")
Back to Journals » Drug Design, Development and Therapy » Volume 9
Progesterone regulates the proliferation of breast cancer cells – in vitro evidence
Authors Azeez J, Sithul H, Hariharan I, Sreekumar S, Prabhakar J, Sreeja S, Pillai M
Received 27 May 2015
Accepted for publication 5 August 2015
Published 9 November 2015 Volume 2015:9 Pages 5987—5999
DOI https://doi.org/10.2147/DDDT.S89390
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Prof. Dr. Wei Duan
Juberiya M Azeez,1 Hima Sithul,1 Indhu Hariharan,1 Sreeja Sreekumar,1 Jem Prabhakar,2 Sreeharshan Sreeja,1 Madhavan Radhakrishna Pillai1
1Cancer Research Program, Rajiv Gandhi Centre for Biotechnology, 2Division of Surgical Oncology, Regional Cancer Centre, Thiruvananthapuram, India
Abstract: Reports state that surgery performed at different phases of the menstrual cycle may significantly affect breast cancer treatment outcome. From previous studies, we identified differentially expressed genes in each menstrual cycle phase by microarray, then subjected them to functional in vitro analyses. Microarray studies disclosed genes that are upregulated in the luteal phase and follicular phase. TOB-1 is a tumor suppressor gene and was expressed exclusively in the luteal phase in our microarray study. Therefore, we further functionally characterized the protein product of TOB-1 in vitro. To our knowledge, no studies have yet been conducted on reactive oxygen species-regulated tumor suppressor interactions in accordance with the biphasic nature of progesterone. This work demonstrates that progesterone can produce reactive oxygen species in MCF-7 cells and that TOB-1 exerts a series of non-genomic interactions that regulate antiproliferative activity by modulating the antioxidant enzyme superoxide dismutase. Furthermore, this study implicates PTEN as an interacting partner for TOB-1, which may regulate the downstream expression of cell cycle control protein p27 via multiple downstream signaling pathways of progesterone through a progesterone receptor, purely in a time- and concentration-dependent manner. These results support the hypothesis that surgery conducted during the luteal phase of the menstrual cycle may facilitate improved patient survival.
Keywords: progesterone, reactive oxygen species, TOB-1, cell growth arrest
Introduction
The importance of the timing of tumor removal in relation to the menstrual cycle and its influence on disease-free survival has been studied by researchers since 1988; however, such work remains exploratory. Although a hypothetical role for the menstrual cycle in the surgical treatment of breast cancer has been proposed by Hortobagyi,1 experimental evidence and supporting data are lacking. Therefore, the molecular mechanisms underlying the timing of breast cancer surgery with respect to menstrual cycle phase are relevant; our studies profiled the gene expression in tissues from different phases of the menstrual cycle. Most of the reflective clinical studies, and two meta-analyses, have reported an average 10-year disease-free survival advantage for breast cancers that are resected during the early luteal phase of the menstrual cycle.2–4 The concept of the timing of surgical intervention to treat breast cancer in premenopausal women based on the different phases of their menstrual cycle originated with a report that women who experienced a mastectomy during their perimenstrual period exhibited a greater metastatic risk than those operated on during their mid-cycle.5–7 The results of numerous studies concerning the consequence of breast cancer surgery timing in relation to the menstrual cycle phase have been conflicting and lacking biological consistency in the survival differences observed as a result of surgery timing in premenopausal women.8–10 In this study, we explored the role of progesterone and the nuclear progesterone receptor (PR) in the progression of steroid hormone receptor-positive breast cancer on patient survival, with indirect implications about surgery timing in relation to the menstrual cycle phase. Progesterone action is mediated by PR, expressed as two functionally different proteins (PRA or PRB) that are controlled by separate promoters.11 Studies on human endometrium have demonstrated that PRA and PRB are either distributed evenly throughout the nucleus or into discrete nuclear foci, the latter of which coincides with the secretory phase of the menstrual cycle, wherein serum progesterone levels are high and PR activity is maximal. Abnormal PR foci formation has been observed in cancer tissue when compared with normal tissue, possibly impacting PR-mediated transcription and following target gene expression.12 Human breast cancer cell-line investigations have provided valuable insights into the paradoxical effects of progestins on cell proliferation, demonstrating clear biphasic effects on cell cycle progression.13 Progestins and growth factors synergistically augment the transcription of key cell cycle components, including cyclin D1, cyclin E, and p21WAF1, indicating crosstalk between these signaling pathways.14 A single treatment of progesterone is growth stimulatory, but after 48 hours cell growth is halted, and a second stimulus fails to restart growth. This growth arrest is accompanied by a gradual increase in p27, leading to cell cycle arrest at the late G1 phase and to initiation of cell differentiation.15–17 Progesterone, the dominant hormone of the luteal phase, impacts the growth inhibition of breast cancer, which should generate research attention. Based on data from real-time and immunohistochemical analyses, we have chosen to study the role of the gene TOB-1, which is overexpressed in the luteal phase, in progesterone-mediated signaling in breast cancer. TOB-1, also known as a transducer of ErbB2, is a tumor suppressor protein that belongs to the B-cell translocation gene (TOB/BTG) family.18 The tumor suppressor activity of TOB-1 resides in its ability to negatively regulate the cell cycle by blocking G1 progression. TOB-1 can serve as a tumor suppressor in MCF-7 breast cancer cells by modulating multiple signaling pathways. Earlier studies have also reported that TOB-1 functions19 by modulating epidermal growth factor receptor and its downstream signaling events through direct or indirect interaction with a key tumor suppressor, PTEN.18 Accumulating evidence has indicated that PTEN exerts its tumor-suppressive behavior through its phosphatase activity and its protein interactions.20 PTEN promotes arrest of the cell cycle G1 phase by downregulating cyclin D1 through its protein phosphatase activity, while upregulating p27 through its lipid phosphatase activity, in breast cancer cells.21 From our studies, we identified certain genes that are solely expressed in each menstrual cycle phase. Additionally, we determined that progesterone – the primary hormone of the luteal phase – regulates TOB-1 function, subsequently inhibiting the expression of the antioxidant enzyme superoxide dismutase 1 (SOD1), which in turn increases the generation of reactive oxygen species (ROS) and leads to cell cycle arrest signaling.
Materials and methods
For immunohistochemical analysis, human tissue section slides were obtained as per the Institutional Human Ethical clearance certificate number IHEC/01/2011/02 from the Rajiv Gandhi Centre for Biotechnology (Thiruvananthapuram, India).
Cell lines, antibodies, and reagents
MCF-7, T47D, SKBR3, and MCF-10A cell lines were purchased from American Type Culture Collection (Manassas, VA, USA) and maintained in Dulbecco’s Modified Eagle’s Medium (DMEM, 10%). For all experiments, cells were starved in DMEM containing 5% charcoal-stripped fetal bovine serum (Invitrogen, Carlsbad, CA, USA). Ro-green fluorescent protein (GFP) MCF-7 cells were obtained as a gift from Dr T R Santhosh Kumar (Rajiv Gandhi Centre for Biotechnology, India). Progesterone and Annexin V-FITC Apoptosis Detection Kits were acquired from Sigma-Aldrich Co. (St Louis, MO, USA). Anti-p27, anti-SOD2, and anti-TOB-1 antibodies were purchased from Santa Cruz Biotechnology Inc. (Santa Cruz, CA, USA). Anti-PRB and anti-SOD1 antibodies were obtained from Cell Signaling Technology (Danvers, MA, USA), anti-p53 antibody was obtained from BD Biosciences (San Diego, CA, USA), and anti-PTEN antibody was obtained from Abcam (Cambridge, UK). All secondary antibodies were from Sigma-Aldrich.
Small interfering RNA transfections
Cells were transfected with TOB-1 (Santa Cruz Biotechnology Inc., Santa Cruz, CA, USA) double-stranded RNA oligonucleotides using the Lipofectamine RNAiMax transfection method (Invitrogen), according to the manufacturer’s protocol. Control small interfering RNA (siRNA) (Santa Cruz Biotechnology Inc.) were used as negative controls for transfection.
Chromatin condensation assay
Apoptotic cell populations were detected using Hoechst 33342 (Life Technologies, Carlsbad, CA, USA) staining. After treatment with progesterone at 25 nM for 24, 48, and 72 hours, cells were fixed with 4% paraformaldehyde for 10 minutes at room temperature, stained with Hoechst 33342 (5 mg/mL) for 20 minutes at 37°C in the dark, and visualized under a fluorescence microscope (Eclipse E-600, Nikon, Melville, NY, USA), utilizing a 350 nm excitation and a 460 nm emission filter.
