")
Back to Journals » Therapeutics and Clinical Risk Management » Volume 15
Profile of tezacaftor/ivacaftor combination and its potential in the treatment of cystic fibrosis
Authors Shiferaw D, Faruqi S
Received 17 March 2019
Accepted for publication 16 June 2019
Published 22 August 2019 Volume 2019:15 Pages 1029—1040
DOI https://doi.org/10.2147/TCRM.S165027
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Garry Walsh
Dejene Shiferaw,* Shoaib Faruqi*
Department of Respiratory Medicine, Hull University Teaching Hospitals NHS Trust, Cottingham HU16 5JQ, UK
*These authors contributed equally to this work
Correspondence: Shoaib Faruqi
Department of Respiratory Medicine, University of Hull and Hull and York Medical School, Hull University Teaching Hospitals NHS Trust, Castle Hill Hospital, Castle Road, Cottingham HU16 5JQ, UK
Tel +44 148 262 6706
Email [email protected]
Abstract: Cystic fibrosis (CF) is a life-limiting autosomal recessive disease caused by dysfunction of the cystic fibrosis transmembrane conductance regulator (CFTR) ion channel. Management of CF has traditionally relied upon managing complications of CFTR protein dysfunction and this has led to a steady improvement in survival of CF patients. However, the landscape of CF care has changed substantially over the last decade with the discovery of CFTR modulators that aim to increase or potentially restore the function of the disease-causing CFTR protein. This narrative review summarizes the development of CFTR therapies so far with emphasis on tezacaftor/ivacaftor combination therapy. We have also summarized the Phase II results of triple combination therapy which promises an effective CFTR modulator therapy for more than 90% of CF patients.
Keywords: cystic fibrosis, CFTR modulators, CFTR potentiators, ivacaftor, tezacaftor, triple therapy
Erratum for this paper has been published
Introduction
Cystic fibrosis (CF) is a life-threatening genetic disease that affects approximately ct 380,000 people worldwide.1 CF is caused by absent or reduced function of the cystic fibrosis transmembrane conductance regulator (CFTR) protein that is encoded by the CFTR gene. It is characterized by pulmonary exacerbations, progressive loss of lung function, respiratory failure, and poor nutritional status. The discovery of the CFTR gene in 1989 has set the stage for understanding the pathogenesis of CF and the development of therapies that target the underlying disease-causing abnormality.2
CFTR is a transmembrane protein, that belongs to the adenosine nucleotide-binding cassette transporters, which functions as an ion channel regulated by cAMP-dependent phosphorylation that conducts chloride and bicarbonate through the apical membrane of epithelial cells.3–5 It also regulates ion transporters such as other chloride channels and epithelial sodium channel (ENaC).6 CFTR is expressed in many epithelia of the body including airway surfaces, sinuses, pancreas, intestine, reproductive system, and sweat glands, which explains the multi-organ pathology of CF.
CF was first described in 1938 as “Cystic fibrosis of the pancreas” by the American Pathologist, Dr. Dorothy Andersen.7 Progressive airway destruction characterized by chronic lung infection and loss of lung function, however, is the predominant cause of morbidity and mortality.7,8 The mechanisms by which CFTR dysfunction in the airway epithelium lead to mucus plugging, chronic inflammation, and impaired antibacterial host defense include: airway surface dehydration due to imbalance between CFTR-dependent chloride secretion and ENaC mediated sodium absorption; and reduced airway surface liquid PH due to impaired bicarbonate secretion which reduces the function of antimicrobial peptides.9
Over 2,000 CFTR gene mutations are described and just over 300 of them are known to cause disease.10,11 However, only a small number of well-studied mutations are responsible for disease in the vast majority of CF patients while many of the other mutations remain uncharacterized. CFTR mutations can lead to decreased quantity or function (and sometimes both) of the CFTR protein at the epithelial cell surface. The overall clinical impact of an individual CFTR mutation depends on the total CFTR activity which in turn depends on the number of CFTR channels at the cell surface and the functional ability of each channel to open (channel gating) and transport (conductance) ions.12 Other non-CFTR genetic, environmental and socio-economic factors are also known to influence overall CF outcome.13
CFTR mutations are traditionally categorized into six functional classes depending on their impact on the CFTR protein production, processing or function (Table 1).
