Back to Journals » Research and Reports in Urology » Volume 13
Profiling of Androgen-Dependent Enhancer RNAs Expression in Human Prostate Tumors: Search for Malignancy Transition Markers
Authors Nishimura K, Mori J, Sawada T, Nomura S, Kouzmenko A, Yamashita K ![]() , Kanemoto Y, Kurokawa T, Hayakawa A, Tokiwa S, Ochi M, Shimmura H, Kato S
, Kanemoto Y, Kurokawa T, Hayakawa A, Tokiwa S, Ochi M, Shimmura H, Kato S ![]()
Received 13 July 2021
Accepted for publication 24 August 2021
Published 14 September 2021 Volume 2021:13 Pages 705—713
DOI https://doi.org/10.2147/RRU.S328661
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Jan Colli
Koichi Nishimura,1,2,* Jinichi Mori,1– 3,* Takahiro Sawada,1,2 Shuhei Nomura,1,4 Alexander Kouzmenko,1 Kaori Yamashita,5 Yoshiaki Kanemoto,1,2 Tomohiro Kurokawa,1,2 Akira Hayakawa,1 Suguru Tokiwa,5 Michihisa Ochi,5 Hiroaki Shimmura,5 Shigeaki Kato1,2
1Research Institute of Innovative Medicine, Tokiwa Foundation, Iwaki, Japan; 2Graduate School of Life Science and Engineering, Iryo Sosei University, Iwaki, Japan; 3Department of Hematology, Jyoban Hospital, Iwaki, Japan; 4Graduate School of Medicine, The University of Tokyo, Tokyo, Japan; 5Department of Urology, Jyoban Hospital, Iwaki, Japan
*These authors contributed equally to this work
Correspondence: Shigeaki Kato Email [email protected]
Introduction: Although the ability of androgens to promote prostate cancer development has been known for decades, the molecular mechanisms of androgen receptor (AR) signaling in the tumorigenesis remain unclear. Enhancer RNAs (eRNAs) transcribed from strong enhancers, or super-enhancers (SEs), have recently emerged as a novel class of regulatory non-coding RNAs (ncRNAs) that facilitate transcription, including that of androgen target genes, through chromatin looping to position enhancers proximate to the promoters. The aim of this study was to assess androgen-dependent transcription in prostate tumors of eRNAs (designated as KLK3eRNAs) from the SE of the KLK3 gene encoding the prostate-specific antigen (PSA) protein, a clinical marker of prostate carcinogenesis.
Materials and Methods: The androgen-induced KLK3eRNAs were identified in the LNCaP human prostate cancer cell line. The expressions of these KLK3eRNAs together with KLK3 and AR mRNA transcripts were assessed by qRT-PCR in prostate tumor samples from five prostate cancer patients.
Results: Androgen-induced KLK3eRNAs have been identified in the LNCaP cells, and their expression was further analyzed in tumors of prostate cancer patients. Transcripts of the tested KLK3eRNAs have been detected in all clinical samples, but their expression patterns differed between individual tumor specimens. We found a statistically significant correlation between the levels of the KLK3 and AR mRNAs with those of the previously reported KLK3eRNAs, while such correlation was not observed for novel KLK3eRNAs described in our recent report.
Conclusion: Presented data suggest that prostate tumor development may associate with epigenetic reorganization in the KLK3 genomic regulatory elements reflected by changes of the KLK3eRNA expression. Our findings support a potential of eRNAs profiling to be used as diagnostic marker.