Detection of apoptotic cells
The Annexin V-FITC Apoptosis Detection Kit (Sigma-Aldrich Co.) was used for the detection of apoptotic cells as per the manufacturer’s protocol. Briefly, cells were treated with progesterone for 48 and 72 hours. After treatment, the cells were washed with cold phosphate-buffered saline (PBS) and then trypsinized. From the cell suspension, 1×106 cells were resuspended in 1× binding buffer and then incubated with 5 μL of Annexin V-FITC and 10 μL of propidium iodide (PI) solution. Finally, they were incubated for 10 minutes in the dark. Fluorescence of the cells was determined using flow cytometry (FACS Aria1; Becton Dickinson, San Jose, CA, USA).
Detection of intracellular ROS
Intracellular ROS were estimated using a fluorescent probe: 2′,7′-dichlorodihydrofluorescein diacetate (Calbiochem, EMD Millipore, Billerica, MA, USA). Cells were seeded on 12-well plates, and at 60% confluence cells were administered a charcoal treated serum treatment, followed by a 25 nM progesterone treatment, along with control cells, for 24, 48, and 72 hours. Cell-permeable fluorogenic probe 2′, 7′-Dichlorodihydrofluorescin diacetate (DCFH-DA) was added to each well at a final concentration of 5 μM, and cells were then incubated for 15 minutes at 37°C with 5% CO2 in the dark. After centrifugation, the cell samples were immediately analyzed via fluorescent dye (2′, 7′-dichlorofluorescein) detection using flow cytometry (FACSAria I, Becton, Dickinson and Company, San Jose, CA, USA).
Redox-sensitive GFP imaging
For redox-sensitive GFP imaging, MCF-7-Ro-GFP cells were cultured in six-well plates and transfected with TOB-1 siRNA. After 24 hours, the cells were treated with progesterone and imaged under an epifluorescent microscope using xenon as the excitation light source (Lamda XL, Sutter Instrument Company, Novato, CA, USA). An emission wavelength of 535/30 nm was collected upon dual excitation using a filter set of 405/20X and 488/20X in a sequential mode. Images were captured with a charge coupled device camera (CoolSNAP HQ, Photometrics, Tucson, AZ, USA). The ratio images were generated by dividing the 405 nm channel by the 488 nm channel on a pixel-by-pixel basis using NIS-Elements software (version 3.21.00, Nikon, Minato-ku, Japan).
Cell cycle analyses
Cell cycle distribution was analyzed by flow cytometry. Control and treated cells were harvested, washed twice with PBS and fixed in 70% ethanol overnight at −20°C. Fixed cells were washed twice with PBS and incubated with 1 mL of PBS containing 50 μg/mL PI and 100 μg/mL RNase A for 30 minutes at 37°C. Stained cells were analyzed using a FACSAria I flow cytometer (Becton, Dickinson and Company).
Western blot analyses
Progesterone-treated and non-treated cells were washed with PBS and lysed using radioimmunoprecipitation assay buffer (150 mM NaCl, 1% nonylphenoxypolyethoxyethanol, 0.5% sodium deoxycholate, 0.1% sodium dodecyl sulfate, 50 mM Tris–HCl, pH 7.4) containing protease inhibitors (Sigma-Aldrich). Briefly, equal amounts of protein, as determined by the Bradford assay, were subjected to sodium dodecyl sulfate polyacrylamide gel electrophoresis, followed by transfer to a nitrocellulose membrane (Millipore, Billerica, MA, USA). The membrane was blocked in a 5% powdered non-fat milk solution for 1 hour. The membrane was then incubated overnight with a primary antibody, followed by incubation with a species-specific horseradish peroxide-conjugated secondary antibody (1:5,000, Sigma-Aldrich) at room temperature for 1 hour. Protein bands were visualized on X-ray film using ECL Plus reagents (Thermo Fisher Scientific, Waltham, MA, USA).
Statistical analyses
Data are presented as the mean ± SD of at least three experiments or are representative of experiments repeated at least three times.
Results
Role of TOB-1 in progesterone signaling