 |
Table 1 Classes of CFTR mutations |
Class I mutations involve premature stop codon, that prevents complete transcription of the CFTR gene, resulting in truncated protein and near complete absence of CFTR protein at the cell surface. This mutation affects 2–5% of the CF population worldwide but is common in certain CF populations like Ashkenazi Jews who carry at least one copy of the mutation in 60% of the cases.14
Class II mutations (including the most common Phe508del[F508del] mutation) lead to an aberrantly folded CFTR protein that is targeted by the cell quality control system for degradation by the proteasome, resulting in near complete absence of mature CFTR protein at the apical surface membrane.
Class III mutations lead to a full-length CFTR protein incorporated into the cell membrane but with defective regulation of chloride transport it leads to markedly reduced channel gating. This is called a gating mutation and Gly551Asp (G551D) is the commonest mutation which is present in approximately 4% of the patients.
Class IV mutations lead to impaired CFTR channel conductance (rate at which ions flow through open channel). The most common mutation is Arg117His (R117H).
Class V mutations result in reduced synthesis of CFTR protein leading to a reduced number of normally functioning CFTR at the cell membrane.
Class VI mutations lead to accelerated turnover of CFTR protein at the cell surface leading to reduced number of functional CFTR.
It is worth noting that these functional classes are not mutually exclusive, and an individual mutation can result in multiple defects spanning multiple classes. For example, the most common disease causing F508del mutation leads to various defects, including reduced folding and trafficking (class II) and defective channel gating (class III) defects amongst others.
Class I, II, and III mutations are associated with no residual CFTR function and patients with these mutations manifest a more severe form of the disease compared to those with class IV, V, and VI mutations that have some residual CFTR function.
Management of CF has traditionally relied upon preventing and controlling complications of CFTR dysfunction. However, over the last decade, the landscape has changed with the discovery of mutation-specific therapy (CFTR modulators) addressing the underlying basic defect causing CF. Here, we review these therapies which are already in clinical use with an emphasis on tezacaftor-ivacftor combination and its potential in the treatment of CF.
Methods
Medline, Embase, The Cochrane Central Register of Controlled Trials and the Web of Science databases were searched for relevant studies. The keywords used were “CF or cystic fibrosis” AND “IVA or ivacaftor” OR “LUM or lumacaftor” OR “TEZ or tezacaftor” OR “VX-659” OR “VX-445” OR “triple therapy”. From this search, appropriate reports on CFTR modulator therapies were identified and applicable cross-references reviewed. Randomized controlled trials evaluating combination therapies with the CFTR corrector tezacaftor (on its own or in combination with VX-659/VX-445) and the CFTR potentiator ivacaftor are analyzed in detail. This is a narrative review of the literature and not a systematic review.
Results and analysis
CFTR modulators
CFTR modulators approved for clinical use include potentiators and correctors.
CFTR potentiators increase the function of the CFTR protein at the cell surface while correctors address the defective protein folding and processing to increase delivery of CFTR protein to the cell surface.