Keywords: enhancer RNA, prostate cancer, androgen receptor, PSA, non-coding RNA, epigenetic modification
Introduction
Prostate cancer development depends on the activation of androgen signaling; hence, the cancer treatment involves androgen deprivation therapies.1,2 Clinical success of such therapies is often limited to a few years of remission followed by patient tumor transition into more malignant hormone-independent stages.1,2 The nuclear androgen receptor (AR) is expressed in hormone-dependent prostate tumors at the levels similar to those in normal tissues,1,2 and AR continues to be expressed at detectable levels even upon cancer transition into the androgen-independent state,3 while an apparent loss of the AR expression has been observed only at more advanced malignancy stages.1–3 Although causative factors in the prostate cancer stage progression remain largely uncovered,4 AR is believed to play a pivotal role in the process, including the transition into androgen-independent tumor state known as a castration-resistant prostate cancer (CRPC).5
AR is a member of the nuclear receptor superfamily and acts as a DNA binding transcription factor controlling expression of target genes.6,7 In prostate cells, the kallikrein-related peptidase 3 gene (KLK3) and its regulatory genomic elements represent an important AR target, since the KLK3 locus encodes the prostate-specific antigen (PSA) protein, a well-characterized and readily detectable in the serum diagnostic marker of prostate cancer.8
It has been established that tumorigenesis associates with a global reconfiguration of the chromatin epigenetic landscape.9,10 Chromatin reorganization has also been shown during prostate cancer progression from androgen-dependency into hormone-independent stages.9,10
For specific chromatin site targeting, AR requires so-called pioneering factors, primarily FOXA111 that function as binding platforms for AR association with chromatin at the target promoters and other regulatory genomic regions even in the absence of ligand. Epigenetic reorganization may associate with change in patterns of chromatin distribution of FOXA1 that results in altered AR genomic association topographies and distinct AR action in prostate cells during transition into CRPC stages.12 Whole-genome-sequencing studies showed that somatic duplications of both AR enhancer and AR protein-encoding sequences associated with increased malignancy of prostate tumors.13 The AR structural gene was found linked by somatic recombination in CRPC cells with a normally distant intergenic enhancer that might be further amplified alone or with the AR encoding locus.14 It has been suggested that increased AR protein expression after somatic enhancer acquisition and/or amplification of the AR cistron may confer androgen-independency at advanced prostate cancer stages.13,14 These findings clearly indicate that AR signaling abnormalities during prostate cancer development can be attributed to structural and epigenetic reorganization at the AR loci.
Chromatin reconfiguration involves action of various epigenetic factors, of which chromatin remodelers and histone modifiers are believed to be the key players.15,16 Recently, non-coding RNAs (ncRNAs) were shown to affect the functional state of chromatin in cooperation with protein epigenetic regulators.17,18
The ncRNAs transcribed from genomic enhancer elements, hence named as enhancer RNAs (eRNAs), are found to be critical for efficient activation of gene expression through spatial proximation of distal enhancer regions with structural gene promoters by chromatin looping.19,20 Although molecular events during the eRNA-induced chromatin looping are not fully understood, they were shown to involve recruitment by eRNAs of cohesin complexes that had been initially known to hold together sister chromatids after replication.19,20 eRNAs are derived from strong or super-enhancers (SEs) comprising arrays of multiple eRNA transcription sites. The levels of eRNA expression reflect the enhancer functional activity.20,21
Recent studies have shown that various transcriptional cofactors function within giant ribonucleoprotein (RNP) condensates formed by a mechanism described as liquid–liquid phase separation (LLPS), and dynamically interact with DNA-bound transcriptional factors, including nuclear hormone receptors, through their low complexity intrinsically disordered regions (IDRs) for efficient coactivation of transcription.22–24 The assembling of such multiunit coactivator condensates at SE and promoter regions has been shown to require eRNAs.22–24 Thus, recent studies emphasize the involvement of eRNAs in variety of regulatory mechanisms affecting chromatin conformation and transcription. It is, therefore, conceivable that development and progression of malignancy may associate with changes in eRNAs’ expression patterns.
The structure and factors regulating the human KLK3 gene have been intensively scrutinized owing to the clinical significance of its expression product, PSA protein, as prostate cancer diagnostic marker.25,26 Previously, KLK3 enhancer-derived eRNAs (designated as K-KLK3eRNAs) were reported to selectively facilitate transcription of AR target genes in LNCaP cells.27 Recently, we found that the human KLK3 genomic locus is located within a registered, yet untested super-enhancer (KLK3-SE) and characterized additional novel KLK3eRNAs (designated as N-KLK3eRNAs) transcribed from this KLK3-SE in the human prostate cancer LNCaP cell line.28 In this study, we analyzed in clinical specimens of prostate tumor expression patterns of the KLK3eRNAs, as potential markers of malignancy development and advanced stage progression.