In view of the contradicting reports of Love et al4 and Hortobagyi,1 our earlier microarray gene expression analyses revealed key genes that are noticeably downregulated in the luteal phase with anti-invasive and anti-metastatic properties. Although estrogen is a major stimulant of mammary cell proliferation, the effect of progesterone remains controversial. As epidemiological data6 have shown that a progesterone-rich phase has a role in surgery timing and patient prognosis, we conducted a detailed study of the stimulatory role of progesterone. Reports regarding the biphasic nature of progesterone also exist.22–24 Notably, PRA and PRB are expressed in most human target cells, suggesting the involvement of alternative mechanisms that control the diversity of progesterone actions.25 As TOB-1 is a well-known tumor suppressor in breast cancer,19 which showed consistent positivity in our clinical samples (Figure 1) in our earlier studies, we focused on the functional characterization of TOB-1 in the presence of a physiological level of progesterone in vitro. The optimum concentration of progesterone in the selected cells was determined by an 3-(4,5-dimethythiazol- 2-yl)-2, 5-diphenyl tetrazolium bromide (MTT)-based cytotoxicity assay (Figure S2) and cell viability assay by fluorescence-activated cell sorting (FACS) (Figure S3) and compared with that reported in previous research. Initial studies of cells treated with progesterone revealed23,24 a time-dependent regulation of TOB-1 in MCF-7 cells (Figure 2).
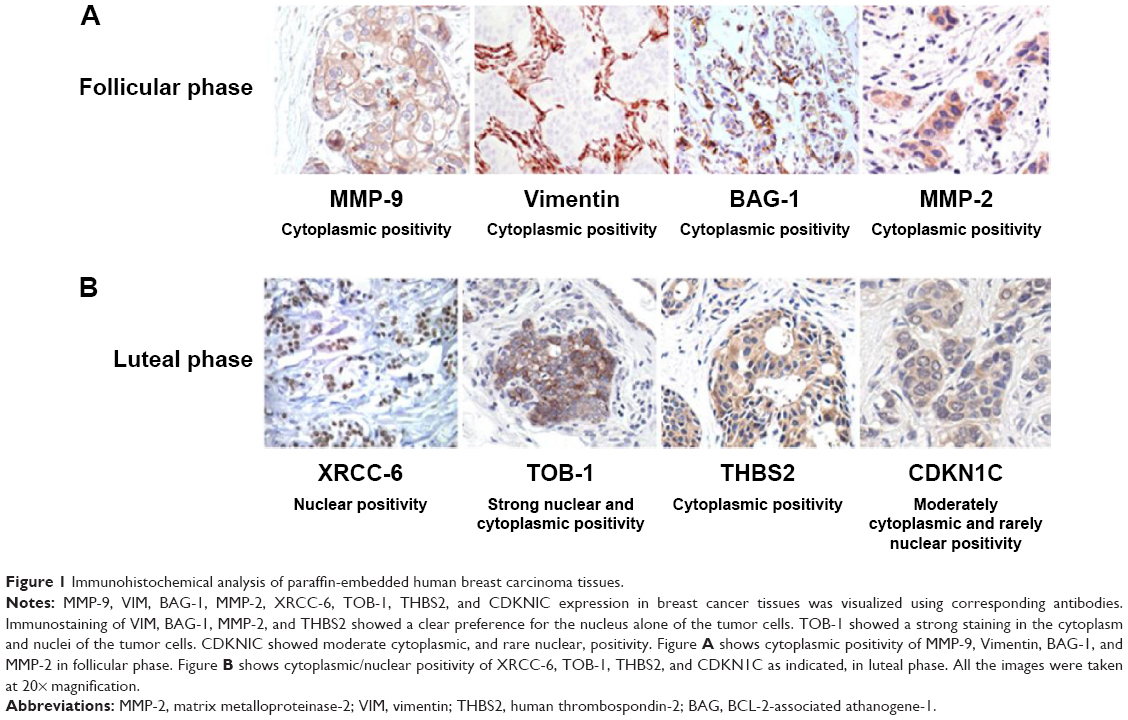 | Figure 1 Immunohistochemical analysis of paraffin-embedded human breast carcinoma tissues. |
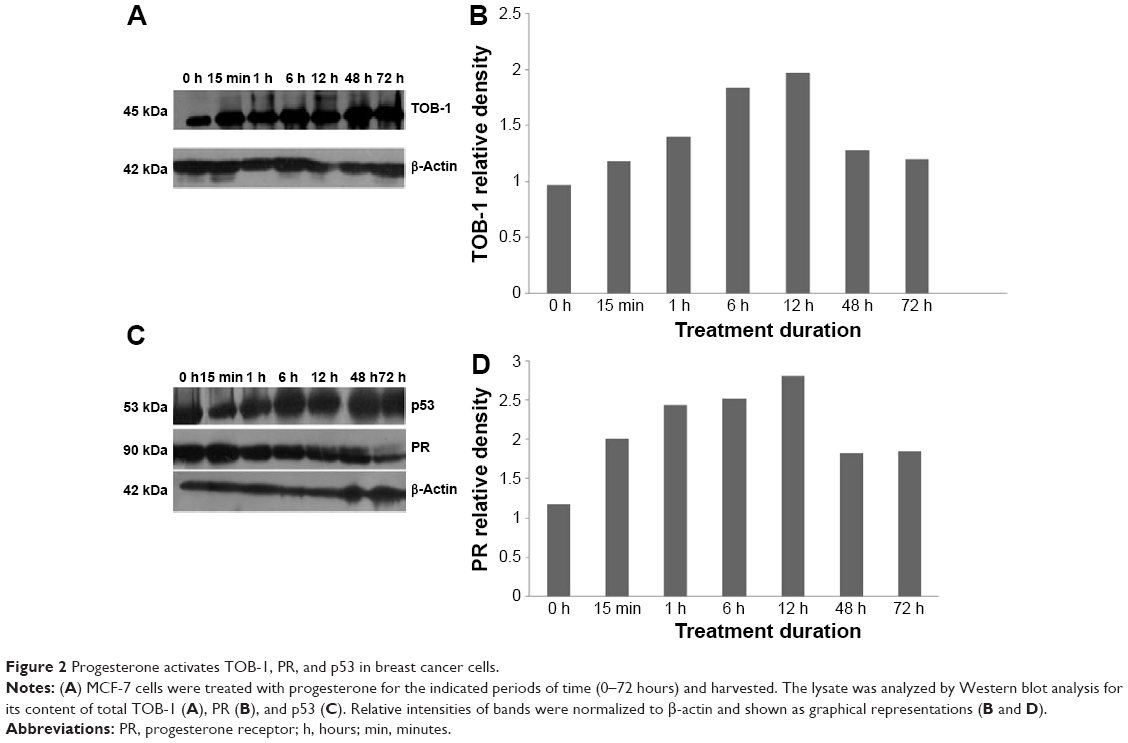 | Figure 2 Progesterone activates TOB-1, PR, and p53 in breast cancer cells. |
Progesterone-induced chromatin condensation and apoptosis
To further understand whether the concentration of progesterone that induced TOB-1 expression in MCF-7 cells simultaneously induced apoptotic changes, chromatin condensation and apoptosis were assessed in progesterone-treated samples by fluorescence-activated cell sorting analyses. As shown in Figure 3A, Hoechst 33342-stained MCF-7 cells treated with progesterone at a significant concentration for 24, 48, and 72 hours showed less significant nuclear chromatin condensation. This result was further confirmed by Annexin V-FITC/PI double staining (Figure 3B). Annexin V-positive/PI-negative cells were regarded as early apoptotic cells, while Annexin V-positive and PI-positive cells were regarded as late apoptotic or necrotic cells. As shown in Figure 3B, we only observed mild apoptosis or necrosis in progesterone-treated cells.
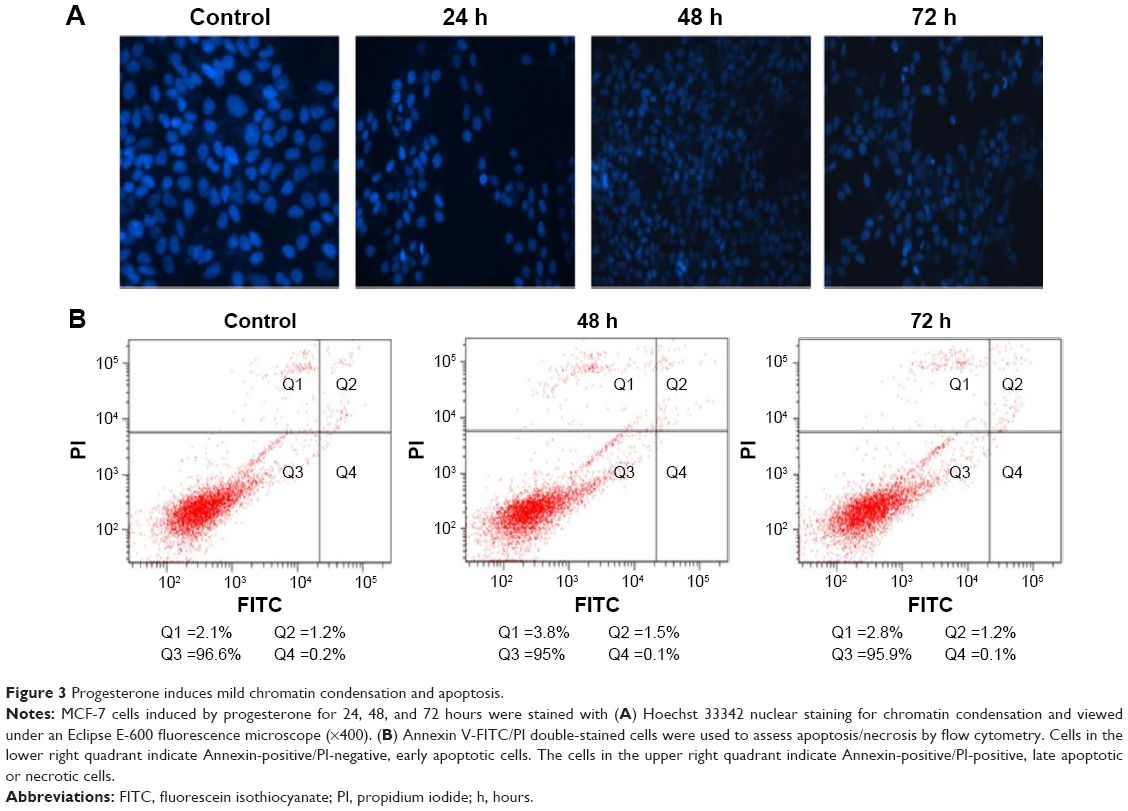 | Figure 3 Progesterone induces mild chromatin condensation and apoptosis. |
Progesterone-induced cell cycle arrest
As progesterone-treated cells showed only mild apoptosis, we confirmed that the progesterone influence on cell growth was due to the cell cycle. Cell cycle analysis by flow cytometry after 48 hours indicated that progesterone-treated cells showed accumulation in the sub-G1 phase (Figure 4). Reports have implicated cell cycle inhibitor p27 as a negative regulator of G1 progression, which in turn controls cyclin D1 expression.26
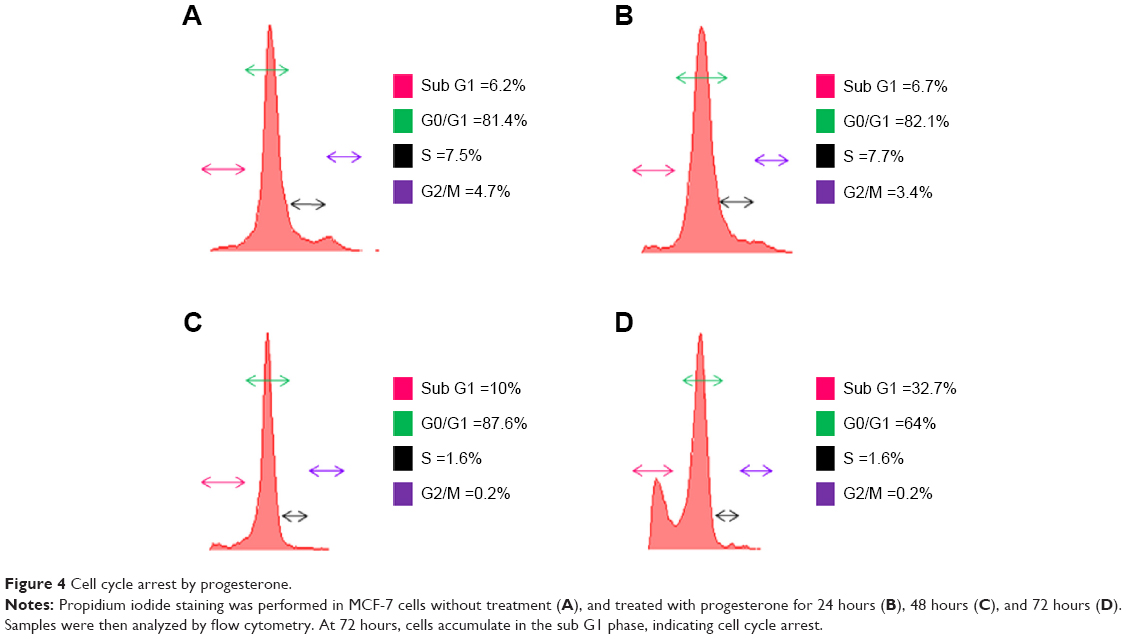 | Figure 4 Cell cycle arrest by progesterone. |
Knockdown of TOB-1 reduced p27 and PTEN activation
