Back to Journals » Cancer Management and Research » Volume 13
Prognostic Alternative Splicing Signatures in Esophageal Carcinoma
Authors Dlamini Z ![]() , Hull R
, Hull R ![]() , Mbatha SZ, Alaouna M
, Mbatha SZ, Alaouna M ![]() , Qiao YL
, Qiao YL ![]() , Yu H
, Yu H ![]() , Chatziioannou A
, Chatziioannou A ![]()
Received 5 February 2021
Accepted for publication 6 April 2021
Published 4 June 2021 Volume 2021:13 Pages 4509—4527
DOI https://doi.org/10.2147/CMAR.S305464
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Eileen O'Reilly
Zodwa Dlamini,1 Rodney Hull,1 Sikhumbuzo Z Mbatha,2 Mohammed Alaouna,1,3 You-Lin Qiao,1,4 Herbert Yu,1,5 Aristotelis Chatziioannou1,6,7
1SAMRC Precision Prevention & Novel Drug Targets for HIV-Associated Cancers Extramural Unit, Pan African Cancer Research Institute, University of Pretoria, Pretoria, South Africa; 2Department of Surgery, Steve Biko Academic Hospital, University of Pretoria, Pretoria, South Africa; 3Department of Internal Medicine, Faculty of Health Sciences, University of the Witwatersrand, Johannesburg, South Africa; 4Cancer Institute/Hospital, Chinese Academy of Medical Sciences and Peking Union Medical College, Beijing, 100021, People’s Republic of China; 5University of Hawaii Cancer Center, Honolulu, HI, USA; 6Center of Systems Biology, Biomedical Research Foundation Academy of Athens, Athens, Greece; 7e-NIOS Applications PC, Kallithea, Athens, 17676, Greece
Correspondence: Zodwa Dlamini
Pan African Cancer Research Institute (PACRI), University of Pretoria, Pretoria, South Africa
Tel +27 761474878
Email [email protected]
Abstract: Alternative splicing (AS) is a method of increasing the number of proteins that the genome is capable of coding for, by altering the pre-mRNA during its maturation. This process provides the ability of a broad range of proteins to arise from a single gene. AS events are known to occur in up to 94% of human genes. Cumulative data have shown that aberrant AS functionality is a major factor in human diseases. This review focuses on the contribution made by aberrant AS functionality in the development and progression of esophageal cancer. The changes in the pattern of expression of alternately spliced isoforms in esophageal cancer can be used as diagnostic or prognostic biomarkers. Additionally, these can be used as targets for the development of new treatments for esophageal cancer.
Keywords: esophageal squamous cell carcinoma, adenocarcinoma, splice variants, cell surface receptors, therapeutic targets, biomarkers
Introduction
Esophageal carcinoma is the ninth most prevalent disease reported in 2017, with about 16,940 newly diagnosed cases and an estimated 15,690 fatalities, making it the sixth leading cause of cancer death worldwide.1,2 The most frequent forms of esophageal cancer can be classified into two major histological subtypes. The most widespread in North and Western Europe, North America and Oceania is esophageal adenocarcinoma. In South and Central Asia, including China, the most widespread form is esophageal squamous cell carcinoma (ESCC).3,4 With numerous advancements in screening and multidisciplinary therapy, the average survival rate for 5 years for esophageal cancer varies between 40 and 59%.5 This high incidence rate and low level of accurate prediction combined with difficulties in diagnosis make markers for cancer of the esophagus a prime area for innovative scientific research. These biomarkers would assist in the early diagnosis and determine the correct treatment for each specific patient.6,7
Alternative splicing (AS) is one of the most important post-transcriptional regulatory pathways8 and plays an essential role to increase protein complexity.9 AS is found to occur in up to 94% of human genes.10 A particular pre-RNA is spliced to give rise to distinct isoforms that are found to have unique expression patterns in multiple tissues and stages of development.11 Dysregulation of AS may therefore affect important biological processes and, consequently, disease-related pathophysiology.12 Common splicing disorders have become more and more apparent and therefore could represent appealing molecular markers of tumorigenesis.13 Invasion and metastasis, apoptosis, hypoxia, alterations in metabolism, angiogenesis, and the escape from immunity take place in a number of oncogenic processes.14,15 Moreover, changes in the expression of several critical splice factors can lead to many changes in the AS of target genes. These target genes act to give cancer cells advantages in growth or survival.16 Aberrant AS can therefore be regarded as another feature of cancer, and research into systemic AS could provide possible biomarkers for malignancies.17
Alternate Splicing in Esophageal Cancer
The Mixture of Isoforms (MISO) online tool uses probability frameworks to quantify the expression level of alternatively spliced genes. In order to perform this function, the tool requires RNA sequence data to assign a probability score that specific reads arose from differently spliced isoforms of the same mRNA species.18 A recent study that aimed to quantify and track changes in alternative splicing that arise during the development and progression of esophageal cancer, made use of the MISO model. By analyzing the alternative splicing events in healthy esophageal tissue from patients and comparing that to alternative splicing events in esophageal cancer tissue, identified 45,439 splicing events in normal esophageal tissue. When comparing this to esophageal cancer samples, changes in the splicing pattern of 13.25% of these alternately spliced genes were detected.19 The authors then performed a similar analysis on an esophageal cancer cell line, comparing it to a normal esophageal cell line, 32,891 splicing events were identified. Of these splicing events, 2.8% showed changes in their splicing profile.19 Another study examining the difference between esophageal cancer tissue and normal esophageal tissue from 79 patients identified 2326 AS events in 1738 genes and the alternate splicing in 1360 genes were determined to be significantly associated with overall survival of esophageal cancer patients.20
Most of the oncogenic splicing events in ESCC seem to be related to the increased expression of isoforms related to increased proliferation, altered cell junction, and increased cell migration.20 These multiple splicing events that are a characteristic of the transition of normal esophageal tissue to esophageal cancer and the subsequent progression and growth of the tumor, have led to many of these splice variants being proposed as viable biomarkers for the diagnosis and prognosis of esophageal cancer. The names of the genes and details of the splicing events are given in Table 1. Examples of some of these splice variants will be discussed in the following sections.
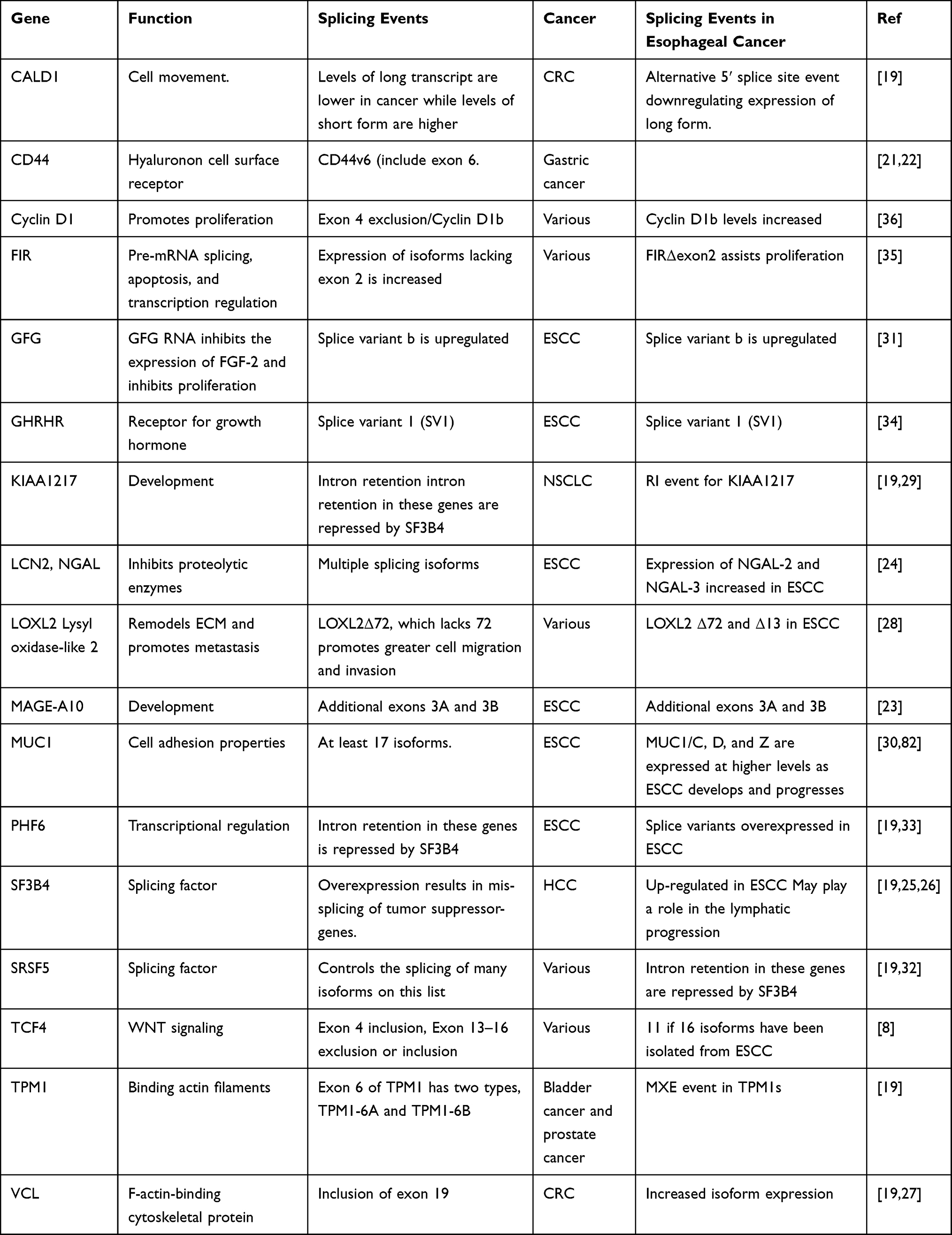 |
Table 1 Splice Variants Which May Be Used as Biomarkers and Therapeutic Targets in Esophageal Cancer |
Alternate Splicing of Cell Surface Receptors in Esophageal Cancer
CD44
CD44 is the dominant cell surface receptor for hyaluronan.37 This receptor can be alternatively spliced to give rise to 19 isoforms. Most of these isoforms arise due to alternate splicing of the extracellular region with up to 16 exons making up this region.38 CD44 variants play a part in the development and progression of malignancies.39 (Figure 1). In particular, different isoforms of CD44 are found to be expressed at higher levels at different stages of various cancers. For example, variants that retain exon 6 were shown to be expressed at higher levels in tumors with increased growth and metastatic abilities.40–42 Increased expression of isoforms retaining exon 9 is associated with increased risk of tumor development and metastatic processes in epidermal tissue.22 Cd44 variants with exons 9 and 6 are present in typical squamous epithelium and esophageal squamous cell carcinoma (ESCC).22 At the same time, almost all patients evaluated recorded elevated levels of EGFR expression in esophageal carcinomas.22
 |
Figure 1 Splice isoforms of CD44. CD44 is alternately spliced to give rise to 19 isoforms. Variants that retain exon 6 are expressed at higher levels in multiple cancers. Additionally, isoforms that also retain exon 9 in addition to retaining exon 6 are found to be expressed at higher levels in squamous epithelial cells. These include isoforms 1, 3, 5, 6, 7, 8, 9, 16 and 17. Those variants containing exon 6 play a role in cancer metastasis and a decrease in the expression of these isoforms inhibits cancer growth. This exon codes for the beginning of the proteins stem structure. A region that is predicted to be modified by glycosylation and is the beginning of the region used to make antibodies against CD44. Only the severely truncated isoform. Isoform 2 lacks the LINK domain. This domain binds to hyaluronic acid and is important in blood cell migration and apoptosis. |
Various studies have been performed on the function of CD44v6 (variants containing exon 6) in human malignancies, which suggest an essential role of these isoforms in the spread of cancers.40–42 Furthermore, the expression of CD44v6 was reduced significantly in irradiated ESCC.22 Decreased expression of CD44v6 and EGFR may inhibit the growth of tumors and reduce the metastatic risk in esophageal cancer patients.22 Previous studies showed that exon 9 containing Cd44 variants were constitutively expressed in normal esophageal mucosal cells.22 Expression of EGFR and CD44v9 isoforms is predicted to be reduced in irradiated esophageal carcinomas. The expression of the EGFR and CD44v6 and v9 molecules might be useful biomarkers for predicting the metastatic potential of upper esophageal tract carcinomas as well as serving as prognostic markers.22
MAGE-A10
Initially, MAGE-A10 (Melanoma Antigen Gene -A10) was detected by a cytotoxic T lymphocyte reactive assay against autologous melanoma cells.43 Indeed, on chromosome Xq28 there are 12 closely related genes in the Mage-a subfamily.44 Members of the MAGE protein family play an important role in the development of germ cells and homologous proteins are present in mice. These genes are expressed in the mouse embryos and are called SMAGE.45 Changes in the expression of MAGE-A1, MAGE-A2, MAGE-A3, MAGE-A4, MAGE-A6, MAGE-A10 and MAGE-A12 have been shown to change in primary tumors and cancer cell lines.46 A highly attractive target of immunotherapy is the tumor-specific expression of some MAGE proteins in cytotoxic lymphocytes (CTL).47–49 MAGE-A3 peptides were induced in clinical trials in 28–55% of metastatic melanoma patients with an immune response and tumor regression,50,51 and MAGE-A10 peptide trials have already been conducted.47 This increased expression of MAGE-A1, MAGEA2b, MAGE-A3, MAGE-A4, MAGE-A6, MAGE-A9, MAGEA10, and MAGE-A12 was demonstrated in esophageal adenocarcinoma through the use of oligonucleotide microarrays.23 A similar protein whose expression changes in tumors is Necdin which is a 325-amino acid protein homolog of MAGE. It shares 30% sequence homology with MAGE. It functions by binding the trans-activated domain of p53 and can inhibit apoptosis under some circumstances.52 The MAGE-A10 gene was originally thought to consist of only four exons.23 However, alternate splice variants with additional exons 3A and 3B were identified using RT-PCR with primers crossing exons 1 through 4, amplifying fragments of 260-and 330-bp23 (Figure 2).