Materials and Methods
Cell Culture and DHT Treatment
The human prostate carcinoma cell line LNCaP (purchased from the RIKEN Cell Bank, Japan; catalogue No RCB2144) cells were seeded at 1.0×106 cells per 10cm dish, pre-incubated with RPMI1640 conditioning media for 48 hours, and then with 10% charcoal-treated (ie, depleted from endogenous androgens) bovine calf serum for further 72 hours before treatment with dihydrotestosterone (DHT). The cells were treated with 10nM DHT (+DHT samples) or equal volumes of the solvent (-DHT, or control samples) in culture media for 5 hours and harvested for RNA extraction.28,29
Quantitative Real-Time PCR (qRT-PCR)
Extraction of total RNA from DHT-treated and untreated LNCaP cells was performed using the RNeasy plus spin column kits (Qiagen, Valencia, CA, USA) according to the manufacturer protocol. Complementary DNAs (cDNAs) were prepared with the iScript cDNA Synthesis Kit (Bio-Rad Laboratories, CA, USA). qRT-PCR analysis was performed using the SYBR Green Master CFX Connect Real-Time PCR System (Bio-Rad Laboratories, CA, USA).28,29 Preparation of complementary DNAs (cDNAs) and qRT-PCR was conducted by the iScript cDNA Synthesis Kit (Bio-Rad Laboratories, CA, USA) and the SYBR Green Master on a CFX Connect Real-Time PCR System (Bio-Rad Laboratories, CA, USA), respectively.29,30 Expression of eRNA transcripts was calculated by the delta–delta Ct method with β-actin expression used as a normalizing internal control. The PCR primer sets used in the study are listed in Supplementary Table 1.
Preparation of Patient Samples
Prostate biopsy samples were collected from 45 patients in our hospital during the period between January 2017 and January 2018. In addition to the excision biopsies, we obtained clinical diagnostic specimens using two bundled biopsy needles, and assigned one specimen for histology (pathology specimen) and the other for RNA extraction (RNA specimen). A pathologist confirmed the boundary between cancer and normal prostate tissue from the section of pathology specimen and marked it on the corresponding RNA specimen.31,32 According to the boundary, RNA specimen was cut into cancer and normal sections. Among the 45 patients, 22 patients were diagnosed with prostate cancer by diagnostic biopsies. Of these 22 prostate cancer specimens, samples from only 5 individual patients (see Table 1) contained sufficient amount of both cancer and normal tissues for RNA analysis. Total RNA was extracted by TRIzol (Invitrogen Carlsbad, CA, USA) and cDNA was synthesized using iScript cDNA Synthesis Kit (Bio-Rad Laboratories, CA, USA).28,29
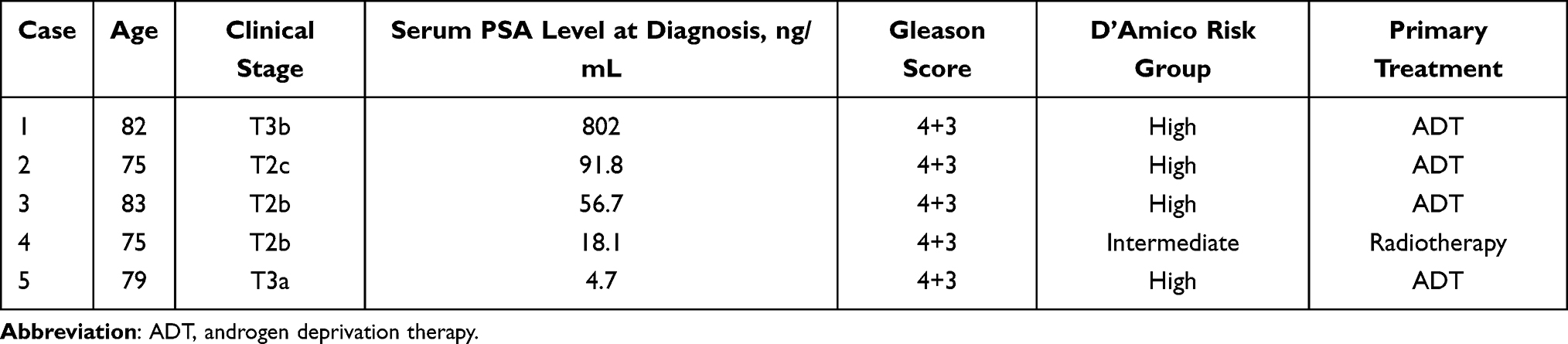 |
Table 1 Patient Characteristics |
Statistical Analysis
Based on the results of qRT-PCR analyses of eRNAs, KLK3 and AR mRNAs, we calculated the ratios of RNA expression in cancer to normal tissue for each individual patient. The raw data are presented in Supplementary Table 2.