The earlier data indicate that progesterone influences cell cycle progression in MCF-7 cells with mild cell death associated with TOB-1 induction. However, whether the observed effect is a direct or indirect consequence of TOB-1 induction remains unclear. Therefore, we first evaluated the expression of the key cell cycle regulator p27 upon progesterone treatment at different time points (Figure 5A and B) and also in TOB-1 silenced cells (Figure 5C and D). In progesterone-treated cells, p27 expression increased to a maximum level within 12 hours (Figure 5A and B), whereas p27 expression decreased in silenced cells (Figure 5E and F). These trends may be due to the time-dependent survival signal of progesterone, as well as the interaction between TOB-1 and p27. Here, progesterone may act as a regulator of the cell cycle through TOB-1 and p27. A recent study showed that the biological activity of TOB-1 is initiated by its interaction with the key tumor suppressor protein PTEN, which inhibits the PI3K/Akt signaling pathway and has a multifunctional role in cell proliferation, migration, and invasion.18,27,28 Interestingly, silencing of TOB-1 resulted in the downregulation of PTEN (Figure 6), suggesting TOB-1-dependent PTEN signaling. Thus, progesterone can modulate the function of TOB-1, subsequently influencing the function of PTEN and ultimately leading to increased p27 expression.
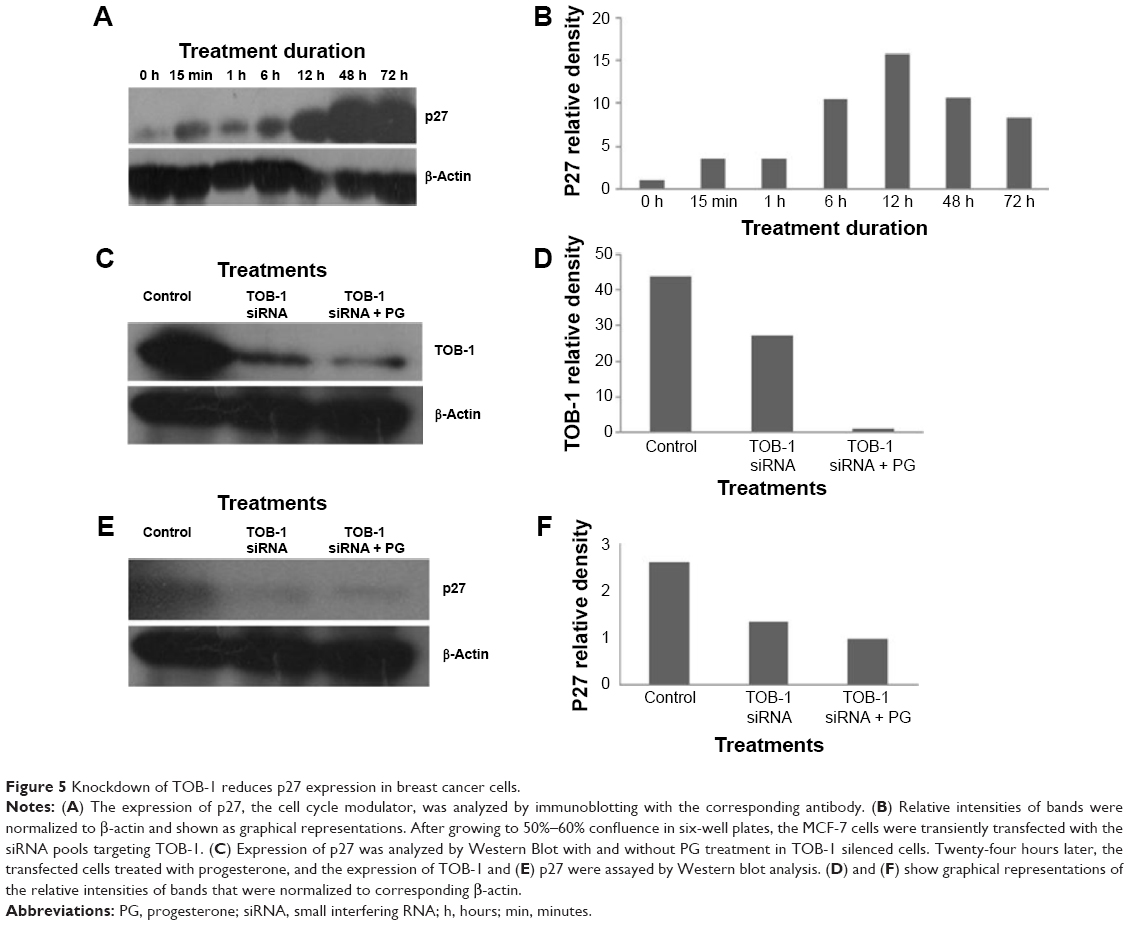 | Figure 5 Knockdown of TOB-1 reduces p27 expression in breast cancer cells. |
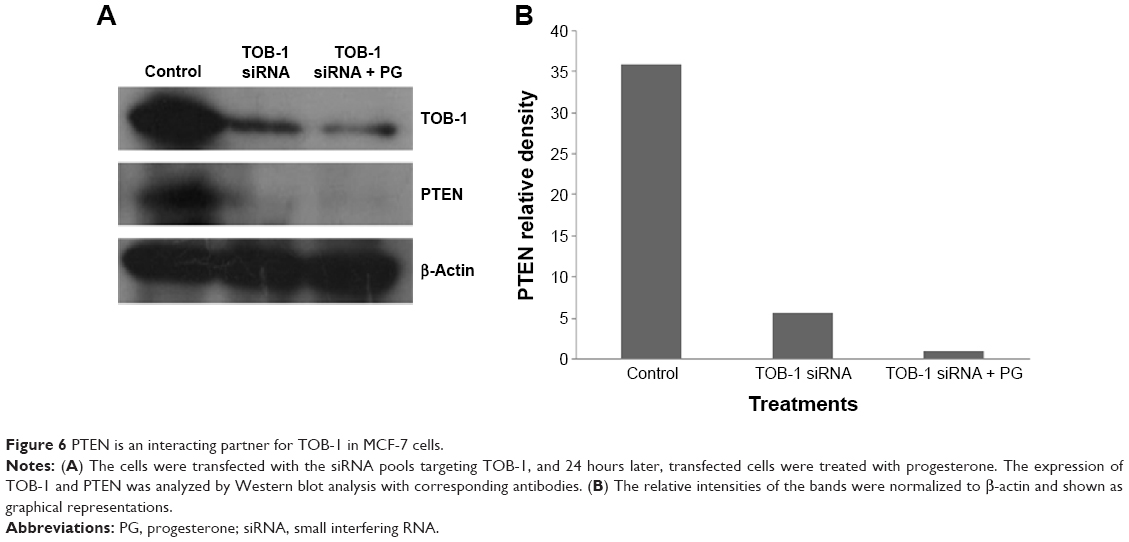 | Figure 6 PTEN is an interacting partner for TOB-1 in MCF-7 cells. |
Progesterone-mediated ROS regulate TOB-1 function
Steroids have been implicated in modulating the redox balance in cells. The ability of estradiol to reduce the generation of ROS has been demonstrated in vitro and in vivo.29 Furthermore, Dabrosin et al30 demonstrated alteration of redox balance in normal cultured human breast epithelial cells by estradiol and progesterone treatment, which was associated with increased cell death. Nguyen and Syed31 reported that progesterone regulates the growth of breast cancer cells by modulating the redox balance. Studies have also suggested the critical role of intracellular redox balance in the progression of the cell cycle.32 We observed an increase in TOB-1 upon progesterone treatment with changes in cell cycle progression. Therefore, experiments were proposed to understand the link between progesterone, TOB-1, and redox balance. Progesterone-induced ROS generation in a time-dependent manner in MCF-7 cells, as indicated by the increase in 2′,7′-dichlorodihydrofluorescein diacetate fluorescence in treated cells when compared with control cells and counterpart MCF-10A cells (Figure 7). These results were further confirmed by calculating the 405/488 nm ratio in MCF-7-Ro-GFP cells (Figure 7B), where TOB-1 silencing reduced the formation of ROS in the presence of progesterone. Another interesting observation is that progesterone treatment does not alter the ROS level in T47D and MCF-10A cells (Figure S1), which may suggest the importance of the estrogen receptor (ER)/PR ratio in different breast cancer cell lines for developing a change in redox balance, in response to progesterone.33 Intracellular ROS are regulated by several antioxidant and pro-oxidant signaling molecules, of which the SOD group of enzymes is prominent; in particular, SOD1 is overexpressed in cancer.34 Thus, we analyzed whether the time-dependent increase in progesterone-induced ROS generation altered the SOD balance. As shown in Figure 7C, we observed a significant reduction in the level of SOD1 in treated cells compared with that of SOD2. SOD1 expression was very low at 24 hours (Figure 7C) compared with control cells. However, SOD2 (Figure 7C) showed reciprocal expression of SOD1. The PR and p53 were considered to be prognostic markers in predicting clinical outcomes of breast cancer patients. We have shown that progesterone treatment causes biphasic activation of the PR. Taken together, all of these experiments suggest that progesterone may alter the redox balance in MCF-7 cells, subsequently triggering tumor suppressor function, which may lead to further cell growth arrest, at specific concentrations.