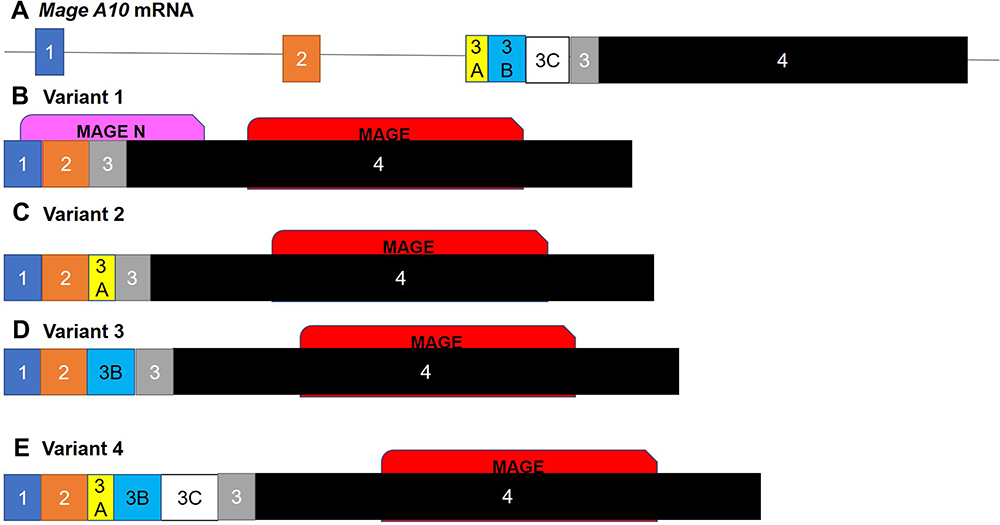 |
Figure 2 Isoforms of MAGE-A10. The alternately spliced isoforms of the (A) MAGE-A10 mRNA. (B) The canonical sequence consists of four exons; the additional variants consist of additional exon 3 variants being included in addition to the original exon 3. (C) Variant 2 contains exon 3A, (D) variant 3 contains exon 3B and (E) variant 4 contains exons 3A, 3B and 3C. The insertion of these extra exons disrupts the folding of the initial N terminal MAGE domain. These domains regulate many developmental processes as well as stress response and the lack of this initial domain may decrease the functionality of these isoforms. |
The sequences in these alternative segments differ from the canonical sequence in the intron section between exons 2 and 3 and results in a number of different expressed sequence tags (ESTs). Searching the EST database with BLAST, using these sequences from MAGE-A2 and MAGE-A6, yielded alternative splice variants that also involve alternative exon variants in the untranslated 5ʹ area. Variant 3, containing exon 3B, shares 83% homology with other MAGE-A2 variants. All MAGE-A10 alternate splice variants seem to play a role in stabilizing mRNA.53
Lipocalin Receptor Neutrophil-Associated Gelatinase (NGAL)
Lipocalins are a varied family of proteins with low overall levels of sequence homology which nonetheless share the same tertiary structural elements. These include the cup-shaped inner ligand binding area with a hydrogen-bonded β-barrel.54,55 This enables lipocalins to combine with a wide number of small, primarily hydrophobic molecules, and to form covalent and non-covalent complexes with other soluble macromolecules.56 One of the members of the Lipocalin family is Lipocalin-2 (LCN2), which is also known as Neutrophil gelatinase-associated lipocalin (NGAL).57 NGAL is expressed in most tissues, aside from neutrophils, and its expression is induced in epithelial cells by inflammation.58,59 NGAL participates in many cellular processes. It interacts with proteolytic enzymes, such as Matrix metallopeptidase 9 (MMP-9), inhibiting the function of these proteins, preventing them from breaking down the extracellular matrix (ECM).60,61 NGAL is also capable of binding iron-binding molecules and are thereby capable of acting as a strong bacteriostatic agent through iron sequestration. It may also be associated with the adaptive immune system and the response to acute infection.62,63 NGAL has also been identified as a survival factor, by preventing apoptosis induced by acute ischemic renal injury.64,65 During the production of primary renal tubular epithelial cells, NGAL may also be important for the delivery of iron to cells.66,67
Previous studies revealed that NGAL is alternately spliced to give rise to multiple isoforms, the expression levels of which vary in different tissues. Additionally, the levels of various NGAL isoforms expressed in carcinoma cells depend on the tissue of origin24. Ngal-2 mRNA was present in precancerous esophageal cells.68 However, it was not the only Ngal isoform present in these cancer cells. A new Ngal isoform, called Ngal-3, was also found to be expressed in esophageal cancer cells. This isoform codes for a 207-amino acid protein, which is similar to NGAL-2, which has a distinct 32-amino-acid C terminus with a 175 amino acid sequence at the N-terminal (Figure 3(I)).24 RT-PCR was used to establish that the levels of the Ngal-3 RNA transcript, were found to be high in cancer cells. High levels of this isoform are also found in the normal tissue surrounding tumor cells.24. The expression of NGAL-3 is up-regulated in 70% of esophageal carcinoma cases compared to normal nearby epithelium, while NGAL- 2/1 was up-regulated in just 55% of cases. This finding indicates that these novel NGAL isoforms may play an important role in esophageal carcinoma.24
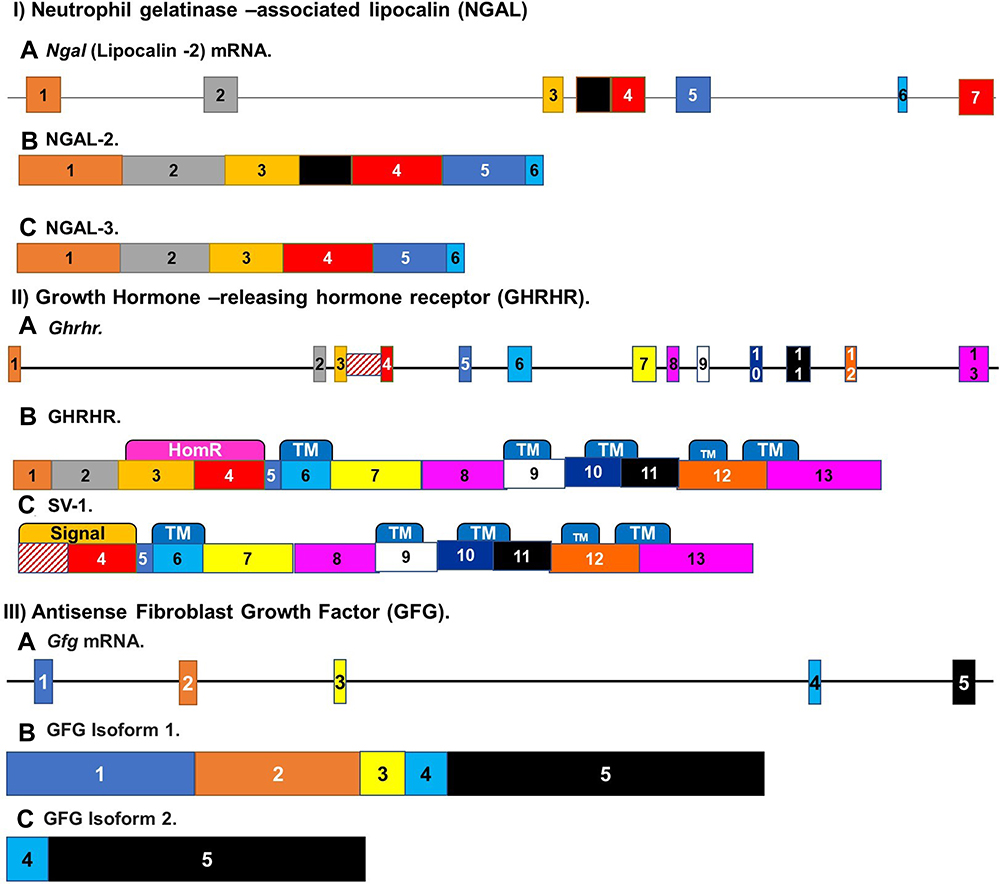 |
Figure 3 Alternate splicing of receptors involved in esophageal cancer. (I) Alternatively, spliced isoforms of Neutrophil gelatinase-associated lipocalin. Ngal mRNA (A) is alternately spliced to give rise to multiple isoforms. Two that are of interest in ESCC are NGALR-2 (B) and NGALR-3 (C). Ngalr-3 codes for a 207-amino acid protein, while Ngalr2 has a distinct 32-amino-acid C terminus with a 175 amino acid sequence at the N-terminal. (II) Alternate splicing of Growth-hormone-releasing hormone receptor (Ghrhr) mRNA is alternately spliced to give rise to multiple variants. In comparison to the (B) canonical protein (C) The Splice variant 1 (SV1) is over-expressed in esophageal squamous cell carcinoma. This variant lacks most of the first three exons apart from a retained fragment of intron 3. (III) Alternate splicing of the Antisense Fibroblast Growth Factor GFG. (A) Gfg mRNA, the antisense FGF, is alternately spliced to give rise to at least six transcripts. (B) The consensus sequence and three other transcripts interact with the FGF-2 transcript and play a part in FGF-2 control. The predominant FGF-AS mRNA expressed in esophageal tumors was (C) isoform 2. |
Structural predictions show that the protein generated by the Ngal-3 isoform consists of four predicted transmembrane domains and an extracellular N-terminal with two potentially N-related glycosylation sites that are essential for the secretion and folding of the protein. This indicates that the new isoform is also localized to the membrane and most likely has a similar activity to other NGAL isoforms.24 The mouse homolog of this isoform, 24p3 was shown to be secreted from the cell through endosome recycling mechanisms.24 This implies that NGAL isoforms may have an endocytosis-like mechanism. Further studies indicate that NGAL and NGAL-3 can form complexes in mammalian cells.24 NGAL and NGAL-2 have also been found to co-localize, indicating that it may interfere with NGAL interactions.24
Growth-Hormone-Releasing Hormone Receptor (GHRHR)
Growth-hormone-releasing hormone receptor (GHRHR) is a G protein coupled receptor whose expression is increased in most cancers. It acts as a receptor for growth hormone. This receptor is alternately spliced to give rise to multiple variants. The Splice variant 1 (SV1) is over-expressed in esophageal squamous cell carcinoma (Figure 3(II)).69 Treatment of these cancers has been successful using GHRHR antagonists since certain forms of tumors have a high pGHRH-R content that reacts to antagonists of GHRH and GHRH-R.70–72 It has been established that these antagonists act on the SV-1 splice variant to inhibit cancer growth and development.69 The expression of this splice variant is also increased in response to hypoxia. The response to hypoxia is able to increase the chances of developing ESCC. The hypoxia-induced increase in GHRHR SV1 expression may help to contribute to the development of esophageal squamous cell carcinoma (ESCC).
