early breast cancer: results from the FinHER trial. Ann Oncol. 2014;25(8):1544–1550. | ||
Li L, Chaudhuri A, Chant J, Tang Z. PADGE: analysis of heterogeneous patterns of differential gene expression. Physiol Genomics. 2007;32(1):154–159. | ||
Alter O, Brown PO, Botstein D. Generalized singular value decomposition for comparative analysis of genome-scale expression data sets of two different organisms. Proc Natl Acad Sci U S A. 2003;100(6):3351–3356. | ||
Tibshirani R, Hastie T, Narasimhan B, Chu G. Diagnosis of multiple cancer types by shrunken centroids of gene expression. Proc Natl Acad Sci U S A. 2002;99(10):6567–6572. | ||
Racila E, Racila DM, Ritchie JM, Taylor C, Dahle C, Weiner GJ. The pattern of clinical breast cancer metastasis correlates with a single nucleotide polymorphism in the C1qA component of complement. Immunogenetics. 2006;58(1):1–8. | ||
Allan AL, George R, Vantyghem SA, et al. Role of the integrin-binding protein osteopontin in lymphatic metastasis of breast cancer. Am J Pathol. 2006;169(1):233–246. | ||
de Silva Rudland S, Martin L, Roshanlall C, et al. Association of S100A4 and osteopontin with specific prognostic factors and survival of patients with minimally invasive breast cancer. Clin Cancer Res. 2006;12(4):1192–1200. | ||
Ali HR, Glont SE, Blows FM, et al. PD-L1 protein expression in breast cancer is rare, enriched in basal-like tumours and associated with infiltrating lymphocytes. Ann Oncol. 2015;26(7):1488–1493. | ||
Nath D, Hartnell A, Happerfield L, et al. Macrophage-tumour cell interactions: identification of MUC1 on breast cancer cells as a potential counter-receptor for the macrophage-restricted receptor, sialoadhesin. Immunology. 1999;98(2):213–219. | ||
Ohnishi K, Komohara Y, Saito Y, et al. CD169-positive macrophages in regional lymph nodes are associated with a favorable prognosis in patients with colorectal carcinoma. Cancer Sci. 2013;104(9):1237–1244. | ||
Ohnishi K, Yamaguchi M, Erdenebaatar C, et al. Prognostic significance of CD169-positive lymph node sinus macrophages in patients with endometrial carcinoma. Cancer Sci. 2016;107(6):846–852. | ||
Shiota T, Miyasato Y, Ohnishi K, et al. The clinical significance of CD169-positive lymph node macrophage in patients with breast cancer. PLoS One. 2016;11(11):e0166680. | ||
Chen J, Yao Y, Gong C, et al. CCL18 from tumor-associated macrophages promotes breast cancer metastasis via PITPNM3. Cancer Cell. 2011;19(4):541–555. |
Supplementary material
Full forms of genes provided in Table 1
STAT1: Signal transducer and activator of transcription 1
IFN: Interferons
IR: Immune response gene
C1QA: Complement C1q subcomponent subunit A
LY9: T-lymphocyte surface antigen Ly-9
TNFRSF17: Tumor necrosis factor receptor superfamily member 17
SPP1: Bone sialoprotein I
XCL2: Chemokine (C motif) ligand 2
HLA-F: HLA class I histocompatibility antigen, alpha chain F
IL2: Interleukin 2
IL12: Interleukin 12
IFNG: Interferon gamma
IL4: Interleukin 4
IL13: Interleukin 13
TGFB: Transforming growth factor beta
UBE2C: Ubiquitin-conjugating enzyme E2 C
PRC1: Protein regulator of cytokinesis 1
CCNB2: G2/mitotic-specific cyclin-B2
CDC20: The cell-division cycle protein 20
CD3D: T-cell surface glycoprotein CD3 delta chain
CD52: Cluster of differentiation 52
CCL19: Chemokine (C-C motif) ligand 19
TRBV20-1: T-cell receptor beta variable 20-1