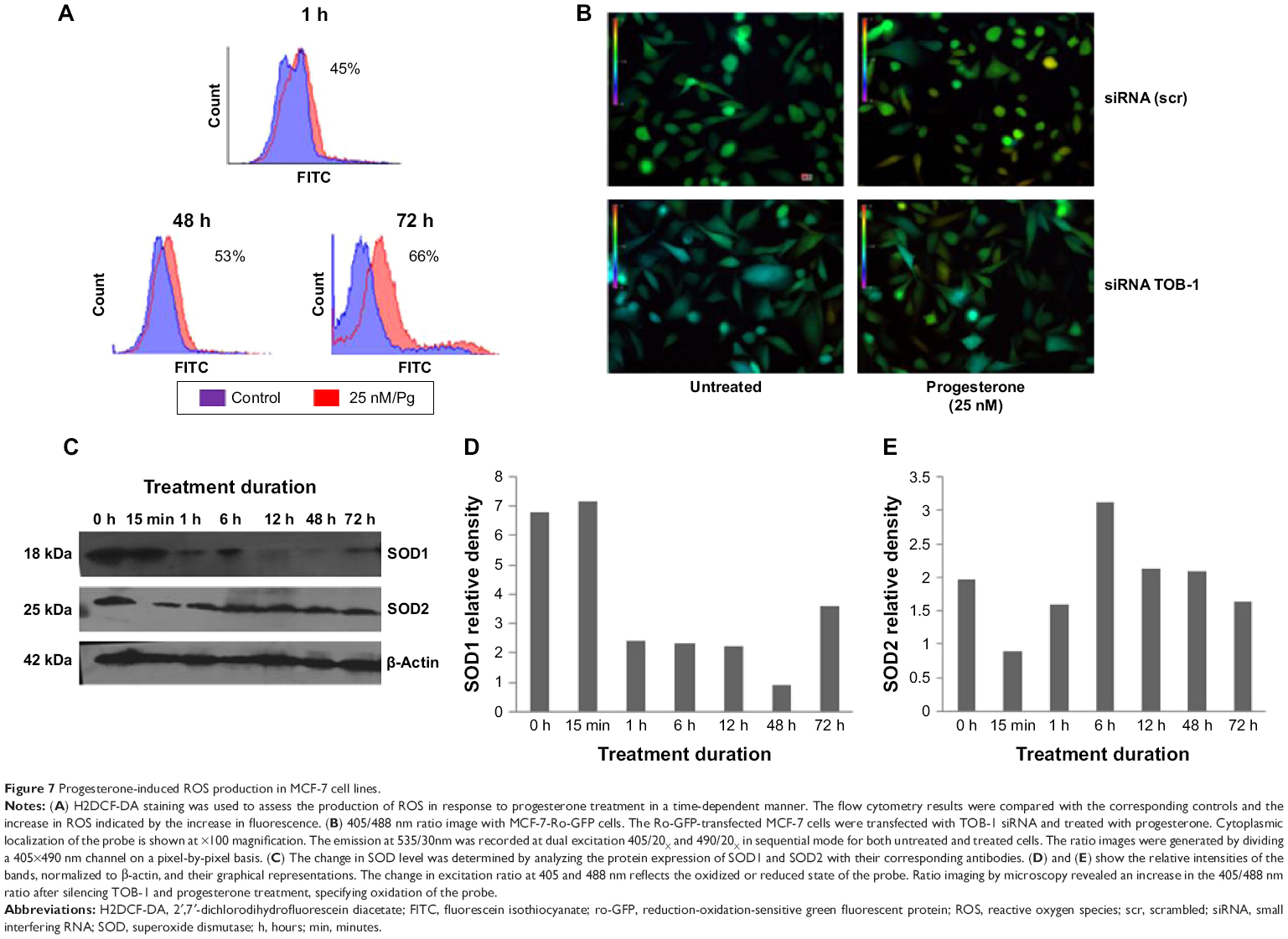 | Figure 7 Progesterone-induced ROS production in MCF-7 cell lines. |
Discussion
Surgical intervention is a treatment option for breast cancer patients.35 In 1989, Hrushesky et al6 proposed that the timing of surgical interventions for breast cancer had a substantial influence on the outcome. The option for surgical intervention mainly depends on the cancer stage.36 Subsequently, a number of controversial reports followed.4 Thus, this study attempted to determine a possible molecular explanation for improved patient survival during breast cancer surgery treatment in the luteal phase. Using microarray-based gene expression analyses, we explored several pro-apoptotic and cell cycle-regulating genes in the luteal phase and anti-apoptotic genes in the follicular phase of the menstrual cycle. In clinical practice, the co-expression of PR with ER is currently assessed as a predictive marker for favorable disease prognosis and for response to hormonal therapy. During the secretory phase of the menstrual cycle, when high circulating levels of progesterone are associated with reduced PR expression, PRA was preferentially reduced, resulting in a distinct predominance of PRB.37 The high posttranslational modification of PR dramatically alters its function, receptor localization, and promoter selectivity. Additionally, the function of PR is complex in the context of tissue-specific effects, isoform-specific actions, timing, and hormone delivery dose.38 In the present study, we demonstrated the expression of PRB (Figure 2C) with a moderate concentration of progesterone in vitro, which most likely reflects the expression of PRs at different phases of the menstrual cycle. For the first time, we showed that the protein product of TOB-1 – TOB-1 – which is identified as one of the differentially expressed genes exclusively present in the luteal phase in our tissue microarray study, exerts ROS-regulated antiproliferative activity, followed by modulation of multiple downstream signaling pathways via PR. Jiao et al18 reported that TOB-1 functions as a tumor suppressor by modulating epidermal growth factor receptor and its downstream signaling pathways through a direct or indirect interaction with the key tumor suppressor PTEN. Loss of PTEN expression occurs commonly in breast cancer, which has been associated with loss of ER39 and resistance to cancer therapies.40 Recent reports have shown the regulatory role of PTEN in breast carcinoma cell growth.41 Correlation of PTEN loss with ROS regulation in progesterone-treated cells in vitro has not been well characterized in the literature, with the exception of a few studies that analyzed prognostic parameters. The findings of these studies were consistent with those of our own study and showed a significant reduction in PTEN expression when the TOB-1 function was silenced. Loss of PTEN expression occurs commonly in breast cancer and correlates with disease-related death, lymph node metastasis, and loss of receptor staining.42 Thus, ROS generated due to progesterone promote a survival signal that may ultimately control the function of PTEN. None of the studies compared survival and disease recurrence in connection with PTEN expression. The tumor suppressor protein p53 is a redox-active transcription factor that organizes and directs cellular responses in the face of a variety of stresses, leading to genomic instability. Both ROS and p53 participate in multiple cellular processes, and interactions between, and pathway intersections of ROS and p53 occur.43 Progesterone, which generates ROS in MCF-7 cells, may activate p53 through antioxidant enzyme interactions (Figure 2C). Vurusaner et al43 reported that manganese superoxide dismutase (MnSOD) is suppressed at the promoter level by p53 activation. Suppression of antioxidant genes by p53 is an alternative way to increase cellular ROS, conferring oxidative stress. Reduced MnSOD activity can favor proliferation owing to increased superoxide and low hydrogen peroxide levels, whereas heightened MnSOD activity drives proliferating cells to transition into quiescence owing to increased hydrogen peroxide generation. In this context, the SOD1 and SOD2 balance determines whether a cell proliferates or transitions to a quiescent state. PTEN and PI3K regulate p27 at a posttranscriptional level, and p27 protein stability also increases in cells expressing PTEN.44 Our study revealed that progesterone can activate PTEN, which may further control the activity of p27 at a transcriptional level. As no significant activity or difference in PI3K phosphorylation occurred upon progesterone treatment, PTEN may be active in progesterone signaling through its phosphatase-independent activity. Several studies have demonstrated that PTEN can activate p53 through direct and indirect protein–protein interactions, including phosphatase-dependent and phosphatase-independent mechanisms.41,45 With all of these observations, we propose a model for the action of progesterone in ER/PR-positive breast cancer via PR in a time- and concentration-dependent manner by modulating signaling pathways that include tumor suppressor pathways (Figure 8). Although many questions remain, the current findings may lead to further investigations regarding the impact of menstrual cycle hormones on breast cancer. A better understanding of breast cancer surgery timing could potentially save the lives of women by preventing recurrence of the disease.