The SV-1 variant lacks most of the first three exons which are substituted by a retained fragment of intron 3 compared to GHRH-R, the remainder of which is the same as the pGHRH-R. The SV1 protein product is differed from the full-length receptor at the N-terminal extracellular domain which could serve as the proposed signal peptide34.
FGF Antisense Gene
Human fibroblast growth factor (fgf) has been mapped to chromosome 4q26, an area that is regularly affected during the development and progression of esophageal cancer.73,74 This gene is bi-directionally transcribed coding for FGF-2 (fibroblast growth factors-2) on the sense strand and FGF-AS/GFG (antisense RNA transcript), henceforth referred to as GFG, on the antisense strand.
FGF-2 plays a role in developmental, anti-apoptotic and survival activities.75 Overexpression of FGF-2 is associated with tumor recurrence and reduced survival after surgical resection of esophageal cancer, and these risks are reduced in tumors co-expressing the FGF antisense gfgRNANA. Gfg RNA participates in the control of the expression of FGF-2 and inhibiting the proliferation of FGF-2 expressing cells.76–79 Evidence from studies in rats indicate that gfg RNA inhibited FGF-2 expression.78
Gfg is also alternately spliced to give rise to at least six transcripts (Figure 3(III)). Four of these (a-d) encode isoforms of new proteins that contain the GFG nudix motif. The Nudix Box motif is found in enzymes that catabolize oxidized nucleotides and other possibly toxic compounds.31,80 The 35 and 25 kDa proteins are coded for by the two longest ORFs (GFGa and GFGb).80 Only GFGc and -d vary in their 3′ untranslated regions and are expected to encode GFG proteins of approximately 18.2 kDa. All four transcripts interact with the FGF-2 transcript and play a part in FGF-2 control.31 Three of these alternatively spliced GFG transcripts encoding GFG/NUDT6 isoforms with distinct N termini were detected in various human tissues including esophageal adenocarcinoma. These isoforms were also found to have different subcellular localizations. hGFGa is localized to mitochondria by an N-terminal targeting sequence (NTS), whereas hGFGb and hGFGc were localized in the cytoplasm and nucleus. The predominant GFG mRNA expressed in esophageal tumors was splice variant b. GFG immunoreactivity was detected in the cytoplasm of all esophageal adenocarcinomas and in 88% of tumor cell nuclei. It has been noted that increased expression of GFG b is associated with a decrease in the growth of esophageal adenocarcinoma cells, along with a decrease in cell growth.31
Muc1 Splice Variants
The Mucin-1 receptor is a soluble cytokine receptor that regulates a broad variety of physiological and pathological disorders.81 MUC1 or polymorphic epithelial mucin (PEM) is considered to be a molecular marker for potential medicinal applications and and increased expressioncan result in the progression of ESCC. Ye et al found that MUC1 helps build a 13-metalloproteinase matrix protein, enhancing ECM breakdown and helps propagate ESCC.82 MUC1 variations have been identified in connection with regional metastases of lymph nodes and in relation to bad overall prognosis.30 Usually, MUC1 is present in most epithelial tissues, but is strongly expressed across a range of malignancies. Mucin1 is alternately spliced to give rise to at least 17 isoforms. Three of these, MUC1/C, D, and Z are expressed at higher levels as ESCC develops and progresses. At the same time, the canonical variant MUC1 b has an anti-cancer effect.30
Transcription Factors
FIR (PUF60)
In several tumors, far upstream binding protein 1 (FUBP1), the c-myc gene transcription activator, is activated.83 FBP interacting repressor (FIR) is a multifunctional protein involved in transcriptional repression of the c-myc-gene.84 The protein is also known as Poly(U)-binding-splicing factor (PUF60). The fi/puf60r mRNA is alternately spliced to give rise to 6 isoforms. The expression of isoforms lacking exon 2 is found to be expressed at higher levels in various cancers84,85. The correct splicing of fir relies on the splicing factor SAP155.85,86 The inhibition of the association of this splicing factor and fir results in an abnormality in splicing of fir that affects the regulation of c-myc leading to proliferation and cell death.87 On the other hand, in hematopoietic tumors, F-box protein and WD repeat domain 7 is also mutated (FBW7).88 FBW7 is a member of the ubiquitin ligase complex of the Skp1-Cull-F-box, which induces degradation via the proteasome of various growth-related proteins such as Notch1, C-Myc and C-Jun and E.86,89,90
The AS of Firs is strongly associated with the inhibition of the binding pocket of FBW7. This was found to be a critical factor for ESCC proliferation and is closely correlated with the AS expression of FIRs.35 The importance of FIRΔexon2 was identified at the mRNA and protein levels in the ESCC tissues (Figure 4).
 |
Figure 4 Alternate splicing of Poly(U)-binding-splicing factor (PUF60). The puf60 mRNA is alternately spliced to give rise to 6 isoforms. Isoforms lacking exon 2 are found to be expressed at higher levels in cancer. |
Human Transcription Factor 4 TCF4
The accumulation of β-catenin in the cytoplasm and nucleus is a common occurrence in ESCC cells.91,92 Normally, the levels of β-catenin within the cytoplasm is very low, being controlled through the degradation of β-catenin through phosphorylation and the activity of degradation complexes.8 TCF factors (T-cell factors), which include lymphoid enhancer-binding factor (LEF1), TCF1, TCF3 and TCF4, transfer signals using Wnt/β-catenin to induce expression of downstream target genes in the canonical Wnt/β-catenin pathway.8 Glycogen synthase Kinase 3 β, casein kinase I and axin may also lead to the activation of the β-catenin/TCF pathway within ESCC through over-expression of lymphoma T-cell-1 or End binding Protein 1.10,93 The activation of the β-catenin/TCF pathway in conjunction with crosstalk between this pathway and the upregulation of the STAT3 pathway induces the development and promotes the progression of ESCC.94. The Wnt/β-catenin signal pathway regulates a range of biological processes. The post-transcriptional modification of mRNAs is one of its key effects in a given cell.8 Many of its target genes are regulated through these modifications. These downstream targets include numerous transcription coactivators, such as TCF/LEF, cAMP/E1A binding protein p300 and Pygopus 2.95
Tcf 4 mRNA transcripts are alternatively spliced, and the expression of these isoforms varies in different forms of cancer. These include colorectal, brain and renal cancers.96–98 Alternate splicing of this gene has also been observed to take place in type 2 diabetes99–101 and in other animal models such as mice and zebrafish.102,103 Sixteen isoforms have been identified, but only 11 have been isolated from ESCC.8
These isoforms differ most widely in three regions. These include the exclusion or inclusion of exon 4, which encodes for a 23 amino acid region.8 The second is the exclusion of exon 13 due to a 3ʹ-end splice donor.104 It includes the N-terminal component of TCF4, and the binding domain for β-catenin. Isoforms lacking exon 13 are a key factor for negative Wnt signaling. The final region is that encoded by exons 13–16, resulting in a highly divergent C-terminal and is found in the TCF4L, TCFM and TCF4S isoforms8 (Figure 5). Transcripts lacking exon 16 have been reported to be prevalent in brain tumors,105 while exon 14A and 14B are mutually exclusive and both are 73bp.8
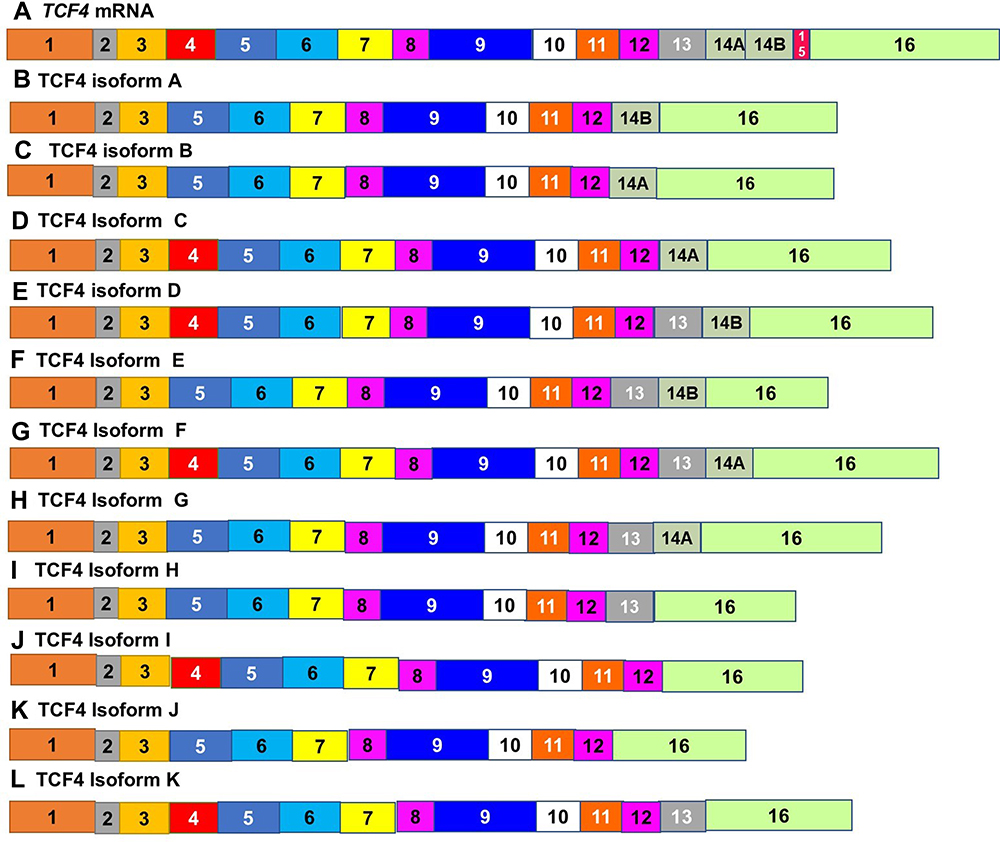 |
Figure 5 Alternately spliced isoforms of Transcription Factor 4. (A) Tcf4 mRNA is alternately spliced to give rise to multiple variants. The expression of 16 isoforms of TCF4 varies in different forms of cancer. Only11 isoforms (B–L) have been isolated from ESCC. These isoforms differ most widely in the inclusion of exon 4 (A–C, F, H, I and K), the exclusion of exon 13 (B–D, J and K) and the region encoded by exons 14~16. |
The N-terminal portion of TCF4 consists of the β-catenin binding domain. In the absence of the HMG-box, TCF4N prevents the activation of promoters due to its inability to bind DNA8 Expression of TCF4N in esophageal cancer has been found to be less regulated than in neighboring tissues. Since the Wnt/ß-catenin pathway is triggered in esophageal cancer. Downregulation of TCF4N can result in tumorigenesis.8 These isoforms can be classified into three different groups based on their domain composition, TCF4E, TCF4M and TCF4S,102 The E-type transcripts contain the entire C-clamp and the two CtBP-binding motifs. The S type transcripts have the partial C-clamp, while the M-types completely lacks any C-clamps.8 Depending on the relationship with other proteins, the TCF4 protein will serve as either transcription activators or repressors.8 Transcriptional activation is partly mediated by β-catenin, which interacts with TCF4 directly through its N-terminal domain.106
Other Genes Alternately Spliced in Esophageal Cancer
Cyclin D1b