We estimated the relationship between the expression ratio of eRNA and KLK3 or AR mRNA in patient samples using a linear regression approach, which included a random intercept for the individual to control for correlation in data from the same individuals.343–36 In the regression models, the dependent variable was the natural logarithm of eRNA and the independent variable was KLK mRNA or AR mRNA. For these analyses, we excluded statistical outliers when the observed value of eRNA was higher than the third quartile plus 1.5 times the interquartile range. The following four sets of relationships were considered and displayed in Figure 1: (A) K-KLK3eRNAs versus KLK3mRNA; (B) N-KLK3eRNAs versus KLK3mRNA; (C) K-KLK3eRNAs versus ARmRNA; and (D) K-KLK3eRNAs versus ARmRNA.
 |
Figure 1 Correlation between expression of KLK3eRNAs and KLK3 or AR mRNA in the patient samples. The ratio of RNA expression level (tumor/normal) of (A and B) KLK3 mRNA in the five patients and (C and D) AR mRNA in the three patients are indicated in the X-axis. The ratio of RNA expression level (tumor/normal) of (A and C) reported and (B and C) novel eRNA were plotted in the Y-axis in each patient. The colors of plots correspond to primer sets shown in Figure 2. A linear regression line was fitted to the plots on the logarithmic Y-axis. Expression levels of AR in cases 4 and 5 were not evaluable. qRT-PCR could not detect RNA in some combinations of sample and primer set due to extremely low expression. If RNA level of either tumor or normal tissues in a patient were not detected using a primer set, we defined this undetectable Ct value as 40, the minimum detectable value of the qRT-PCR assay. P-values in the figure are the regression outputs. P-value less than 0.05 was considered statistically significant in this study, meaning that there was a statistically significant relationship in (A, C and D). |
Ethics
Patient samples collection has been conducted in accordance with the Declaration of Helsinki and relevant national regulations. A signed Informed Consent Form has been obtained from each individual patient. Clinical samples handling and assessment protocols have been approved by the Jyoban Hospital Institutional Review Board.
Results
KLK3 Super-Enhancer Encodes Androgen-Inducible and Non-Inducible eRNAs
By in silico analysis of the dbSUPER database, we identified an SE registered in the vicinity of the human KLK3 locus that might control the KLK3 expression. Previously, several androgen-inducible eRNA transcripts were reported to support androgen-dependent KLK3 mRNA expression.27 These known eRNAs have been designated as K-KLK3eRNAs to distinguish them from a recently reported by our group novel N-KLK3eRNAs.28 We analyzed by qRT-PCS individual transcript expression of the K-KLK3eRNAs (sense strand primer sets S1-S7; antisense strand primer sets AS1-AS4) and the N-KLK3eRNAs (sense strand primer sets S8-S9; antisense strand primer sets AS5-AS8).
In the LNCaP cells, we observed a DHT-dependent induction of all the K-KLK3eRNAs and S9, AS5 and AS8 N-KLK3eRNAs, while the expression of the S8, AS6 and AS7 N-KLK3eRNAs did not change after the treatment with androgen (Figure 2). Thus, the KLK3eRNA expression profiling in the LNCaP prostate cancer cells revealed heterogeneity of the KLK3eRNAs species, including their differential response to androgens. This finding suggests distinct biological roles of the eRNAs transcribed from the KLK3 SE with possible different impacts on the androgen-dependent expression of the PSA protein.