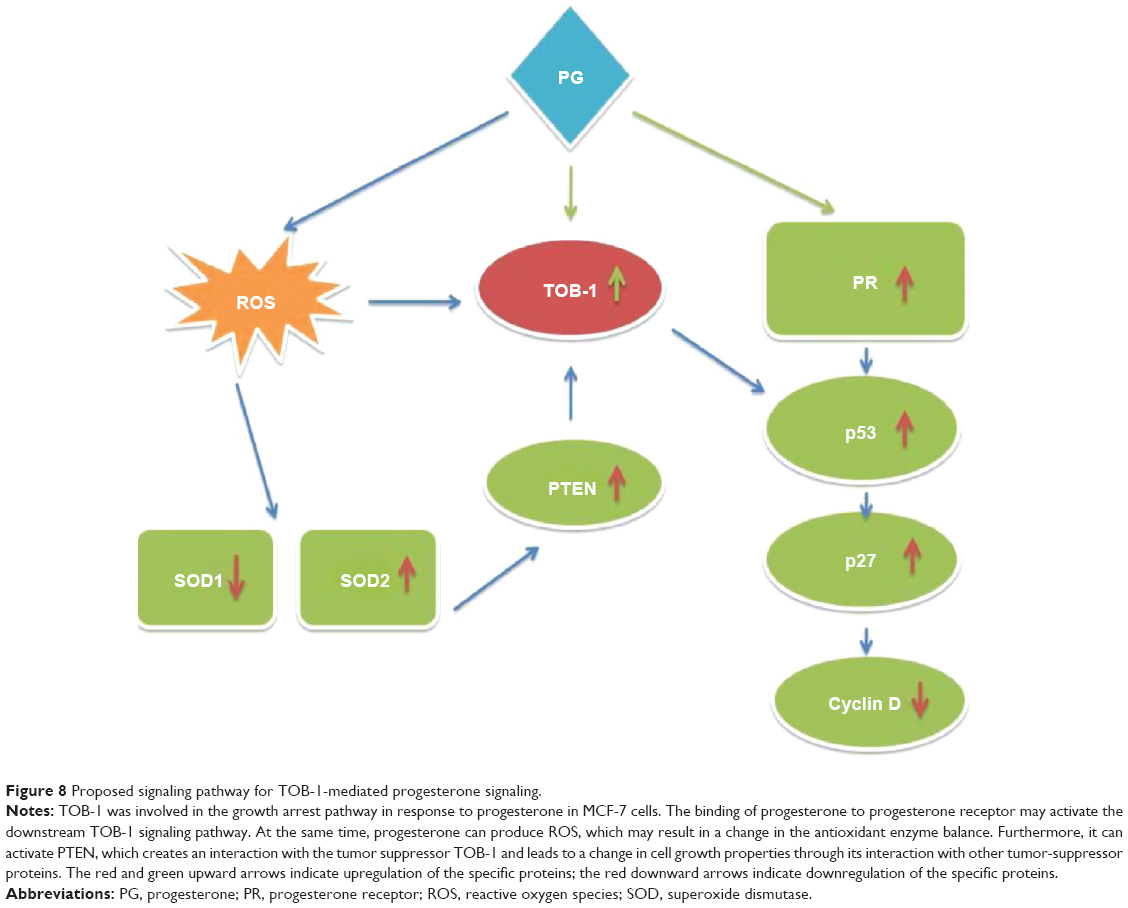 | Figure 8 Proposed signaling pathway for TOB-1-mediated progesterone signaling. |
Conclusion
Different studies have demonstrated a biphasic progesterone response in breast cancer cells, consisting of an initial proliferative burst, followed by sustained growth arrest. The present study identified TOB-1 as a target for progesterone-mediated signaling through the PR, significantly influencing p53, PTEN, and p27 expression and downregulating antioxidant enzymes. The unique cascade identified from this study appears to result in the inhibition of breast cancer growth. Our results support, mechanistically, how ROS modulate and trigger the function of TOB-1 and PTEN, promoting crosstalk and subsequent signaling to cancer cells in optimum progesterone-rich conditions. The results clearly show that the luteal phase hormone has a role in the inhibition of breast cancer growth, which in turn may influence patient outcome, depending on the phase of the menstrual cycle during surgical breast cancer treatment. The timing of surgical treatment during a particular menstrual cycle phase plays a significant role in the survival of pre-menopausal breast cancer and could perhaps prolong and/or save a great number of patients’ lives.
Acknowledgments
We are grateful to the Regional Cancer Centre for providing samples. We would also like to thank Santhi Achuthan, Sherin, and Dr TR Santhosh Kumar, from the Rajiv Gandhi Centre for Biotechnology, for helping us with the experiments. This study was supported by a grant from the Kerala State Council for Science Technology and Environment (KSCSTE), the Government of Kerala, India, and the Rajiv Gandhi Centre for Biotechnology.
Disclosure
The authors report no conflicts of interest in this work.