Cyclin D1 interacts with Cyclin Dependent Kinase (CDK) to stimulate the progression of the cell cycle, allowing cells to enter the S phase. It has been found to be overexpressed in a number of cancers including cancer of the esophagus, breast, and pancreas.107–112 Its function is negatively regulated through its nuclear export. Human cyclin D1 undergoes alternating splicing that creates a specific D1 transcript, Cyclin D1b (Figure 6).109 The resulting protein lacks the necessary COOH-terminus sequence that regulates the export of nuclear cyclin D1.36 Cyclin D1b mRNA aligned with G/A polymorphism at the exon 4/intron 4 boundary at codon 870.36,109,110,113 The splice-donor, splice-acceptor chain will change this polymorphism and thus affect the frequency of intron excision. Cyclin D1 normally acts to decrease proliferation, while Cyclin D1b appears to act against Cyclin D1. Cyclin d1b mRNA is normally found at low levels in a number of cell types.36 Cancer cells with increased expression of Cyclin D1b are subject to aberrant proliferation.36
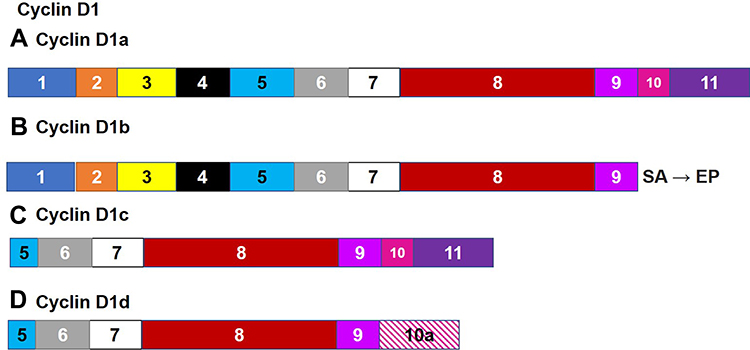 |
Figure 6 Alternate Splicing of Cyclin D1. Human cyclin D1 undergoes alternating splicing that creates a specific D1 transcript, cyclin d1b (B), the resulting protein lacks the motif that regulates the export of nuclear cyclin D1 encoded by exons 10 and 11. This region is found in the canonical isoform (A) as well as Cyclin D1c (C). Cyclin D1 (A, C and D) normally act to decrease proliferation, while cyclin D1b appears to act against Cyclin D1. |
Cyclin D1b is able to impede its own nuclear export as the lack of the C terminus sequence prevents its association with and consequent GSK-3-and CRM1-dependent nuclear export.114 This allows Cyclin D1b to remain active in the nucleus leading to continued cell growth and cellular transformation. Additionally, constitutive nuclear cyclin D1b/CDK4 complexes can stimulate transformation by disrupting the phosphorylation/dephosphorylation state of retinoblastoma protein (RB) standard.36
hTERT
Telomerase is a ribonucleoprotein enzyme which prevents the loss of telomere DNA ends during DNA replication by adding TTAGG to telomeric ends. This results in delayed cell replicative senescence. The reactivation of telomerase plays a significant role in carcinogenic cell immortalization.115 Mammalian telomerase is a holoenzyme composed of three primary units. Studies suggest that the expression of hTERT mRNA is the key determinant of telomerase activity in certain cancers.116–118 The transcription of human telomerase is initiated by upstream signaling molecules such as C-Myc, estrogen and progesterone.119–121 Repressors of transcription include Wilms tumor 1 suppressor,122 mitotic arrest deficient (MAD) and P53.123–125 Research has demonstrated the correlation between the increased level of htert mRNA and telomerase in Wilms tumor,126 urothelial cancer,127 skin tumors,128 primary central nervous system malignant lymphoma,129 hepatocellular carcinoma,117,130 colon carcinoma,118 cervical cancer,127 and ovarian tumors.131,132
Telomerase levels were observed to be 46-fold higher in esophageal adenocarcinoma compared to regular mucosa.115 Telomerase expression was slightly higher in esophageal adenocarcinoma than in Barrett’s esophagus.115 In general, telomerase expression was reactivated after the Barrett esophagus stage, but there is overlap between the Barrett esophagus stage and the progression to esophageal adenocarcinoma in terms of telomerase function.115 It has been established that telomerase activity was 3 times lower in the Barrett's esophagus in contrast to normal mucosa.133 Quantitative RT-PCR showed that htert mRNA levels rise to those observed in esophageal adenocarcinoma in a stepwise fashion that mirrors the progression of cancer stage.115,134 The inhibition of telomerase through ribonucleases activity inhibits the development and progression of esophageal and gastric adenocarcinomas.115,135
The alternate splicing of htert has been observed in a number of cell lines and tissues.115 Two splice variants possess changes in the region coding for the reverse transcriptase domain. The α-deletion variant has a deletion at the 5ʹ end of exon 6115 (Figure 7). The mRNA of human telomerase contains seven conserved reverse transcriptase motifs that are known as 1, 2, 3, A, B, C, D and E. The β variant lacks 182 bp due to an exclusion of exon 7 or 8, and this isoform only contains the B, C and D reverse transcriptase motifs.115 One of the splice variants codes for a non-functional protein that results in the lack of the any telomerase activity. Or inhibits the activity of any functional active telomerase.115 The overexpression of this variant in immortal cell lines telomerases and tumor cell lines in telomerases inhibits the production of endogenous telomerases.136,137
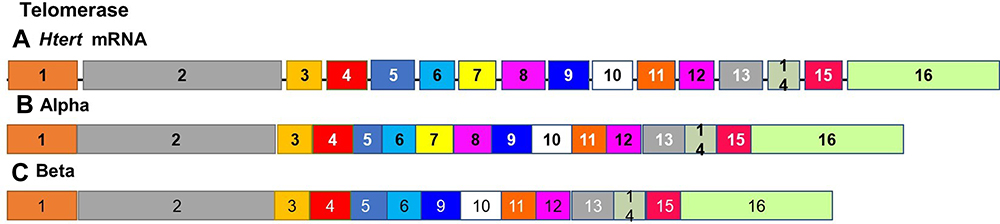 |
Figure 7 Alternate splicing of Telomerase. The alternate splicing of htert mRNA gives rise to the canonical variant (A) and at least two additional splice variants possessing changes in the region coding for the reverse transcriptase domain. The α-deletion variant (B) has a deletion at the 5ʹ end of exon 6 The β variant (C) lacks 182 bp due to an exclusion of exon 7 or 8, and this isoform only contains the B, C and D reverse transcriptase motifs and codes for a non-functional protein that results in the lack of any telomerase activity. |
However, various studies have shown that the levels of htert mRNA in esophageal adenocarcinoma and Barrett’s esophagus do not dictate the level of telomerase activity.115 It is not clear if htert mRNA splicing plays a role in telomerase regulation.115 Key regulatory mechanisms for the production of telomerase are likely to be post-translational and may involve phosphorylation of hTP1 or hTERT.138,139
Catenin Beta 1 Splice Variants and Downstream Targets
Catenin beta 1 (CTNNB1) is associated with cell signaling where it plays a role as a component of the adherens junction, through signaling via the Adenomatous Polyposis coli (APC) and through the Wnt tumor suppressor pathway. β-catenin carries out its effects by binding epithelial cadherins, the APC tumor suppressor, TCF, AXIN, GSK3b and a-catenin.140.
Many of the functions of CTNNB1 may involve mediating cell development and the associated processes such as embryogenesis, injury healing and tumor metastases. CTNNB1 interacts with TCF and stimulates target transcription. These targets include myc and wap1.141,142 B-catenin degradation requires an APC-Gsk3b serine/threonine kinase complex consisting of multiple proteins. In cancer cells this degradation can be prevented through mutations and splice variants of ctnnb1. These include mutations that typically involve Exon 3 of the ctnnb1 gene.143
Additionally, different isoforms of CTNNB1 have been isolated that differ, based on the inclusion or absence of a 159bp region, in exon 16. The expression of these two isoforms is distinctly different between normal esophagus epithelia, squamous dysplasia and invasive ESCC.140 These isoforms are named CTNNB1 (16A), which retains this region while CYNNB1 (16B) lacks this region. Both of these splice variants are present in normal and cancerous esophageal epithelial cells. However, the ratio of these isoforms was found to change in esophageal cancer cells. The level of 16A was shown to decrease while that of 16B increases as a tumor develops and progresses. The ratio of Myc proto-oncogene protein (MYC)/and the cyclin-dependent kinase (WAF1) increases, with increased expression of the MYC proto-oncogene.140,144
Lysyl Oxidase, LOXL2
The LOX family of proteins has a retained C terminal and a more variable amino terminal.145 The C end contains the cofactor and copper-binding motif essential for protein conformation and catalytic action. The N-terminus of LOXL2, LOXL3, and LOXL4 consists of four cysteine receptor domains which mediate protein-protein and cell signal interactions.145 LOXL2 mainly reshapes the micro-environment of the tumor, enhancing proliferation, penetration, vasculogenesis and metastasis of cancers.146–148 Studies indicate that LOXL2 is expressed at higher levels in various types of cancer cells including breast, pancreatic, colorectal, pulmonary and gastric cancer cells.149 These cancer cells had lower LOX activity. LOXL2 is also closely associated to the site of lymph node metastasis in ESCC.150 Two splice variants for LOXL2 were identified, LOXL2 Δ72 and Δ13 in ESCC. LOXL2 Δ72 has a 72 bp deletion resulting in a loss of 24 amino acids.151
LncRNA-uc002yug.2 and RUNX1
LncRNAs (Long noncoding RNAs) are implicated in various human diseases.152 The modified expressions of many lncRNAs have been observed in many different human tumors.153–156 For protein–chromatin interactions, lncRNAs may provide modular scaffolds for the assembly of molecular complexes.157 Moreover, several long-term antisense intergenic non-coding RNAs, silence genes.158 The association between altered transcription of lncRNAs and poor ESCC prognosis was assessed in cancer patients in China. This indicates that long-non-coding RNAs have a key regulatory role in cancer biology.152 One of the lncRNAs that have been identified as playing an important role in cancer is lncRNA-uc002yug.2. This lncRNA is typically overexpressed in ESCC. LncRNA-uc002yug.2 expression levels in ESCC can be a predictor of patient survival .152 One of the targets of this lncRNA is the Runt-related transcription factor 1 (RUNX1).