 |
Figure 2 Expression of eRNAs in the LNCaP cells. Genomic location of a super-enhancer (SE) in the human KLK3 locus (A). The location of the KLK3 (PSA) gene (by RefSeq genes) and the adjacent super-enhancer (SE) registered in dpSUPER database in LNCaP cells is shown in the upper panel. Acetylated histone H3 at lysine 27 (H3K27ac) registered for LNCaP cells is used as the known active enhancer histone marker, with AR binding sites in LNCaP cells documented in ChIP atlas database in the middle panel. The region harboring consensus AR binding elements (ARE) is shown as KLK3 ARE as an orange box. Pink and sky blue boxes are amplicons of previously reported sense (S1–8) and antisense (AS1–6) eRNAs, respectively. (B) The expressions of KLK3 mRNA, AR, and previously reported and newly identified KLK3eRNAs in LNCaP cells were tested by RT-PCR at least three times and one of the data set is representatively shown. *P<0.05. |
Analysis of KLK3eRNA, KLK3 mRNA and AR mRNA Transcripts in Human Prostate Tumor Samples
After characterization of the KLK3eRNAs expression in the LNCaP cells, we investigated their expression in human prostate cancer specimens together with expression of the KLK3 and AR mRNAs. Among the collected tumor biopsy samples, we were able to select specimens from only five individual patients (Table 1) with reliably distinguishable and dissectible normal and malignant tissue areas, as the sets of the two sections from the other 17 patients were pathologically unclear to judge the tissues as tumors or normal tissues. The reliably distinguishable sets from the five patients were used for histological analysis and RNA isolation (Figure 3). (Table 1) that had reliably distinguishable and dissectible areas of normal and malignant tissue samples in quantities sufficient for histological analysis and RNA isolation (Figure 3). Among the tested specimens, cases 1–3 were derived from the patients with high serum levels of PSA (Table 1).
 |
Figure 3 Histology of normal prostate and prostate tumor tissues in the patients. The tissues were stained with hematoxylin and eosin (×200) for histopathological analysis to diagnose tumors. The representative sections of the several data sets from each tissues are shown. |
All the K- and N-KLK3RNAs expressed in the LNCaP cells (Figure 2) were also detectable in the human prostate samples (Supplementary Table 2). However, there was significant variation in expression levels of these eRNAs among the tested specimens, and between normal and malignant areas of individual sample tissues.
Correlation Between KLK3eRNAs and KLK3 or AR mRNAs Expression in Prostate Tumors
Considering that the K-KLK3eRNAs were originally reported to facilitate DHT-inducible expression of the KLK3 gene,27 we assessed the correlation between the expression levels of the KLK3eRNAs and KLK3 mRNA and AR mRNA in the patient samples using the regression approach,33–36 and also evaluated the tumor/normal tissue ratio of these RNA expression levels in each individual patient specimen (Figure 1).
The expression levels of the K-KLK3eRNAs (Figure 1A and C) were higher in the tumor areas in comparison to that in the normal sample counterparts, but somewhat opposite expression ratio patterns were observed for the N-KLK3eRNAs (Figure 1B and D). Furthermore, there was a statistically significant correlation between expression levels of K-KLK3eRNAs and those of the KLK3 mRNA (Figure 1A) and AR mRNA (Figure 1C). However, no such correlation was observed for the N-KLK3eRNAs (Figure 1B and D and Table 2).
 |
Table 2 Correlation of RNA Expression Ratio (Tumor/Normal) Between eRNAs and KLK3, and eRNAs and AR |
Thus, the differences in the KLK3eRNA expression between individual specimens suggest that the chromatin environment at the KLK genomic locus may vary in prostate tumor cells. It would be important to further investigate whether the KLK3eRNAs expression profiles may reflect the progression of prostate tumors into more malignant stages.