References
Hortobagyi GN. The influence of menstrual cycle phase on surgical treatment of primary breast cancer: have we made any progress over the past 13 years? J Natl Cancer Inst. 2002;94(9):641–643. | ||
Hrushesky WJM. Breast cancer, timing of surgery, and the menstrual cycle: call for prospective trial. J Womens Health. 1996;5:555–565. | ||
Hrushesky WJM. Mastectomy and oophorectomy by menstrual cycle phase in women with operable breast cancer (letter to the editor). J Natl Cancer Inst. 2002;94(22):1720–1721. | ||
Love RR, Ba Duc N, Van Dinh N, et al. Mastectomy and oophorectomy by menstrual cycle phase in women with operable breast cancer. J Natl Cancer Inst. 2002;94(9):662–669. | ||
Hrushesky WJ, Gruber SA, Sothern RB. Natural killer cell activity: age, estrous- and circadian-stage dependence and inverse correlation with metastatic potential. J Natl Cancer Inst. 1988;80(15):1232–1237. | ||
Hrushesky WJ, Bluming AZ, Gruber SA, Sothern RB. Menstrual influence on surgical cure of breast cancer. Lancet. 1989;2(8669):949–952. | ||
Ratajczak HV, Sothern RB, Hrushesky WJM. Estrous influence on surgical cure of a mouse breast cancer. J Exp Med. 1988;168(1):73–83. | ||
Badwe RA, Gregory WM, Chaudary MA, et al. Timing of surgery during menstrual cycle and survival of premenopausal women with operable breast cancer. Lancet. 1991;337(8752):1261–1264. | ||
Hrushesky WJ. Timing of surgery in breast cancer (letter). Lancet. 1991;337:1603–1604. | ||
Senie R, Rosen P, Rhodes P, Lesser M. Timing of breast cancer excision during the menstrual cycle influences duration of disease-free survival. Ann Intern Med. 1991;115(5):337–342. | ||
Kastner P, Krust A, Turcotte B, et al. Two distinct estrogen-regulated promoters generate transcripts encoding the two functionally different human progesterone receptor forms A and B. EMBO J. 1990;9:1603–1614. | ||
Arnett-Mansfield RL, DeFazio A, Mote PA, Clarke CL. Subnuclear distribution of progesterone receptors A and B in normal and malignant endometrium. J Clin Endocrinol Metab. 2004;89(3):1429–1442. | ||
Sutherland RL, Prall OWJ, Watts CKW, Musgrove EA. Estrogen and progestin regulation of cell cycle progression. J Mammary Gland Biol Neoplasia. 1988;3(1):63–72. | ||
Lange CA, Richer JK, Shen T, Horwitz KB. Convergence of progesterone and epidermal growth factor signaling in breast cancer. Potentiation of mitogen-activated protein kinase pathways. J Biol Chem. 1998;273:31308–31316. | ||
Clarke CL, Sutherland RL. Progestin regulation of cellular proliferation. Endocr Rev. 1990;11(2):266–302. | ||
Horwitz KB. Mechanisms of hormone resistance in breast cancer. Breast Cancer Res Treat. 1993;26(2):119–130. | ||
Santen RJ, Manni A, Harvey H, Redmond C. Endocrine treatment of breast cancer in women. Endocr Rev. 1990;11(2):221–265. | ||
Jiao Y, Sun KK, Zhao L, Xu JY, Wang LL, Fan SJ. Suppression of human lung cancer cell proliferation and metastasis in vitro by the transducer of ErbB-2.1 (TOB-1). Acta Pharmacol Sin. 2012;33(2):250–260. | ||
O’Malley S, Su H, Zhang T, Ng C, Ge H, Tang CK. TOB suppresses breast cancer tumorigenesis. Int J Cancer. 2009;125(8):1805–1813. | ||
Yamada KM, Araki M. Tumor suppressor PTEN: modulator of cell signaling, growth, migration and apoptosis. J Cell Sci. 2001;114(Pt 13):2375–2382. | ||
Weng LP, Brown JL, Eng C. PTEN coordinates G1 arrest by down-regulating cyclin D1 via its protein phosphatase activity and up-regulating p27 via its lipid phosphatase activity in a breast cancer model. Hum Mol Genet. 2001;10:599–604. | ||
Groshong SD, Owen GI, Grimison B, et al. Biphasic regulation of breast cancer cell growth by progesterone: role of the cyclin-dependent kinase inhibitors, p21 and p27(Kip1). Mol Endocrinol. 1997;11(11):1593–1607. | ||
Lange CA, Richer JK, Horwitz KB. Hypothesis: progesterone primes breast cancer cells for cross-talk with proliferative or antiproliferative signals. Mol Endocrinol. 1999;13(6):829–836. | ||
Owen GI, Richer JK, Tung L, Takimoto G, Horwitz KB. Progesterone regulates transcription of the p21(WAF1) cyclin-dependent kinase inhibitor gene through Sp1 and CBP/p300. J Biol Chem. 1998;273(17):10696–10701. | ||