LYN: LYN Proto-oncogene, Src family tyrosine kinase
STK17A: Serine/threonine kinase 17a
CSF1R: Colony-stimulating factor 1 receptor
PIM2: Pim-2 proto-oncogene, serine/threonine kinase
LCK: LCK proto-oncogene, Src family tyrosine kinase
STK17B: Serine/threonine kinase 17b
BTK: Bruton tyrosine kinase
HCK: HCK proto-oncogene, Src family tyrosine kinase
PRKCB1: Protein kinase C beta
PRKCQ: Protein kinase C theta
FYN: FYN proto-oncogene, Src family tyrosine kinase
ITK: IL2 Inducible T-cell kinase
ZAP70: Zeta chain of T-cell receptor associated protein kinase 70
MAP4K1: Mitogen-activated protein kinase 1
STK10: Serine/threonine kinase 10
B/P metagenes: B/P cluster consisted of genes (mostly immunoglobulin-encoding genes)
M/D metagenes: Genes of M/D cluster (including major histocompatibility complex class II and myeloid-specific markers)
T/NK metagenes: Expression of genes in T/NK cluster (such as components of the T cell receptor–cluster of differentiation 3 complex and granzymes)
CXCL13: C-X-C motif chemokine ligand 13
HAPLN1: Hyaluronan and proteoglycan link protein 1
HRBL: Arf-GAP domain and FG repeats-containing protein 2
MATN1: Matrilin 1, cartilage matrix protein
PRTN3: Proteinase 3
SSX3: SSX family member 3
RPS28: Ribosomal protein S28
EXOC7: Exocyst complex component 7
ABO: Alpha 1-3-N-acetylgalactosaminyltransferase and alpha 1-3-galactosyltransferase
CLIC5: Chloride intracellular channel 5
RFXDC2: Regulatory factor X domain containing 2
ZNF3: Zinc finger protein 3
PRRG3: Proline rich and Gla domain 3
CD247: CD247 molecule
CD3E: CD3e molecule
CD4: CD4 molecule
CD28: CD28 molecule
CTLA4: Cytotoxic T-lymphocyte associated protein 4
CXCL11: C-X-C motif chemokine ligand 11
CXCL13: C-X-C motif chemokine ligand 13
CXCL9: C-X-C motif chemokine ligand 9
CXCR3: C-X-C motif chemokine receptor 3
HLA–DQA1: Major histocompatibility complex, Class II, DQ alpha 1
IL1B: Interleukin 1 beta
ITK: IL2 Inducible T-cell kinase
LAG3: Lymphocyte activating 3
LCK: LCK Proto-oncogene, Src family tyrosine kinase
NFATC2: Nuclear factor of activated T-cells 2
PPIA: Peptidylprolyl isomerase A
PTPRC: Protein tyrosine phosphatase, receptor type C
SLAMF6: SLAM family member 6
SLAMF7: SLAM family member 7
SLAMF9: SLAM family member 9
TCRA: T-cell receptor alpha locus
RELT: RELT, TNF receptor
CD27: CD27 molecule
VTCN1: V-set domain containing T-cell activation inhibitor 1
ATRN: Attractin
CD69: CD69 molecule
ICOS: Inducible T-cell costimulator
IL7R: Interleukin 7 receptor
B2M: Beta-2-microglobulin
CD8B: CD8b molecule
GZMB: Granzyme B
IL12B: Interleukin 12B
LILRB3: Leukocyte immunoglobulin like receptor B3
TIA1: TIA1 Cytotoxic granule associated RNA binding protein
CD72: CD72 molecule
IGHG1: Immunoglobulin heavy constant gamma 1 (G1m marker)
IGHM: Immunoglobulin heavy constant Mu
IGHG2: Immunoglobulin heavy constant gamma 2 (G2m marker)
IGJ: A J chain is a protein component of the antibodies IgM and IgA.
C1S: Complement C1s subcomponent
C3: Complement C3
PTPRC: Protein tyrosine phosphatase, receptor type C
CD53: CD53 molecule
LCP2: Lymphocyte cytosolic protein 2
LAPTM5: Lysosomal protein transmembrane 5
DOCK2: Dedicator of cytokinesis 2
IL10RA: Interleukin 10 receptor subunit alpha
CYBB: Cytochrome B-245 beta chain
CD48: CD48 molecule
ITGB2: Integrin subunit beta 2
EVI2B: Ecotropic viral integration site 2B
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.