Discussion
Mortality of prostate cancer patients often associates with tumor progression into CRPC stages. Significantly, the transition of prostate tumors into fatal stages may take many years.1–3 Therefore, discovery of markers reliably predicting malignancy advancement rates would improve diagnosis efficiency, may help to optimize therapeutic strategies and ultimately, reduce mortality of the patients.37,38
Currently, the serum concentration of PSA is the most widely used non-invasive marker of prostate cancer. PSA protein is a product of the KLK3 gene. Therefore, analysis of upstream factors affecting KLK3 gene expression may lead to identification of prostate cancer diagnostic markers at earlier stages than those reflected by the PSA levels. The KLK3 gene promoter bears functional AR binding sites acting as androgen-responsive enhancer elements (AREs).8 In addition, several eRNAs have been identified to be transcribed in LNCaP cells from the KLK3 gene-associated cluster of enhancer elements in the androgen-dependent manner, which promote KLK3 expression.27,28 In this study, we have initially confirmed the expression in LNCaP cells of the previously described eRNAs (K-KLK3eRNAs)27 as well as novel N-KLK3eRNAs recently identified by our group,28 and analyzed the inducibility of their transcription by DHT (Figure 2). We then analyzed the expression of the KLK3eRNAs in prostate tissue samples. Transcripts of the K- and N-KLK3eRNA were detectable in the tumors as well as normal sections of prostate samples from the same patients (Figure 1). The KLK3 mRNA expression levels exhibited statistically significant, albeit moderate correlation with those of the K-KLK3eRNAs in the analyzed malignant and normal prostate tissues (Figure 1), supporting the conclusion that KLK3eRNAs might facilitate the KLK3 expression.27,29 However, the expression levels of the K-KLK3eRNAs and KLK3 mRNA were diverse among the individual patient tissues, suggesting that the chromatin epigenetic landscape at the KLK3 locus and its genomic regulatory regions may differ at different stages of prostate cancer progression and/or prostate cell environment in the tissue.
Consistent with earlier reports,27,28 we observed the AR mRNA expression in our LNCaP cells, and the DHT-dependent induction of KLK3 mRNA and K-KLK3eRNAs expression correlated with the DHT-dependent proliferation of these cells. In the analyzed prostate tissues, levels of the AR mRNA expression were different between individual patient samples and between adjacent malignant and normal sections of the same patient sample (Figure 1). These findings are consistent with the previous reports1–3 showing that the levels and distribution of AR expression were quite diverse in prostatic tumors, depending on tumor progression stages and cell-types. Most notably, we did not observe correlation between the N-KLK3eRNAs expression and that of KLK3 mRNA or AR mRNA in the tumor samples. Also, there was no apparent correlation between the KLK3 mRNA and AR mRNA levels in the tested clinical samples (Table 2). Thus, it appears that the expression of the KLK3 mRNA and subsets of the KLK3eRNAs in the prostate tumors exhibit divergent androgen-dependency regardless of the levels of AR expression. This suggests that other factor(s) or signaling pathway(s) might be also involved in the control of the KLK3 mRNA and eRNAs transcription during prostate tumorigenesis,9,16,28,39,40 and that AR signaling may not be the only defining factor in activation of the KLK3 gene expression reflected by elevation of circulated PSA in prostate cancer patients.
Conclusion
We have found in the LNCaP cells that KLK3eRNAs consist of heterogeneous transcripts regulated by diverse signaling pathways, including androgen-inducible and non-inducible eRNAs. The KLK3eRNAs described in the LNCaP cells are also expressed in human prostate tumors. However, the levels of the KLK3eRNAs were found to be different in individual tumor samples and also differed between normal and malignant areas from the same tissue samples.
Although activated AR signaling or increased androgen sensitivity has been inferred in developed prostate tumors,1–5 we have not observed in the analyzed prostate samples a clear correlation between the AR expression and the levels of the KLK3 mRNA and KLK3eRNAs transcripts. Furthermore, we showed that the transcription of a subset of the KLK3eRNAs was not dependent on androgens.
Thus, our findings clearly indicate that the activity of super-enhancer elements supporting the KLK3 gene expression might be also controlled by androgen-independent regulators and/or signaling pathways. Further analysis of larger numbers of prostate tumor samples may reveal consistent patterns of changes in the KLK3eRNAs expression associated with prostate carcinogenesis and point to novel regulatory factors involved in malignant transformation of prostate cells.
Acknowledgments
The authors thank the members of the Jyoban Hospital staff who supported this study, and Ms Mai Hirata for manuscript preparation. This study is financially supported by the Tokiwa Foundation research grant.
Disclosure
The authors declare no conflicts of interest.
References
1. Attard G, Parker C, Eeles RA, et al. Prostate cancer. Lancet. 2016;387:70–82. doi:10.1016/S0140-6736(14)61947-4
2. Li Z, Alyamani M, Li J, et al. Redirecting abiraterone metabolism to fine-tune prostate cancer anti-androgen therapy. Nature. 2016;533:547–551. doi:10.1038/nature17954
3. Henshall S, Quinn DI, Lee CS, et al. Altered expression of androgen receptor in the malignant epithelium and adjacent stroma is associated with early relapse in prostate cancer. Cancer Res. 2001;61:423–427.