Ivacaftor, the only licensed CFTR potentiator, was approved by FDA in January 2012 based on studies which demonstrated sustained increase in percent predicted FEV1 (ppFEV1) of 10.6 percentage points and a 55% reduction in the rate of pulmonary exacerbations in patients with at least one copy of G551D mutation (STRIVE study). Patients on ivacaftor had improved quality of life, significant weight gain, and a decrease in sweat chloride level compared to those in the placebo group.15 Subsequently, the ENVISION study showed similar benefit in 6–11 years-old children with CF, with G551D mutation, despite milder degree of lung disease at baseline.16 Patients who switched from placebo to ivacaftor in the long-term PERSIST study showed sustained improvement in FEV1, weight and exacerbation rate, through to 96 weeks, which were similar to those observed in the active group during the 48 weeks prior to the study (Table 2) 10.17
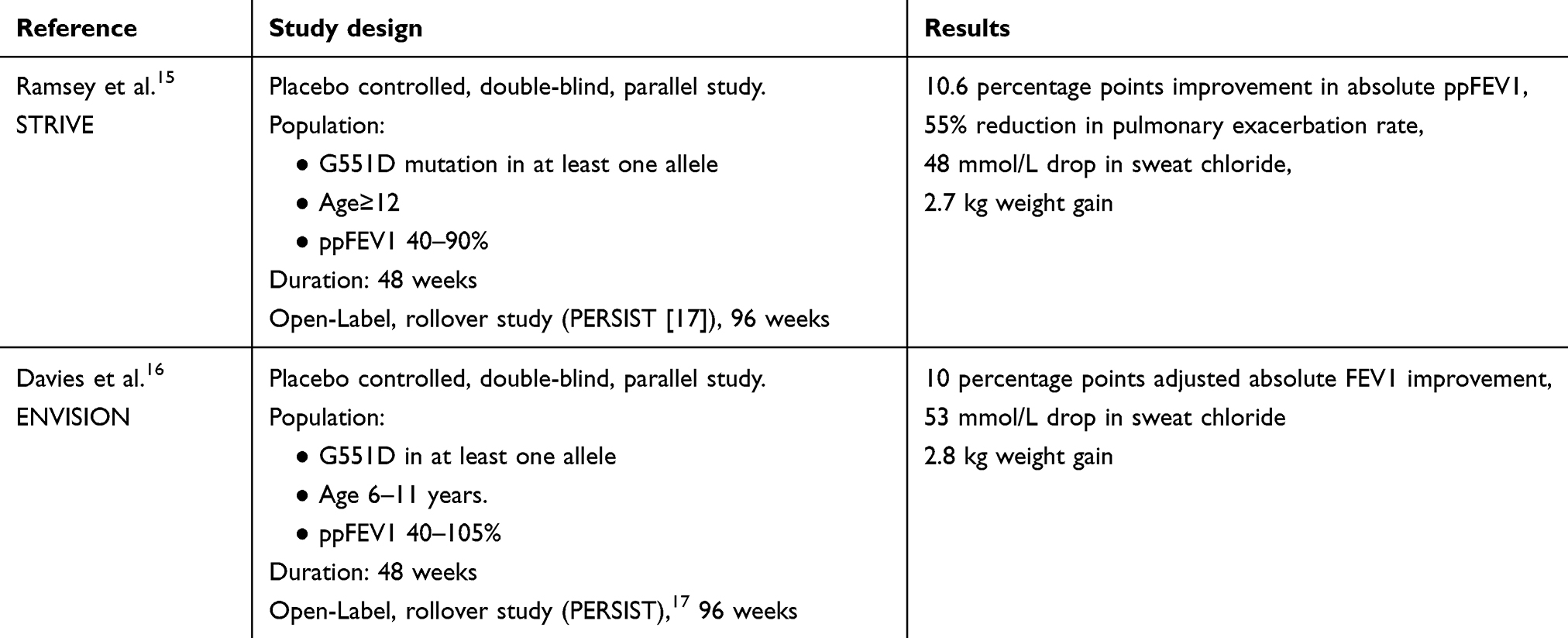 |
Table 2 Clinical trials of ivacaftor monotherapy in CF |
Observational data from the US and UK patient registries have demonstrated clinically favorable results for patients treated with ivacaftor including a lower prevalence of CF-related complications and improved lung function.18 Ivacaftor is also demonstrated to significantly improve objectively measured cough as well as extra-oesophageal manifestations of reflux.19,20
Ivacaftor is now indicated for several mutations within classes III, IV, and V based on clinical and in vitro data using Fisher rat thyroid cell lines.21 However, it is worth noting that only around 4% of CF patients carry the G551D mutation and even with the extended indication only 10–15% of CF patients will benefit from Ivacaftor.1,10
To improve CFTR function in the majority of the CF population, targeting the most common F508del CFTR mutation is required. Approximately 50% of CF patients are homozygous and around 90% of the CF population carry at least one copy of this mutation. Unlike G551D mutation, F508del mutation is far more complex to correct as it results in defective CFTR folding and processing in the endoplasmic reticulum, resulting in minimal amounts of CFTR protein delivered to the cell surface. Furthermore, the small amount of F508del-CFTR protein at the cell surface exhibits defective channel gating and turnover.