Scarpin KM, Graham JD, Mote PA, Clarke CL. Progesterone action in human tissues: regulation by progesterone receptor (PR) isoform expression, nuclear positioning and coregulator expression. Nucl Recept Signal. 2009;31(7):e009. | ||
Huang S, Christopher SC, Donald EI. Control of cyclin D1, p27Kip1, and cell cycle progression in human capillary endothelial cells by cell shape and cytoskeletal tension. Mol Biol Cell. 1998;9:3179–3193. | ||
Tamura M, Gu J, Takino T, Yamada KM. Tumor suppressor PTEN inhibition of cell invasion, migration, and growth: differential involvement of focal adhesion kinase and p130Cas. Cancer Res. 1999;59(2):442–449. | ||
Chetram MA, Hinton CV. PTEN regulation of ERK1/2 signaling in cancer. J Recept Signal Transduct Res. 2012;32(4):190–195. | ||
Neugarten J. Estrogen and oxidative stress. Gend Med. 2007;4(1):31–32. | ||
Dabrosin C, Hammar M, Ollinger K. Impact of oestradiol and progesterone on antioxidant activity in normal human breast epithelial cells in culture. Free Radic Res. 1998;28(3):241–249. | ||
Nguyen H, Syed V. Progesterone inhibits growth and induces apoptosis in cancer cells through modulation of reactive oxygen species. Gynecol Endocrinol. 2011;27(10):830–806. | ||
Liou GY, Storz P. Reactive oxygen species in cancer. Free Radic Res. 2010;44(5):479–496. | ||
Nadal-Serrano M, Sastre-Serra J, Pons DG, Miró AM, Oliver J, Roca P. The ERalpha/ERbeta ratio determines oxidative stress in breast cancer cell lines in response to 17beta-estradiol. J Cell Biochem. 2012;113(10):3178–3185. | ||
Papa L, Manfredi G, Germain D. SOD1, an unexpected novel target for cancer therapy. Genes Cancer. 2014;5(1–2):15–21. | ||
Ruiterkamp J, Ernst MF. The role of surgery in metastatic breast cancer. Eur J Cancer. 2011;47(Suppl 3):S6–S22. | ||
Jatoi I. Timing of surgery for primary breast cancer with regard to the menstrual cycle phase and prognosis. Breast Cancer Res Treat. 1998;52(1–3):217–225. | ||
Mote PA, Balleine RL, McGowan EM, Clarke CL. Colocalization of progesterone receptors A and B by dual immunofluorescent histochemistry in human endometrium during the menstrual cycle. J Clin Endocrinol Metab. 1999;84(8):2963–2971. | ||
Hagan CR, Lange C. Molecular determinants of context dependent progesterone receptor action in breast cancer. BMC Med. 2014;12:32. | ||
Depowski PL, Rosenthal SI, Ross JS. Loss of expression of the PTEN gene protein product is associated with poor outcome in breast cancer. Mod Pathol. 2001;14(7):672–676. | ||
Pandolfi PP. Breast cancer – loss of PTEN predicts resistance to treatment. N Engl J Med. 2004;351(22):2337–2338. | ||
Ghosh AK, Grigorieva I, Steele R, Hoover RG, Ray RB. PTEN transcriptionally modulates c-myc gene expression in human breast carcinoma cells and is involved in cell growth regulation. Gene. 1999;235(1–2):85–91. | ||
Perren A, Weng LP, Boag AH, et al. Immunohistochemical evidence of loss of PTEN expression in primary ductal adenocarcinomas of the breast. Am J Pathol. 1999;155(4):1253–1260. | ||
Vurusaner B, Poli G, Basaga H. Tumor suppressor genes and ROS: complex networks of interactions. Free Radic Biol Med. 2012;52(1):7–18. | ||
Mamillapalli R, Gavrilova N, Mihaylova VT, et al. PTEN regulates the ubiquitin-dependent degradation of the CDK inhibitor p27(KIP1) through the ubiquitin E3 ligase SCF(SKP2). Curr Biol. 2001;11(4):263–267. | ||
Freeman DJ, Li AG, Wei G, et al. PTEN tumor suppressor regulates p53 protein levels and activity through phosphatase-dependent and -independent mechanisms. Cancer Cell. 2003;3(2):117–130. |
Supplementary materials
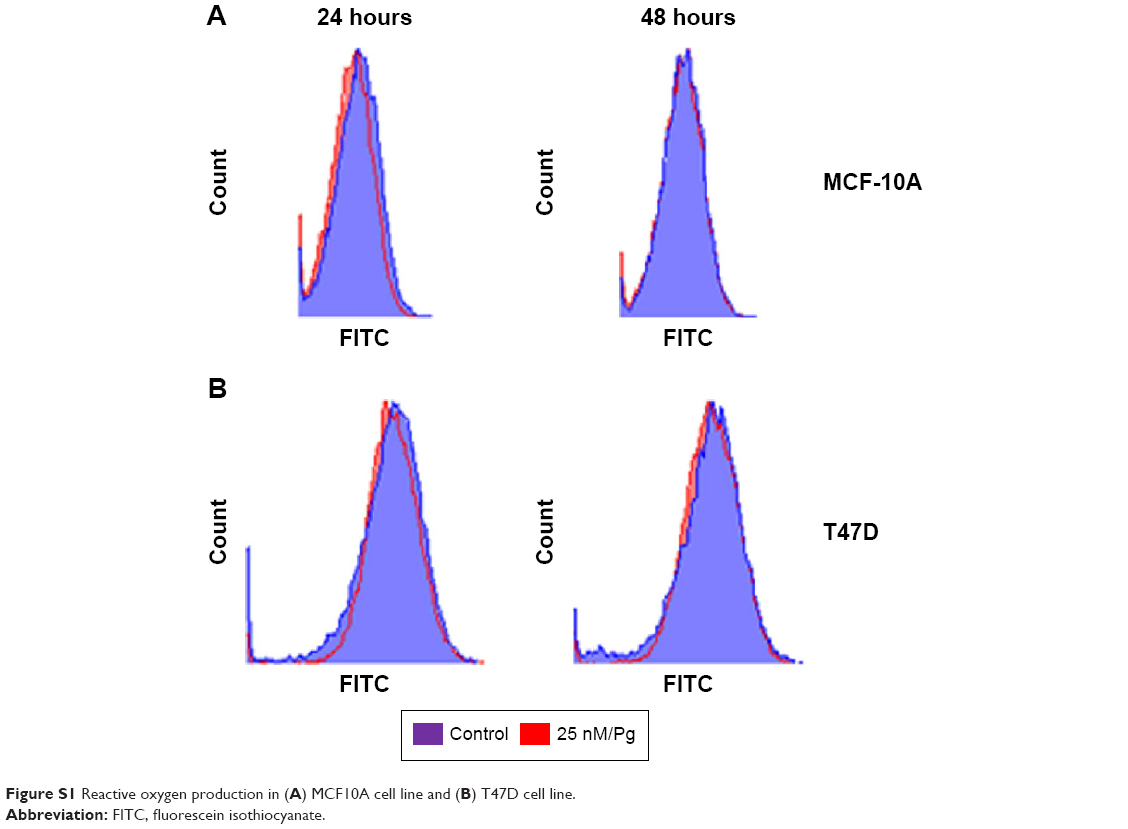 | Figure S1 Reactive oxygen production in (A) MCF10A cell line and (B) T47D cell line. |
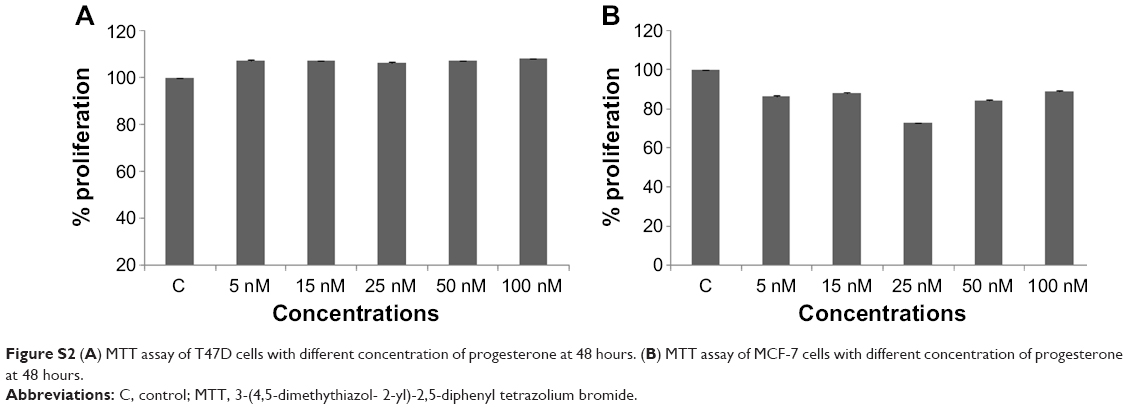 | Figure S2 (A) MTT assay of T47D cells with different concentration of progesterone at 48 hours. (B) MTT assay of MCF-7 cells with different concentration of progesterone at 48 hours. |
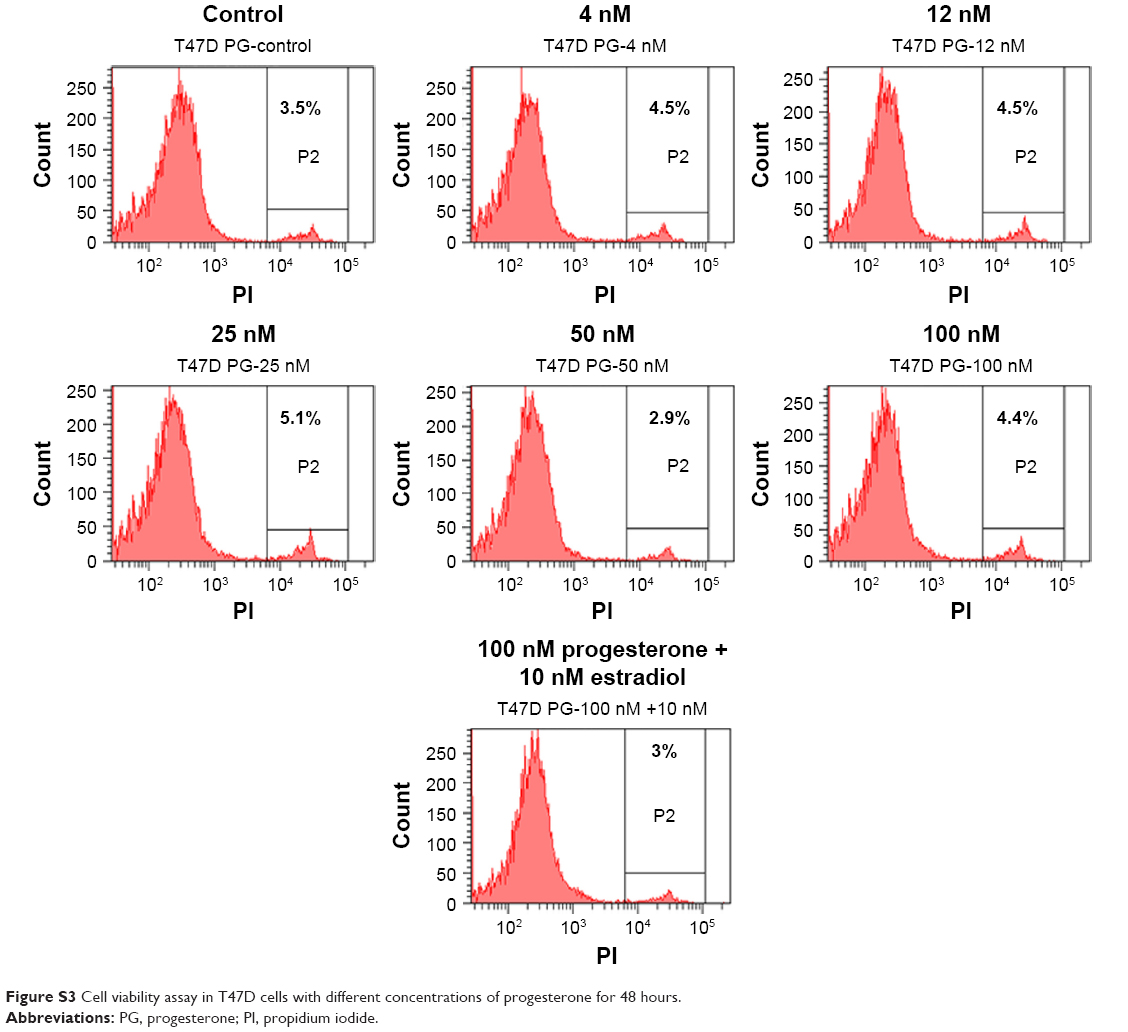 | Figure S3 Cell viability assay in T47D cells with different concentrations of progesterone for 48 hours. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.