4. Scher HI, Fizazi K, Saad F, et al. Increased survival with enzalutamide in prostate cancer after chemotherapy. N Engl J Med. 2012;367:1187–1197. doi:10.1056/NEJMoa1207506
5. Sharifi N. Mechanisms of androgen receptor activation in castration-resistant prostate cancer. Endocrinology. 2013;154:4010–4017. doi:10.1210/en.2013-1466
6. Mangelsdorf DJ, Thummel C, Beato M, et al. The nuclear receptor superfamily: the second decade. Cell. 1995;83:835–839. doi:10.1016/0092-8674(95)90199-X
7. Kawano H, Sato T, Yamada T, et al. Suppressive function of androgen receptor in bone resorption. Proc Natl Acad Sci U S A. 2003;100:9416–9421. doi:10.1073/pnas.1533500100
8. Qin J, Liu X, Laffin B, et al. The PSA(-/lo) prostate cancer cell population harbors self-renewing long-term tumor-propagating cells that resist castration. Cell Stem Cell. 2012;10:556–569. doi:10.1016/j.stem.2012.03.009
9. Wang L, Lonard DM, O’Malley BW. The role of steroid receptor coactivators in hormone dependent cancers and their potential as therapeutic targets. Horm Cancer. 2016;7:229–235. doi:10.1007/s12672-016-0261-6
10. Chen T, Dent SY. Chromatin modifiers and remodellers: regulators of cellular differentiation. Nat Rev Genet. 2014;15:93–106. doi:10.1038/nrg3607
11. Lupien M, Eeckhoute J, Meyer CA, et al. FoxA1 translates epigenetic signatures into enhancer-driven lineage-specific transcription. Cell. 2008;132:958–970. doi:10.1016/j.cell.2008.01.018
12. Wang Q, Li W, Zhang Y, et al. Androgen receptor regulates a distinct transcription program in androgen-independent prostate cancer. Cell. 2009;138:245–256. doi:10.1016/j.cell.2009.04.056
13. Viswanathan SR, Ha G, Hoff AM, et al. Structural alterations driving castration-resistant prostate cancer revealed by linked-read genome sequencing. Cell. 2018;174:433–447. doi:10.1016/j.cell.2018.05.036
14. Takeda DY, Spisak S, Seo JH, et al. A somatically acquired enhancer of the androgen receptor is a noncoding driver in advanced prostate cancer. Cell. 2018;174:422–432. doi:10.1016/j.cell.2018.05.037
15. Greer EL, Shi Y. Histone methylation: a dynamic mark in health, disease and inheritance. Nat Rev Genet. 2012;13:343–357. doi:10.1038/nrg3173
16. Rosenfeld MG, Lunyak VV, Glass CK. Sensors and signals: a coactivator/corepressor/epigenetic code for integrating signal-dependent programs of transcriptional response. Genes Dev. 2006;20:1405–1428. doi:10.1101/gad.1424806
17. Cech TR, Steitz JA. The noncoding RNA revolution-trashing old rules to forge new ones. Cell. 2014;157:77–94. doi:10.1016/j.cell.2014.03.008
18. Li W, Notani D, Ma Q, et al. Functional roles of enhancer RNAs for oestrogen-dependent transcriptional activation. Nature. 2013;498:516–520. doi:10.1038/nature12210
19. Li W, Notani D, Rosenfeld MG. Enhancers as non-coding RNA transcription units: recent insights and future perspectives. Nat Rev Genet. 2016;17:207–223. doi:10.1038/nrg.2016.4
20. Guan D, Xiong Y, Borck PC, et al. Diet-induced circadian enhancer remodeling synchronizes opposing hepatic lipid metabolic processes. Cell. 2018;174:831–842. doi:10.1016/j.cell.2018.06.031
21. Kim YH, Marhon SA, Zhang Y, Steger DJ, Won KJ, Lazar MA. Rev-erbalpha dynamically modulates chromatin looping to control circadian gene transcription. Science. 2018;359:1274–1277. doi:10.1126/science.aao6891
22. Boija A, Klein IA, Sabari BR, et al. Transcription factors activate genes through the phase-separation capacity of their activation domains. Cell. 2018;175:1842–1855. doi:10.1016/j.cell.2018.10.042
23. Chong S, Dugast-Darzacq C, Liu Z, et al. Imaging dynamic and selective low-complexity domain interactions that control gene transcription. Science. 2018;361:eaar2555. doi:10.1126/science.aar2555
24. Alberti S, Gladfelter A, Mittag T. Considerations and challenges in studying liquid-liquid phase separation and biomolecular condensates. Cell. 2019;176:419–434. doi:10.1016/j.cell.2018.12.035
25. Lawrence MG, Lai J, Clements JA. Kallikreins on steroids: structure, function, and hormonal regulation of prostate-specific antigen and the extended kallikrein locus. Endocr Rev. 2010;31:407–446.