A Phase II clinical trial has shown that Ivacaftor alone is not an effective therapeutic option for patients homozygous for the F508del mutation.22 To enhance F508del-CFTR protein activity drugs that increase the delivery of functional CFTR to the cell surface are required; these are termed CFTR correctors.
Lumacaftor (VX-809) is the first corrector to advance through clinical trials based on in vitro data; using a human bronchial epithelium (HBE) cell model an increase in CFTR quantity has been demonstrated.23 In cultured HBE from F508del homozygous patients, lumacaftor-enhanced chloride secretion to approximately 14% of non-CF HBE. However, monotherapy with lumacaftor in F508del homozygous patients failed to demonstrate significant clinical benefit.24
Adding ivacaftor to potentiate the function of the lumacaftor-rescued CFTR was examined in two randomized control trials termed TRAFFIC and TRANSPORT.25 In these randomized controlled trials, involving patients ≥12 years of age and homozygous for F508del mutation, combination therapy with lumacaftor and ivacaftor (Orkambi) showed a modest but statistically significant and sustained increase in ppFEV1 (2.6 percentage points improvement in absolute ppFEV1) and a 34% reduction in pulmonary exacerbations compared to placebo.
Safety and efficacy of long-term treatment with lumacaftor/ivacaftor combination therapy were studied in a Phase III extension study (PROGRESS) (Table 3). This showed that long-term treatment with lumacaftor/ivacaftor was associated with a 42% slower rate of decline in FEV1 as compared to matched registry control.26
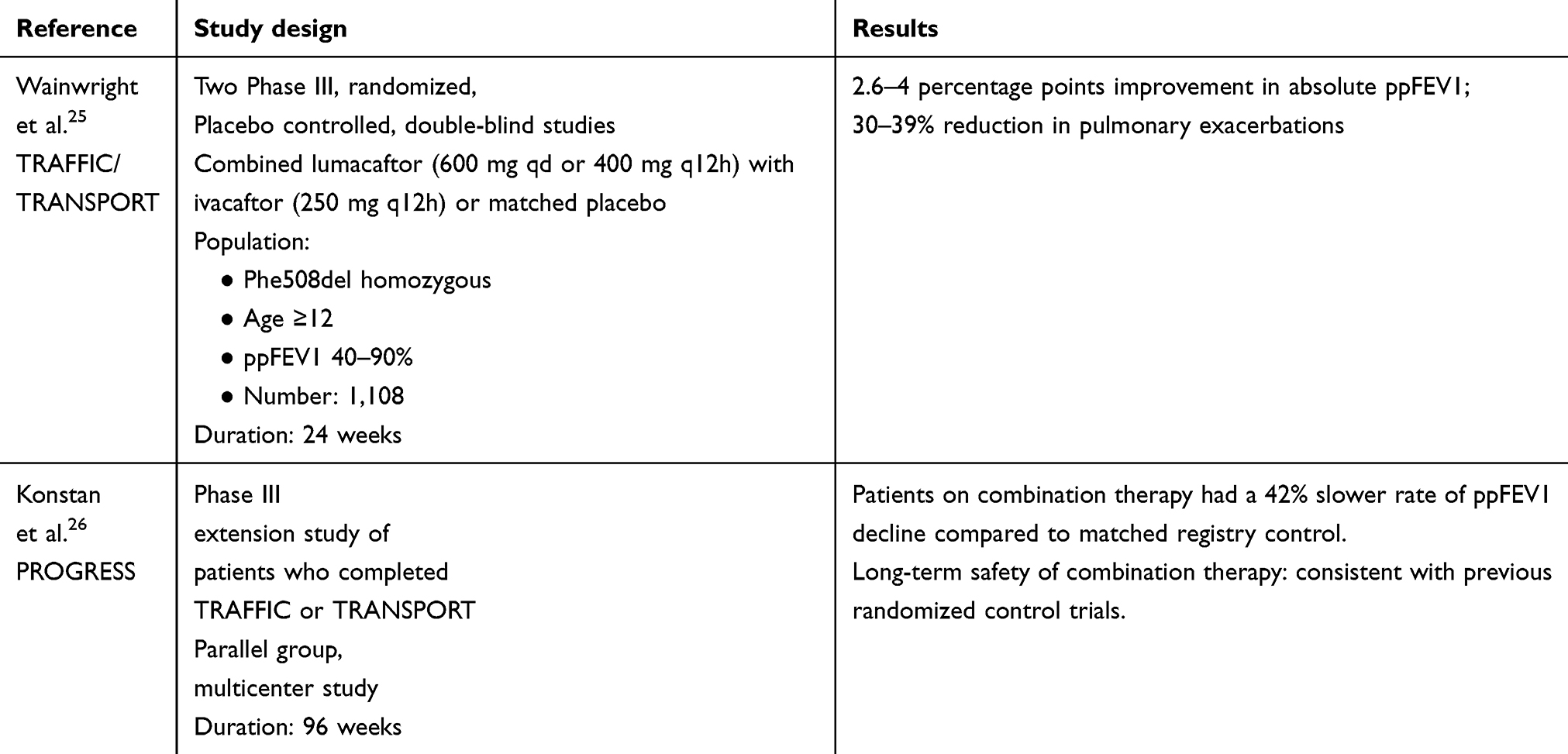 |
Table 3 Clinical trials of lumacaftor and ivacaftor combination therapy in CF |
This combination treatment was not effective in F508del heterozygous patients, and some patients were not able to tolerate the treatment due to adverse effects of chest tightness and acute drop in lung function.27 In addition, significant drug-to-drug interaction of lumacaftor limited the use of combination therapy. Therefore, a more effective CFTR modulator with less drug-to-drug interaction is needed. Tezacaftor and other newer-generation correctors have the potential to overcome these limitations. In this narrative review, combination therapy with tezacaftor/ivacaftor and newer-generation correctors/tezacaftor/ivacaftor is appraised.
Tezacaftor