RUNX1 is a transcription factor that plays a role in hematopoietic cell differentiation, the development of pain transmitting neurons and forms heterodimers with binding proteins to increase DNA binding and initiation of transcription.159 Several studies have demonstrated the significance of CEBP5-007 in stopping a variety of forms of cell proliferation and tumor suppression.152 CEBPα has been shown to specifically interact with cyclin-dependent kinase 2/cyclin-dependent kinase 4 and arrest proliferation of cells.160 RUNX1 plays a significant role in multiple cancers, including breast cancer161 and epithelial cancer.162 LncRNA-uc002yug.2 is able to influence alternate splicing of Runx1, by changing the function of splicing factors. The splicing factor influenced by lncRNA-uc002yug.2 is SFRS1. This is an SR template protein involved in constituent expression of MBNL1, a member of the ES cell-specific AS family of muscle-like RNA binding proteins.163 LincRNAuc002yug.2 was shown to encourage RUNX1 AS by modulation of the affinity between the AS and RUNX1 factors, to minimize RUNX1 expression and to raise RUNX1a expression, resulting in fewer expression, and more help for tumor and cell proliferation.152 Three alternate splice variants for human RUNX1 have been identified, RUNX1a, RUNX1b and RUNX1c.Splicing of runx1 mRNA to form the short Runx1a isoform, which functions to inhibit RUNX 1b and RUNX1c. This leads to a decrease in the expression of CCAAT/enhancer-binding protein-α (CEBP).152 RUNX1 deletion have been observed in some esophageal tumors, suggesting that RUNX1 has the ability to suppress esophageal cancer.164–166
Targeting Alternative Splicing for the Development of Therapeutic Targets and Biomarkers
Pathologically altered AS can promote the development and progression of multiple diseases including cancer. These aberrant changes in alternative splicing can promote cancer and therefore, may serve as therapeutic targets. Despite esophageal cancer itself not being currently targeted by methods that influence alternate splicing, the process of alternate splicing can be easily targeted since it relies on multiple protein/protein, protein/RNA interactions and post-translational modifications. Many small molecules have been isolated that can target splicing factors such as SF3B and other components of the U2 snRNP. This can prevent spliceosome assembly.167 Apart from inhibiting the core spliceosome, another target for inhibition is splicing regulatory proteins. These proteins promote oncogenesis when they are over-expressed168 or their function is altered.169 An example of this is the serine arginine splicing promoting proteins. These proteins require phosphorylation in order to perform their function. Inhibiting phosphorylation of these proteins can modulate splicing.170 A final means of altering splicing for therapeutic purposes is through the use of oligonucleotide-based therapies that target individual splicing events. These oligonucleotides are designed to have complementary sequences allowing them to hybridize to target mRNA and alter splicing.171
The specific splicing patterns present in individual types of cancer vary based on the stage of the cancer. Therefore, by developing assays based on detecting the types of splice variants present can be used to stratify patients based on the stage of the cancer and severity of the disease. This will allow for specific treatments, more accurate diagnosis and finally a better indication of the patient’s prognosis. A recent analysis of splicing alterations in esophageal carcinoma identified 2389 AS events that occurred in esophageal cancer and were related to patient outcomes.172 Another study characterized the predictive power of different splicing events with esophageal cancer patient outcomes. It was found that 83% of alternative acceptor site splicing events could be associated with patient survival, 99% of alternative donor site events and 97% of alternative terminator site events.173
Conclusion and Prospects
Esophageal cancer is an important health concern especially in poorer developing countries and regions and the most rapid and possibly easiest way to change gene expression is through changes to alternative splicing of mRNA. These isoforms can allow for the changes in the behavior of epidermal esophageal cells, leading them to become more prone to developing into esophageal cancer. Alternatively, some isoforms can act as anticancer variants that act to inhibit the development of esophageal cancer. Therapeutically, alternative splicing can be targeted by promoting or inhibiting the formation of some of these splice variants. Any of those variants discussed in Alternate Splicing in Esophageal Cancer of this paper are already being suggested as potential targets for treatments that affect alternate splicing. For instance, the esophageal cancer promoting activities of the SV-1 splice variant of GHRHR, can potentially be blocked using the growth hormone-releasing hormone receptor antagonist MIA-602.69 Additionally, all of the alternately spliced genes discussed in this review can be targeted using antisense oligonucleotide to silence those variants that promote cancer. At the same time studies have indicated that changes in alternate splicing associated with esophageal cancer, closely correlate with patient survival, suggesting the usefulness of alternate splicing profiles as prognostic biomarkers. Finally, the differences in the alternative splicing profiles between esophageal adenocarcinoma and esophageal squamous cell carcinoma, indicate that alternate splicing profiles can be used to diagnose patients rapidly and accurately, in summary the changes in alternate splicing that occur in esophageal carcinoma are promising targets for the identification of new therapeutic targets and the development of new biomarker assays.
However, despite the promise of these splicing patterns serving as biomarkers and individual splice variants serving as therapeutic targets there are questions as to how accurately this can be achieved. Firstly, specifically targeting the alternate splicing in esophageal cancer with new therapeutic drugs may prove difficult. This is because many of the strategies, such as the inhibition of splicing factors, would interfere with splicing in normal tissues. However, other strategies such as antisense and splicing switch oligonucleotides do not have this problem as these can be used to target specific variants which can promote carcinogenesis. Secondly, the difference between the splicing profiles between healthy and cancerous tissue is different enough that they can reliably be used as diagnostic markers. However, this would require far more intensive research into the splicing profiles in order for these profiles to serve as an accurate diagnostic tool. Lastly the proven association between some splicing profiles and patient survival implies that these profiles can be used as a prognostic tool. Once again this would require further research to increase the accuracy of these diagnostic tests. These tests can be carried out using PCR arrays that target specific isoforms. An array of genes in these assays would allow for predictions to be made with confidence.
Acknowledgment
The authors would like to thank the South African Medical Research Council (SAMRC) for funding this research.
Disclosure
Aristotelis Chatziioannou reports being the CEO of e-NIOS APPLICATIONS PC, during the conduct of the study; and that as CEO and CSO they direct the research activities of e-NIOS APPLICATIONS PC, outside the submitted work. The authors report no other potential conflicts of interest in this work.
References
1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA Cancer J Clin. 2016;66(1):7–30. doi:10.3322/caac.21332
2. Fitzmaurice C, Dicker D, Pain A, et al. The global burden of cancer 2013. JAMA Oncol. 2015;1(4):505–527. doi:10.1001/jamaoncol.2015.0735
3. Arnold M, Soerjomataram I, Ferlay J, et al. Global incidence of oesophageal cancer by histological subtype in 2012. Gut. 2015;64(3):381–387. doi:10.1136/gutjnl-2014-308124
4. Chen W, Zheng R, Baade PD, et al. Cancer statistics in China, 2015. CA Cancer J Clin. 2016;66(2):115–132. doi:10.3322/caac.21338
5. Pennathur A, Gibson MK, Jobe BA, et al. Oesophageal carcinoma. Lancet. 2013;381(9864):400–412. doi:10.1016/S0140-6736(12)60643-6
6. Napier KJ, Scheerer M, Misra S. Esophageal cancer: a review of epidemiology, pathogenesis, staging workup and treatment modalities. World J Gastrointest Oncol. 2014;6(5):112–120. doi:10.4251/wjgo.v6.i5.112
7. Smyth EC, Lagergren J, Fitzgerald RC, et al. Oesophageal cancer. Nat Rev Dis Primers. 2017;3:17048.
8. Xiong HY, Alipanahi B, Lee LJ, et al. RNA splicing. The human splicing code reveals new insights into the genetic determinants of disease. Science. 2015;347(6218):1254806. doi:10.1126/science.1254806
9. Matera AG, Wang Z. A day in the life of the spliceosome. Nat Rev Mol Cell Biol. 2014;15(2):108–121. doi:10.1038/nrm3742
10. Wang ET, Sandberg R, Luo S, et al. Alternative isoform regulation in human tissue transcriptomes. Nature. 2008;456(7221):470–476. doi:10.1038/nature07509
11. Kalsotra A, Cooper TA. Functional consequences of developmentally regulated alternative splicing. Nat Rev Genet. 2011;12(10):715–729. doi:10.1038/nrg3052
12. Gamazon ER, Stranger BE. Genomics of alternative splicing: evolution, development and pathophysiology. Hum Genet. 2014;133(6):679–687. doi:10.1007/s00439-013-1411-3
13. Song X, Zeng Z, Wei H, et al. Alternative splicing in cancers: from aberrant regulation to new therapeutics. Semin Cell Dev Biol. 2018;75:13–22. doi:10.1016/j.semcdb.2017.09.018
14. David CJ, Manley JL. Alternative pre-mRNA splicing regulation in cancer: pathways and programs unhinged. Genes Dev. 2010;24(21):2343–2364. doi:10.1101/gad.1973010
15. Oltean S, Bates DO. Hallmarks of alternative splicing in cancer. Oncogene. 2014;33(46):5311–5318. doi:10.1038/onc.2013.533
16. Sveen A, Kilpinen S, Ruusulehto A, et al. Aberrant RNA splicing in cancer; expression changes and driver mutations of splicing factor genes. Oncogene. 2016;35(19):2413–2427.
17. Ladomery M. Aberrant alternative splicing is another hallmark of cancer. Int J Cell Biol. 2013;2013:463786. doi:10.1155/2013/463786
18. Katz Y, Wang ET, Airoldi EM, et al. Analysis and design of RNA sequencing experiments for identifying isoform regulation. Nat Methods. 2010;7(12):1009–1015. doi:10.1038/nmeth.1528
19. Ding J, Li C, Cheng Y, et al. Alterations of RNA splicing patterns in esophagus squamous cell carcinoma. Cell Biosci. 2021;11(1):36. doi:10.1186/s13578-021-00546-z
20. Mao S, Li Y, Lu Z, et al. Survival-associated alternative splicing signatures in esophageal carcinoma. Carcinogenesis. 2019;40(1):121–130. doi:10.1093/carcin/bgy123
21. Li Y, Yuan Y. Alternative RNA splicing and gastric cancer. Mutat Res. 2017;773:263–273. doi:10.1016/j.mrrev.2016.07.011
22. Koyama S, Maruyama T, Adachi S. Expression of epidermal growth factor receptor and CD44 splicing variants sharing exons 6 and 9 on gastric and esophageal carcinomas: a two-color flow-cytometric analysis. J Cancer Res Clin Oncol. 1999;125(1):47–54. doi:10.1007/s004320050241
23. Lin J, Lin L, Thomas DG, et al. Melanoma-associated antigens in esophageal adenocarcinoma: identification of novel MAGE-A10 splice variants. Clin Cancer Res. 2004;10(17):5708–5716. doi:10.1158/1078-0432.CCR-04-0468
24. Fang WK, Xu L-Y, Lu XF, et al. A novel alternative spliced variant of neutrophil gelatinase-associated lipocalin receptor in oesophageal carcinoma cells. Biochem J. 2007;403(2):297–303. doi:10.1042/BJ20060836
25. Shen Q, Nam SW. SF3B4 as an early-stage diagnostic marker and driver of hepatocellular carcinoma. BMB Rep. 2018;51(2):57–58. doi:10.5483/BMBRep.2018.51.2.021
26. Kidogami S, Iguchi T, Sato K, et al. SF3B4 plays an oncogenic role in esophageal squamous cell carcinoma. Anticancer Res. 2020;40(5):2941–2946. doi:10.21873/anticanres.14272
27. Bisognin A, Pizzini S, Perilli L, et al. An integrative framework identifies alternative splicing events in colorectal cancer development. Mol Oncol. 2014;8(1):129–141. doi:10.1016/j.molonc.2013.10.004
28. Zou HY, Lv G-Q, Dai L-H, et al. A truncated splice variant of human lysyl oxidase-like 2 promotes migration and invasion in esophageal squamous cell carcinoma. Int J Biochem Cell Biol. 2016;75:85–98. doi:10.1016/j.biocel.2016.04.003
29. Dickinson A, Saraswat M, Mäkitie A, et al. Label-free tissue proteomics can classify oral squamous cell carcinoma from healthy tissue in a stage-specific manner. Oral Oncol. 2018;86:206–215. doi:10.1016/j.oraloncology.2018.09.013
30. Song ZB, Gao SS, Yi XN, et al. Expression of MUC1 in esophageal squamous-cell carcinoma and its relationship with prognosis of patients from Linzhou city, a high incidence area of northern China. World J Gastroenterol. 2003;9(3):404–407. doi:10.3748/wjg.v9.i3.404
31. Bessman MJ, Frick DN, O’Handley SF. The MutT proteins or “Nudix” hydrolases, a family of versatile, widely distributed, “housecleaning” enzymes. J Biol Chem. 1996;271(41):25059–25062. doi:10.1074/jbc.271.41.25059
32. Yang S, Jia R, Bian Z. SRSF5 functions as a novel oncogenic splicing factor and is upregulated by oncogene SRSF3 in oral squamous cell carcinoma. Biochim Biophys Acta Mol Cell Res. 2018;1865(9):1161–1172. doi:10.1016/j.bbamcr.2018.05.017
33. Yu Q, Yin L, Jian Y, et al. Downregulation of PHF6 inhibits cell proliferation and migration in hepatocellular carcinoma. Cancer Biother Radiopharm. 2019;34(4):245–251. doi:10.1089/cbr.2018.2671
34. Rekasi Z, Varga JL, Schally AV, et al. Antagonists of growth hormone-releasing hormone and vasoactive intestinal peptide inhibit tumor proliferation by different mechanisms: evidence from in vitro studies on human prostatic and pancreatic cancers 1. Endocrinology. 2000;141(6):2120–2128. doi:10.1210/endo.141.6.7511
35. Ogura Y, Hoshino T, Tanaka N, et al. Disturbed alternative splicing of FIR (PUF60) directed cyclin E overexpression in esophageal cancers. Oncotarget. 2018;9(33):22929–22944. doi:10.18632/oncotarget.25149
36. Lu F, Gladden AB, Diehl JA. An alternatively spliced cyclin D1 isoform, cyclin D1b, is a nuclear oncogene. Cancer Res. 2003;63(21):7056–7061.