26. Gallee MP, Visser-de Jong E, van der Korput JA, et al. Variation of prostate-specific antigen expression in different tumour growth patterns present in prostatectomy specimens. Urol Res. 1990;18:181–187. doi:10.1007/BF00295844
27. Hsieh CL, Fei T, Chen Y, et al. Enhancer RNAs participate in androgen receptor-driven looping that selectively enhances gene activation. Proc Natl Acad Sci U S A. 2014;111:7319–7324. doi:10.1073/pnas.1324151111
28. Sawada T, Nishimura K, Mori J, et al. Androgen-dependent and DNA binding-independent association of androgen receptor with chromatic regions coding androgen-induced non-coding RNAs. Biosci Biotec Biochem. Epub 2021 Jul 23. doi:10.1093/bbb/zbab135
29. Takahashi S, Watanabe T, Okada M, et al. Noncanonical Wnt signaling mediates androgen-dependent tumor growth in a mouse model of prostate cancer. Proc Natl Acad Sci U S A. 2011;108:4938–4943. doi:10.1073/pnas.1014850108
30. Kamoshida Y, Fujiyama-Nakamura S, Kimura S, et al. Ecdysone receptor (EcR) suppresses lipid accumulation in the Drosophila fat body via transcription control. Biochem Biophys Res Commun. 2012;421:203–207. doi:10.1016/j.bbrc.2012.03.135
31. Yamashita K, Shimmura H, Tokiwa S, Kato S. Painless vascular leiomyoma found after incision of the vaginal wall: a case report. Urol Case Rep. 2018;17:19–21. doi:10.1016/j.eucr.2017.12.005
32. Tokiwa S, Shimmura H, Nomura S, et al. Degarelix treatment is compatible with diabetes and antithrombotic therapy in patients with prostate cancer. Res Rep Urol. 2017;9:225–232.
33. Dobin A, Davis CA, Schlesinger F, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi:10.1093/bioinformatics/bts635
34. Limpert E, Stahel WA, Abbt M. Log-normal distributions across the sciences: keys and clues. BioScience. 2001;51:341–352. doi:10.1641/0006-3568(2001)051[0341:LNDATS]2.0.CO;2
35. Moore DS, McCabe GP, Craig BA. Introduction to the Practice of Statistics.
36. Fox J. Applied Regression Analysis and Generalized Linear Models. SAGE Publications; 2015.
37. Shariat SF, Semjonow A, Lilja H, Savage C, Vickers AJ, Bjartell A. Tumor markers in prostate cancer I: blood-based markers. Acta Oncol. 2011;50(Suppl 1):61–75. doi:10.3109/0284186X.2010.542174
38. Crawford ED, Ventii K, Shore ND. New biomarkers in prostate cancer. Oncology. 2014;28:135–142.
39. Vo BT, Morton D, Komaragiri S, Millena AC, Leath C, Khan SA. TGF-beta effects on prostate cancer cell migration and invasion are mediated by PGE2 through activation of PI3K/AKT/mTOR pathway. Endocrinology. 2013;154:1768–1779. doi:10.1210/en.2012-2074
40. Vanhara P, Hampl A, Kozubik A, Soucek K. Growth/differentiation factor-15: prostate cancer suppressor or promoter? Prostate Cancer Prostatic Dis. 2012;15:320–328. doi:10.1038/pcan.2012.6
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.