Tezacaftor: mechanism of action
Tezacaftor is a CFTR corrector without some of the problems associated with lumacaftor therapy. The chemical structure and mechanism of action of tezacaftor are very similar to that of lumacaftor. Therapy leads to enhanced processing and trafficking of F508del-CFTR resulting in demonstrable increase in chloride transport. In an in-vitro study using an Ussing chamber and utilizing F508del/F508del‐HBE cells derived from six donors, enhanced chloride transport was demonstrated with both tezacaftor monotherapy as well as in combination with ivacaftor.28 Twenty-four hour treatment with tezacaftor improved chloride transport from a baseline of 2.5 to 8.1% of normal levels. Combination treatment with ivacaftor for 24 hrs further improved this to 15.7% of non‐CF airway cells. Treatment of these cultured cells for 72 hrs also increased fluid transport and ciliary beat frequency to levels greater than those observed with either agent alone. This in-vitro data suggest that the combination of tezacaftor and ivacaftor may lead to an improvement in chloride transport, fluid transport, and ciliary beat frequency in F508del/F508del‐HBE cells. Subsequently in both Phase II and Phase III clinical trials, a decrease in sweat chloride levels was demonstrable with tezacaftor and tezacaftor/ivacaftor combination therapy.29–33
Tezacaftor: pharmacological profile
The pharmacokinetic profile of tezacaftor monotherapy and tezacaftor/ivacaftor combination therapy has been reported in a Phase II trial.29 In subjects homozygous for F508del, tezacaftor was rapidly absorbed after oral administration and reached steady state level in around 2 weeks. Exposures of tezacaftor and its metabolites were similar with both tezacaftor monotherapy as well as with tezacaftor/ivacaftor combination therapy. Similarly, exposures of ivacaftor and its metabolites were unaffected by concomitant tezacaftor therapy. Steady-state area under the curve estimates of tezacaftor was similar with both the 50 mg every 12 hrs and the 100 mg daily doses. The recommended dose of tezacaftor/ivacaftor combination therapy is 100 mg every day of tezacaftor and 150 mg every 12 hrs for ivacaftor. At this dose, steady-state exposures of tezacaftor and ivacaftor in F508del/G551D compound heterozygous subjects were demonstrated to be very similar to that in subjects homozygous for F508del.