37. Peach RJ, Hollenbaugh D, Stamenkovic I, et al. Identification of hyaluronic acid binding sites in the extracellular domain of CD44. J Cell Biol. 1993;122(1):257–264. doi:10.1083/jcb.122.1.257
38. Fox SB, Fawcett J, Jackson DG, et al. Normal human tissues, in addition to some tumors, express multiple different CD44 isoforms. Cancer Res. 1994;54(16):4539–4546.
39. Albelda SM. Role of integrins and other cell adhesion molecules in tumor progression and metastasis. Lab Invest. 1993;68(1):4–17.
40. Wielenga VJ, Heider KH, Offerhaus GJ, et al. Expression of CD44 variant proteins in human colorectal cancer is related to tumor progression. Cancer Res. 1993;53(20):4754–4756.
41. Mulder JW, Sewnath M, Offerhaus G, et al. Colorectal cancer prognosis and expression of exon-v6-containing CD44 proteins. Lancet. 1994;344(8935):1470–1472. doi:10.1016/S0140-6736(94)90290-9
42. Kaufmann M, von Minckwitz G, Heider K-H, et al. CD44 variant exon epitopes in primary breast cancer and length of survival. Lancet. 1995;345(8950):615–619. doi:10.1016/S0140-6736(95)90521-9
43. van der Bruggen P, Traversari C, Chomez P, et al. A gene encoding an antigen recognized by cytolytic T lymphocytes on a human melanoma. Science. 1991;254(5038):1643–1647. doi:10.1126/science.1840703
44. Chomez P, De Backer O, Bertrand M, et al. An overview of the MAGE gene family with the identification of all human members of the family. Cancer Res. 2001;61(14):5544–5551.
45. Chomez P, Williams R, De Backer O, et al. The SMAGE gene family is expressed in post-meiotic spermatids during mouse germ cell differentiation. Immunogenetics. 1996;43(1–2):97–100. doi:10.1007/BF00186613
46. Van den Eynde BJ, van der Bruggen P. T cell defined tumor antigens. Curr Opin Immunol. 1997;9(5):684–693. doi:10.1016/S0952-7915(97)80050-7
47. Dutoit V, Rubio-Godoy V, Dietrich PY, et al. Heterogeneous T-cell response to MAGE-A10(254–262): high avidity-specific cytolytic T lymphocytes show superior antitumor activity. Cancer Res. 2001;61(15):5850–5856.
48. Tatsumi T, Kierstead LS, Ranieri E, et al. MAGE-6 encodes HLA-DRbeta1*0401-presented epitopes recognized by CD4+ T cells from patients with melanoma or renal cell carcinoma. Clin Cancer Res. 2003;9(3):947–954.
49. Rimoldi D, Salvi S, Reed D, et al. cDNA and protein characterization of human MAGE-10. Int J Cancer. 1999;82(6):901–907. doi:10.1002/(SICI)1097-0215(19990909)82:6<901::AID-IJC21>3.0.CO;2-X
50. Marchand M, van Baren N, Weynants P, et al. Tumor regressions observed in patients with metastatic melanoma treated with an antigenic peptide encoded by gene MAGE-3 and presented by HLA-A1. Int J Cancer. 1999;80(2):219–230. doi:10.1002/(SICI)1097-0215(19990118)80:2<219::AID-IJC10>3.0.CO;2-S
51. Thurner B, Haendle I, Röder C, et al. Vaccination with mage-3A1 peptide-pulsed mature, monocyte-derived dendritic cells expands specific cytotoxic T cells and induces regression of some metastases in advanced stage IV melanoma. J Exp Med. 1999;190(11):1669–1678. doi:10.1084/jem.190.11.1669
52. Ohman Forslund K, Nordqvist K. The melanoma antigen genes–any clues to their functions in normal tissues? Exp Cell Res. 2001;265(2):185–194. doi:10.1006/excr.2001.5173
53. Rousseau P, Le Discorde M, Mouillot G, et al. The 14 bp deletion-insertion polymorphism in the 3ʹ UT region of the HLA-G gene influences HLA-G mRNA stability. Hum Immunol. 2003;64(11):1005–1010. doi:10.1016/j.humimm.2003.08.347
54. Flower DR. Beyond the superfamily: the lipocalin receptors. Biochim Biophys Acta. 2000;1482(1–2):327–336. doi:10.1016/S0167-4838(00)00169-2
55. Flower DR. The lipocalin protein family: structure and function. Biochem J. 1996;318(Pt 1):1–14. doi:10.1042/bj3180001
56. Flower DR, North AC, Sansom CE. The lipocalin protein family: structural and sequence overview. Biochim Biophys Acta. 2000;1482(1–2):9–24. doi:10.1016/S0167-4838(00)00148-5
57. Kjeldsen L, Johnsen AH, Sengeløv H, et al. Isolation and primary structure of NGAL, a novel protein associated with human neutrophil gelatinase. J Biol Chem. 1993;268(14):10425–10432. doi:10.1016/S0021-9258(18)82217-7
58. Nielsen BS, Borregaard N, Bundgaard JR, et al. Induction of NGAL synthesis in epithelial cells of human colorectal neoplasia and inflammatory bowel diseases. Gut. 1996;38(3):414–420. doi:10.1136/gut.38.3.414
59. Friedl A, Stoesz SP, Buckley P, et al. Neutrophil gelatinase-associated lipocalin in normal and neoplastic human tissues. Cell type-specific pattern of expression. Histochem J. 1999;31(7):433–441. doi:10.1023/A:1003708808934
60. Yan L, Borregaard N, Kjeldsen L, et al. The high molecular weight urinary matrix metalloproteinase (MMP) activity is a complex of gelatinase B/MMP-9 and neutrophil gelatinase-associated lipocalin (NGAL). Modulation of MMP-9 activity by NGAL. J Biol Chem. 2001;276(40):37258–37265. doi:10.1074/jbc.M106089200
61. Tschesche H, Zölzer V, Triebel S, et al. The human neutrophil lipocalin supports the allosteric activation of matrix metalloproteinases. Eur J Biochem. 2001;268(7):1918–1928. doi:10.1046/j.1432-1327.2001.02066.x
62. Goetz DH, Holmes MA, Borregaard N, et al. The neutrophil lipocalin NGAL is a bacteriostatic agent that interferes with siderophore-mediated iron acquisition. Mol Cell. 2002;10(5):1033–1043. doi:10.1016/S1097-2765(02)00708-6
63. Flo TH, Smith KD, Sato S, et al. Lipocalin 2 mediates an innate immune response to bacterial infection by sequestrating iron. Nature. 2004;432(7019):917–921. doi:10.1038/nature03104
64. Mori K, Lee HT, Rapoport D, et al. Endocytic delivery of lipocalin-siderophore-iron complex rescues the kidney from ischemia-reperfusion injury. J Clin Invest. 2005;115(3):610–621. doi:10.1172/JCI23056
65. Tong Z, Wu X, Ovcharenko D, et al. Neutrophil gelatinase-associated lipocalin as a survival factor. Biochem J. 2005;391(Pt 2):441–448. doi:10.1042/BJ20051020
66. Yang J, Mori K, Li JY, et al. Iron, lipocalin, and kidney epithelia. Am J Physiol Renal Physiol. 2003;285(1):F9–18. doi:10.1152/ajprenal.00008.2003
67. Gwira JA, Wei F, Ishibe S, et al. Expression of neutrophil gelatinase-associated lipocalin regulates epithelial morphogenesis in vitro. J Biol Chem. 2005;280(9):7875–7882. doi:10.1074/jbc.M413192200
68. Li EM, Xu L-Y, Cai W-J, et al. [Functions of neutrophil gelatinase-associated lipocalin in the esophageal carcinoma cell line SHEEC]. Sheng Wu Hua Xue Yu Sheng Wu Wu Li Xue Bao. 2003;35(3):247–254. Chinese.
69. Xiong X, Ke X, Wang L, et al. Splice variant of growth hormone-releasing hormone receptor drives esophageal squamous cell carcinoma conferring a therapeutic target. Proc Natl Acad Sci U S A. 2020;117(12):6726–6732. doi:10.1073/pnas.1913433117
70. Rekasi Z, Czompoly T, Schally AV, et al. Isolation and sequencing of cDNAs for splice variants of growth hormone-releasing hormone receptors from human cancers. Proc Natl Acad Sci U S A. 2000;97(19):10561–10566. doi:10.1073/pnas.180313297
71. Szalontay L, Schally AV, Popovics P, et al. Novel GHRH antagonists suppress the growth of human malignant melanoma by restoring nuclear p27 function. Cell Cycle. 2014;13(17):2790–2797. doi:10.4161/15384101.2015.945879
72. Klukovits A, Schally AV, Szalontay L, et al. Novel antagonists of growth hormone-releasing hormone inhibit growth and vascularization of human experimental ovarian cancers. Cancer. 2012;118(3):670–680. doi:10.1002/cncr.26291
73. Rumpel CA, Powell SM, Moskaluk CA. Mapping of genetic deletions on the long arm of chromosome 4 in human esophageal adenocarcinomas. Am J Pathol. 1999;154(5):1329–1334. doi:10.1016/S0002-9440(10)65386-2
74. Barnas C, Henn T, Stark M, et al. Detection of genetic alterations in cancers of the esophagus and esophagogastric junction by comparative genomic hybridization: frequent involvement of chromosome 4q.
75. Powers CJ, McLeskey SW, Wellstein A. Fibroblast growth factors, their receptors and signaling. Endocr Relat Cancer. 2000;7(3):165–197. doi:10.1677/erc.0.0070165
76. Murphy PR, Knee RS. Identification and characterization of an antisense RNA transcript (gfg) from the human basic fibroblast growth factor gene. Mol Endocrinol. 1994;8(7):852–859. doi:10.1210/mend.8.7.7984147
77. Knee R, Li AW, Murphy PR. Characterization and tissue-specific expression of the rat basic fibroblast growth factor antisense mRNA and protein. Proc Natl Acad Sci U S A. 1997;94(10):4943–4947. doi:10.1073/pnas.94.10.4943
78. Li AW, Murphy PR. Expression of alternatively spliced FGF-2 antisense RNA transcripts in the central nervous system: regulation of FGF-2 mRNA translation. Mol Cell Endocrinol. 2000;170(1–2):233–242. doi:10.1016/S0303-7207(00)00440-8
79. Asa SL, Ramyar L, Murphy PR, et al. The endogenous fibroblast growth factor-2 antisense gene product regulates pituitary cell growth and hormone production. Mol Endocrinol. 2001;15(4):589–599. doi:10.1210/mend.15.4.0626
80. Zhang SC, Barclay C, Alexander LA, et al. Alternative splicing of the FGF antisense gene: differential subcellular localization in human tissues and esophageal adenocarcinoma. J Mol Med (Berl). 2007;85(11):1215–1228. doi:10.1007/s00109-007-0219-9
81. Kausar T, Sharma R, Hasan MR, et al. Overexpression of a splice variant of oncostatin M receptor beta in human esophageal squamous carcinoma. Cell Oncol. 2011;34(3):177–187. doi:10.1007/s13402-011-0011-2
82. Ye Q, Yan Z, Liao X, et al. MUC1 induces metastasis in esophageal squamous cell carcinoma by upregulating matrix metalloproteinase 13. Lab Invest. 2011;91(5):778–787. doi:10.1038/labinvest.2011.12
83. Liu J, He L, Collins I, et al. The FBP interacting repressor targets TFIIH to inhibit activated transcription. Mol Cell. 2000;5(2):331–341. doi:10.1016/S1097-2765(00)80428-1
84. Matsushita K, Tomonaga T, Shimada H, et al. An essential role of alternative splicing of c-myc suppressor FUSE-binding protein–interacting repressor in carcinogenesis. Cancer Res. 2006;66(3):1409–1417. doi:10.1158/0008-5472.CAN-04-4459
85. Matsushita K, Kajiwara T, Tamura M, et al. SAP155-mediated splicing of FUSE-binding protein-interacting repressor serves as a molecular switch for c-myc gene expression. Mol Cancer Res. 2012;10(6):787–799. doi:10.1158/1541-7786.MCR-11-0462
86. Rahmutulla B, Matsushita K, Nomura F. Alternative splicing of DNA damage response genes and gastrointestinal cancers. World J Gastroenterol. 2014;20(46):17305–17313. doi:10.3748/wjg.v20.i46.17305
87. Matsushita T, Ohyabu N, Fujitani N, et al. Site-specific conformational alteration induced by sialylation of MUC1 tandem repeating glycopeptides at an epitope region for the anti-KL-6 monoclonal antibody. Biochemistry. 2013;52(2):402–414. doi:10.1021/bi3013142
88. Song JH, Schnittke N, Zaat A, et al. FBXW7 mutation in adult T-cell and B-cell acute lymphocytic leukemias. Leuk Res. 2008;32(11):1751–1755. doi:10.1016/j.leukres.2008.03.040
89. Zhang W, Koepp DM. Fbw7 isoform interaction contributes to cyclin E proteolysis. Mol Cancer Res. 2006;4(12):935–943. doi:10.1158/1541-7786.MCR-06-0253
90. Cheng Y, Li G. Role of the ubiquitin ligase Fbw7 in cancer progression. Cancer Metastasis Rev. 2012;31(1–2):75–87. doi:10.1007/s10555-011-9330-z
91. Zhang G, Zhou X, Xue L, et al. Accumulation of cytoplasmic beta-catenin correlates with reduced expression of E-cadherin, but not with phosphorylated Akt in esophageal squamous cell carcinoma: immunohistochemical study. Pathol Int. 2005;55(6):310–317. doi:10.1111/j.1440-1827.2005.01840.x
92. Lv J, Cao X-F, Ji L, et al. Association of β-catenin, Wnt1, Smad4, Hoxa9, and Bmi-1 with the prognosis of esophageal squamous cell carcinoma. Med Oncol. 2012;29(1):151–160. doi:10.1007/s12032-010-9816-5
93. Yan S, Zhou C, Zhang W, et al. beta-Catenin/TCF pathway upregulates STAT3 expression in human esophageal squamous cell carcinoma. Cancer Lett. 2008;271(1):85–97. doi:10.1016/j.canlet.2008.05.035
94. Wang Y, Zhou X, Zhu H, et al. Overexpression of EB1 in human esophageal squamous cell carcinoma (ESCC) may promote cellular growth by activating beta-catenin/TCF pathway. Oncogene. 2005;24(44):6637–6645. doi:10.1038/sj.onc.1208819
95. Ring A, Kim YM, Kahn M. Wnt/catenin signaling in adult stem cell physiology and disease. Stem Cell Rev Rep. 2014;10(4):512–525. doi:10.1007/s12015-014-9515-2
96. Shiina H, Igawa M, Breault J, et al. The human T-cell factor-4 gene splicing isoforms, Wnt signal pathway, and apoptosis in renal cell carcinoma. Clin Cancer Res. 2003;9(6):2121–2132.
97. Tsedensodnom O, Koga H, Rosenberg SA, et al. Identification of T-cell factor-4 isoforms that contribute to the malignant phenotype of hepatocellular carcinoma cells. Exp Cell Res. 2011;317(7):920–931. doi:10.1016/j.yexcr.2011.01.015
98. Duval A, Rolland S, Tubacher E, et al. The human T-cell transcription factor-4 gene: structure, extensive characterization of alternative splicings, and mutational analysis in colorectal cancer cell lines. Cancer Res. 2000;60(14):3872–3879.
99. Le Bacquer O, Shu L, Marchand M, et al. TCF7L2 splice variants have distinct effects on beta-cell turnover and function. Hum Mol Genet. 2011;20(10):1906–1915. doi:10.1093/hmg/ddr072
100. Hansson O, Zhou Y, Renström E, et al. Molecular function of TCF7L2: consequences of TCF7L2 splicing for molecular function and risk for type 2 diabetes. Curr Diab Rep. 2010;10(6):444–451. doi:10.1007/s11892-010-0149-8
101. Prokunina-Olsson L, Kaplan LM, Schadt EE, et al. Alternative splicing of TCF7L2 gene in omental and subcutaneous adipose tissue and risk of type 2 diabetes. PLoS One. 2009;4(9):e7231. doi:10.1371/journal.pone.0007231
102. Weise A, Bruser K, Elfert S, et al. Alternative splicing of Tcf7l2 transcripts generates protein variants with differential promoter-binding and transcriptional activation properties at Wnt/beta-catenin targets. Nucleic Acids Res. 2010;38(6):1964–1981. doi:10.1093/nar/gkp1197
103. Young RM, Reyes AE, Allende ML. Expression and splice variant analysis of the zebrafish tcf4 transcription factor. Mech Dev. 2002;117(1–2):269–273. doi:10.1016/S0925-4773(02)00180-6
104. Kennell JA, O’Leary EE, Gummow BM, et al. T-cell factor 4N (TCF-4N), a novel isoform of mouse TCF-4, synergizes with beta-catenin to coactivate C/EBPalpha and steroidogenic factor 1 transcription factors. Mol Cell Biol. 2003;23(15):5366–5375. doi:10.1128/MCB.23.15.5366-5375.2003
105. Howng SL, Huang F-H, Hwang S-L, et al. Differential expression and splicing isoform analysis of human Tcf-4 transcription factor in brain tumors. Int J Oncol. 2004;25(6):1685–1692.
106. Cuilliere-Dartigues P, El-Bchiri J, Krimi A, et al. TCF-4 isoforms absent in TCF-4 mutated MSI-H colorectal cancer cells colocalize with nuclear CtBP and repress TCF-4-mediated transcription. Oncogene. 2006;25(32):4441–4448. doi:10.1038/sj.onc.1209471
107. Bani-Hani K, Martin IG, Hardie LJ, et al. Prospective study of cyclin D1 overexpression in Barrett’s esophagus: association with increased risk of adenocarcinoma. J Natl Cancer Inst. 2000;92(16):1316–1321. doi:10.1093/jnci/92.16.1316
108. Diehl JA. Cycling to cancer with cyclin D1. Cancer Biol Ther. 2002;1(3):226–231. doi:10.4161/cbt.72
109. Betticher DC, Thatcher N, Altermatt HJ, et al. Alternate splicing produces a novel cyclin D1 transcript. Oncogene. 1995;11(5):1005–1011.
110. Hibberts NA, Simpson DJ, Bicknell JE, et al. Analysis of cyclin D1 (CCND1) allelic imbalance and overexpression in sporadic human pituitary tumors. Clin Cancer Res. 1999;5(8):2133–2139.
111. Kong S, Amos CI, Luthra R, et al. Effects of cyclin D1 polymorphism on age of onset of hereditary nonpolyposis colorectal cancer. Cancer Res. 2000;60(2):249–252.
112. Zheng Y, Shen H, Sturgis EM, et al. Cyclin D1 polymorphism and risk for squamous cell carcinoma of the head and neck: a case-control study. Carcinogenesis. 2001;22(8):1195–1199. doi:10.1093/carcin/22.8.1195
113. Simpson DJ, Frost SJ, Bicknell JE, et al. Aberrant expression of G(1)/S regulators is a frequent event in sporadic pituitary adenomas. Carcinogenesis. 2001;22(8):1149–1154. doi:10.1093/carcin/22.8.1149
114. Alt JR, Cleveland JL, Hannink M, et al. Phosphorylation-dependent regulation of cyclin D1 nuclear export and cyclin D1-dependent cellular transformation. Genes Dev. 2000;14(24):3102–3114. doi:10.1101/gad.854900
115. Barclay JY, Morris A, Nwokolo CU. Telomerase, hTERT and splice variants in Barrett’s oesophagus and oesophageal adenocarcinoma. Eur J Gastroenterol Hepatol. 2005;17(2):221–227. doi:10.1097/00042737-200502000-00014
116. Bodnar AG, Ouellette M, Frolkis M, et al. Extension of life-span by introduction of telomerase into normal human cells. Science. 1998;279(5349):349–352. doi:10.1126/science.279.5349.349
117. Nakayama J, Tahara H, Tahara E, et al. Telomerase activation by hTRT in human normal fibroblasts and hepatocellular carcinomas. Nat Genet. 1998;18(1):65–68. doi:10.1038/ng0198-65
118. Meyerson M, Counter CM, Eaton EN, et al. hEST2, the putative human telomerase catalytic subunit gene, is up-regulated in tumor cells and during immortalization. Cell. 1997;90(4):785–795. doi:10.1016/S0092-8674(00)80538-3
119. Wang J, Xie LY, Allan S, et al. Myc activates telomerase. Genes Dev. 1998;12(12):1769–1774. doi:10.1101/gad.12.12.1769
120. Kyo S, Kanaya T, Takakura M, et al. Expression of human telomerase subunits in ovarian malignant, borderline and benign tumors. Int J Cancer. 1999;80(6):804–809. doi:10.1002/(SICI)1097-0215(19990315)80:6<804::AID-IJC2>3.0.CO;2-B
121. Wang Z, Kyo S, Takakura M, et al. Progesterone regulates human telomerase reverse transcriptase gene expression via activation of mitogen-activated protein kinase signaling pathway. Cancer Res. 2000;60(19):5376–5381.
122. Shay JW, Bacchetti S. A survey of telomerase activity in human cancer. Eur J Cancer. 1997;33(5):787–791. doi:10.1016/S0959-8049(97)00062-2
123. Oh S, Song Y, Yim J, et al. The Wilms’ tumor 1 tumor suppressor gene represses transcription of the human telomerase reverse transcriptase gene. J Biol Chem. 1999;274(52):37473–37478. doi:10.1074/jbc.274.52.37473
124. Oh S, Song Y-H, Yim J, et al. Identification of Mad as a repressor of the human telomerase (hTERT) gene. Oncogene. 2000;19(11):1485–1490. doi:10.1038/sj.onc.1203439
125. Kanaya T, Kyo S, Hamada K, et al. Adenoviral expression of p53 represses telomerase activity through down-regulation of human telomerase reverse transcriptase transcription. Clin Cancer Res. 2000;6(4):1239–1247.
126. Dome JS, Chung S, Bergemann T, et al. High telomerase reverse transcriptase (hTERT) messenger RNA level correlates with tumor recurrence in patients with favorable histology Wilms’ tumor. Cancer Res. 1999;59(17):4301–4307.
127. Takakura M, Kyo S, Kanaya T, et al. Expression of human telomerase subunits and correlation with telomerase activity in cervical cancer. Cancer Res. 1998;58(7):1558–1561.
128. Wu A, Ichihashi M, Ueda M. Correlation of the expression of human telomerase subunits with telomerase activity in normal skin and skin tumors. Cancer. 1999;86(10):2038–2044. doi:10.1002/(SICI)1097-0142(19991115)86:10<2038::AID-CNCR22>3.0.CO;2-A
129. Harada K, Kurisu K, Arita K, et al. Telomerase activity in central nervous system malignant lymphoma. Cancer. 1999;86(6):1050–1055.
130. Toshikuni N, Nouso K, Higashi T, et al. Expression of telomerase-associated protein 1 and telomerase reverse transcriptase in hepatocellular carcinoma. Br J Cancer. 2000;82(4):833–837. doi:10.1054/bjoc.1999.1008
131. Kyo S, Takakura M, Kanaya T, et al. Estrogen activates telomerase. Cancer Res. 1999;59(23):5917–5921.
132. Park TW, Riethdorf S, Riethdorf L, et al. Differential telomerase activity, expression of the telomerase catalytic sub-unit and telomerase-RNA in ovarian tumors. Int J Cancer. 1999;84(4):426–431. doi:10.1002/(SICI)1097-0215(19990820)84:4<426::AID-IJC17>3.0.CO;2-1
133. Bachor C, Bachor OA, Boukamp P. Telomerase is active in normal gastrointestinal mucosa and not up-regulated in precancerous lesions. J Cancer Res Clin Oncol. 1999;125(8–9):453–460. doi:10.1007/s004320050302
134. Lord RV, Salonga D, Danenberg KD, et al. Telomerase reverse transcriptase expression is increased early in the Barrett’s metaplasia, dysplasia, adenocarcinoma sequence. J Gastrointest Surg. 2000;4(2):135–142. doi:10.1016/S1091-255X(00)80049-9
135. Usselmann B, Portsmouth D, Barclay J, et al. Inhibition of telomerase by site-specific ribonucleases in gastric and esophageal adenocarcinoma. Dig Dis Sci. 2001;46(12):2666–2672. doi:10.1023/A:1012767127278
136. Colgin LM, Wilkinso C, Englezou A, et al. The hTERTalpha splice variant is a dominant negative inhibitor of telomerase activity. Neoplasia. 2000;2(5):426–432. doi:10.1038/sj.neo.7900112
137. Yi X, White DM, Aisner DL, et al. An alternate splicing variant of the human telomerase catalytic subunit inhibits telomerase activity. Neoplasia. 2000;2(5):433–440. doi:10.1038/sj.neo.7900113
138. Li H, Zhao L, Yang Z, et al. Telomerase is controlled by protein kinase Calpha in human breast cancer cells. J Biol Chem. 1998;273(50):33436–33442. doi:10.1074/jbc.273.50.33436
139. Kharbanda S, Kumar V, Dhar S, et al. Regulation of the hTERT telomerase catalytic subunit by the c-Abl tyrosine kinase. Curr Biol. 2000;10(10):568–575. doi:10.1016/S0960-9822(00)00483-8
140. Roth MJ, Hu N, Johnson LL, et al. beta-Catenin splice variants and downstream targets as markers for neoplastic progression of esophageal cancer. Genes Chromosomes Cancer. 2005;44(4):423–428. doi:10.1002/gcc.20251
141. He TC, Sparks AB, Rago C, et al. Identification of c-MYC as a target of the APC pathway. Science. 1998;281(5382):1509–1512. doi:10.1126/science.281.5382.1509
142. Bièche I, Laurendeau I, Tozlu S, et al. Quantitation of MYC gene expression in sporadic breast tumors with a real-time reverse transcription-PCR assay. Cancer Res. 1999;59(12):2759–2765.
143. Behrens J. Control of beta-catenin signaling in tumor development. Ann N Y Acad Sci. 2000;910:21–33. doi:10.1111/j.1749-6632.2000.tb06698.x
144. Bitzer M, Stahl M, Arjumand J, et al. C-myc gene amplification in different stages of oesophageal squamous cell carcinoma: prognostic value in relation to treatment modality. Anticancer Res. 2003;23(2b):1489–1493.
145. Lv GQ, Zou H-Y, Liao L-D, et al. Identification of a novel lysyl oxidase-like 2 alternative splicing isoform, LOXL2 Δe13, in esophageal squamous cell carcinoma. Biochem Cell Biol. 2014;92(5):379–389. doi:10.1139/bcb-2014-0046
146. Barry-Hamilton V, Spangler R, Marshall D, et al. Allosteric inhibition of lysyl oxidase-like-2 impedes the development of a pathologic microenvironment. Nat Med. 2010;16(9):1009–1017. doi:10.1038/nm.2208
147. Barker HE, Chang J, Cox TR, et al. LOXL2-mediated matrix remodeling in metastasis and mammary gland involution. Cancer Res. 2011;71(5):1561–1572. doi:10.1158/0008-5472.CAN-10-2868
148. Quail DF, Joyce JA. Microenvironmental regulation of tumor progression and metastasis. Nat Med. 2013;19(11):1423–1437. doi:10.1038/nm.3394
149. Barker HE, Cox TR, Erler JT. The rationale for targeting the LOX family in cancer. Nat Rev Cancer. 2012;12(8):540–552. doi:10.1038/nrc3319
150. Li TY, Xu L-Y, Wu ZY, et al. Reduced nuclear and ectopic cytoplasmic expression of lysyl oxidase-like 2 is associated with lymph node metastasis and poor prognosis in esophageal squamous cell carcinoma. Hum Pathol. 2012;43(7):1068–1076. doi:10.1016/j.humpath.2011.07.027
151. Wu BL, Zou HY, Lv GQ, et al. Protein-protein interaction network analyses for elucidating the roles of LOXL2-delta72 in esophageal squamous cell carcinoma. Asian Pac J Cancer Prev. 2014;15(5):2345–2351. doi:10.7314/APJCP.2014.15.5.2345
152. Wu H, Zheng J, Deng J, et al. LincRNA-uc002yug.2 involves in alternative splicing of RUNX1 and serves as a predictor for esophageal cancer and prognosis. Oncogene. 2015;34(36):4723–4734. doi:10.1038/onc.2014.400
153. Huarte M, Rinn JL. Large non-coding RNAs: missing links in cancer? Hum Mol Genet. 2010;19(R2):R152–61. doi:10.1093/hmg/ddq353
154. Gibb EA, Brown CJ, Lam WL. The functional role of long non-coding RNA in human carcinomas. Mol Cancer. 2011;10:38. doi:10.1186/1476-4598-10-38
155. Prensner JR, Chinnaiyan AM. The emergence of lncRNAs in cancer biology. Cancer Discov. 2011;1(5):391–407. doi:10.1158/2159-8290.CD-11-0209
156. Ji P, Diederichs S, Wang W, et al. MALAT-1, a novel noncoding RNA, and thymosin β 4 predict metastasis and survival in early-stage non-small cell lung cancer. Oncogene. 2003;22(39):8031–8041. doi:10.1038/sj.onc.1206928
157. Tsai MC, Manor O, Wan Y, et al. Long noncoding RNA as modular scaffold of histone modification complexes. Science. 2010;329(5992):689–693. doi:10.1126/science.1192002
158. Yap KL, Li S, Muñoz-Cabello AM, et al. Molecular interplay of the noncoding RNA ANRIL and methylated histone H3 lysine 27 by polycomb CBX7 in transcriptional silencing of INK4a. Mol Cell. 2010;38(5):662–674. doi:10.1016/j.molcel.2010.03.021
159. Okuda T, Nishimura M, Nakao M, et al. RUNX1/AML1: a central player in hematopoiesis. Int J Hematol. 2001;74(3):252–257. doi:10.1007/BF02982057
160. Wang H, Iakova P, Wilde M, et al. C/EBPalpha arrests cell proliferation through direct inhibition of Cdk2 and Cdk4. Mol Cell. 2001;8(4):817–828. doi:10.1016/S1097-2765(01)00366-5
161. Chimge N-O, Frenkel B. The RUNX family in breast cancer: relationships with estrogen signaling. Oncogene. 2013;32(17):2121–2130. doi:10.1038/onc.2012.328
162. Scheitz CJ, Lee TS, McDermitt DJ, et al. Defining a tissue stem cell-driven Runx1/Stat3 signalling axis in epithelial cancer. EMBO j. 2012;31(21):4124–4139. doi:10.1038/emboj.2012.270
163. Han H, Irimia M, Ross PJ, et al. MBNL proteins repress ES-cell-specific alternative splicing and reprogramming. Nature. 2013;498(7453):241–245. doi:10.1038/nature12270
164. Dulak AM, Schumacher SE, van Lieshout J, et al. Gastrointestinal adenocarcinomas of the esophagus, stomach, and colon exhibit distinct patterns of genome instability and oncogenesis. Cancer Res. 2012;72(17):4383–4393. doi:10.1158/0008-5472.CAN-11-3893
165. Ito Y. Oncogenic potential of the RUNX gene family: ‘overview’. Oncogene. 2004;23(24):4198–4208. doi:10.1038/sj.onc.1207755
166. Tsuzuki S, Hong D, Gupta R, et al. Isoform-specific potentiation of stem and progenitor cell engraftment by AML1/RUNX1. PLoS Med. 2007;4(5):e172. doi:10.1371/journal.pmed.0040172
167. Nakajima H, Hori Y, Terano H, et al. New antitumor substances, FR901463, FR901464 and FR901465. II. Activities against experimental tumors in mice and mechanism of action. J Antibiot. 1996;49(12):1204–1211. doi:10.7164/antibiotics.49.1204
168. Karni R, Hippo Y, Lowe SW, et al. The splicing-factor oncoprotein SF2/ASF activates mTORC1. Proc Natl Acad Sci U S A. 2008;105(40):15323–15327. doi:10.1073/pnas.0801376105
169. Zhang J, Lieu YK, Ali AM, et al. Disease-associated mutation in SRSF2 misregulates splicing by altering RNA-binding affinities. Proc Natl Acad Sci U S A. 2015;112(34):E4726–34. doi:10.1073/pnas.1514105112
170. Wang Z, Burge CB. Splicing regulation: from a parts list of regulatory elements to an integrated splicing code. Rna. 2008;14(5):802–813. doi:10.1261/rna.876308
171. Castanotto D, Stein CA. Antisense oligonucleotides in cancer. Curr Opin Oncol. 2014;26(6):584–589. doi:10.1097/CCO.0000000000000127
172. Sun JR, Kong CF, Lou YN, et al. Genome-wide profiling of alternative splicing signature reveals prognostic predictor for esophageal carcinoma. Front Genet. 2020;11:796. doi:10.3389/fgene.2020.00796
173. Xie ZC, Wu H-Y, Ma F-C, et al. Prognostic alternative splicing signatures and underlying regulatory network in esophageal carcinoma. Am J Transl Res. 2019;11(7):4010–4028.
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.