Reports suggest that lumafctor may be an inducer of cytochrome (CYP) drug metabolizing enzymes.29,34,35 Lumacaftor being an inducer of CYP3A4 and ivacaftor being a substrate, a clinically significant interaction between the two drugs is possible. However, tezacaftor is not an inducer of CYP3A4 enzymes which makes drug–drug interactions of less concern. It has been demonstrated that the metabolism of drugs which are CYP3A substrates, including hormonal contraceptives, is unlikely to be affected by tezacaftor/ivacaftor combination therapy.35
Tezacaftor/ivacaftor combination therapy: clinical efficacy data
In subjects homozygous for F508del or compound heterozygous for F508del and G551D, the safety and efficacy of tezacaftor monotherapy and tezacaftor/ivacaftor combination therapy was evaluated in a multi-center, double-blind, placebo-controlled Phase II trial.29 In homozygous patients varying doses of tezacaftor in a dose escalation phase as well as in a dosage regimen testing phase were used, on its own or in combination with ivacaftor. The heterozygotes received tezacaftor in addition to regular physician-prescribed ivacaftor. The primary end points of this trial were safety through the study (56 days) and change in sweat chloride levels from baseline through day 28. Secondary end-point included change in ppFEV1 through day 28. Pharmacokinetics data were also reported.
Eligible subjects included in this study met standard diagnostic criteria for CF and had ppFEV1 between 40% and 90%; body weight ≥40 kg; and a body mass index (BMI) ≥18.5 kg/m2. The F508del homozygous subjects were ≥18 years of age and those heterozygous for F508del and G551D ≥12 years of age. Subjects were randomly assigned in a 4:1 ratio to four different doses of tezacaftor (10, 30, 100, and 150 mg od.) on its own and in combination with ivacaftor 150 mg q 12 hourly in the dose escalation phase. In the dosage regimen testing phase, three doses of tezacaftor/ivacaftor (tezacaftor 100 mg/ivacaftor 150 mg 12 hourly; tezacaftor 100 mg/ivacaftor 50 mg 12 hourly; and tezacaftor 50 mg 12 hourly/ivacaftor 150 mg 12 hourly) were evaluated. In the F508del and G551D group tezacaftor 100 mg od/ivacaftor 150 mg 12 hourly was used. In this group, the comparator was placebo/ivacaftor. Thus, there were 14 arms to the study, including placebo only arms. Subjects received medications for 28 days with a further 28-day washout period and follow-up at 56 days.
One hundred and eighty-five of 190 subjects (mean age 30 years, 56% males) overall completed the study. The incidence of adverse events was found to be similar across all the study arms. Though adverse events reported were common (90.9% with tezacaftor monotherapy, 86.8% with tezacaftor/ivacaftor, and 90.9% in the placebo arm) the majority of these (81.4%) were mild to moderate in nature. The most common adverse events reported were infective pulmonary exacerbation of CF, cough, increased sputum, nausea, diarrhea, headache, and fatigue. All study-attributable serious adverse effects were pulmonary exacerbations and were reported in 7% of the subjects in an active drug arm (10 of 139) and 15% in the placebo arm (5 of 33).
Significant within-group decrease in sweat chloride levels from baseline to day 28 in the F508del homozygous group was observed with the two higher doses of tezacaftor (100 and 150 mg od) and 10, 30, and 100 mg of tezacaftor in combination with ivacaftor. Decrease in sweat chloride levels with the tezacaftor 100 mg od and ivacaftor 150 mg hourly dose (6.04 mmol/L) were significant as compared with placebo as well. The largest improvement in ppFEV1 (within-group increase from baseline of 3.75 percentage points; treatment effect vs placebo 3.89 percentage points, p<0.05) was with tezacaftor 100 mg od and ivacaftor 150 mg 12 hourly.
In the F508del and G551D compound heterozygous group, though the within group decrease (7.02 mmol/L) in sweat chloride levels was not significant, as compared to placebo this was significant (−17.20, p<0.05). Statistically significant increase in absolute and relative within-group ppFEV1 through day 28 was observed (4.6 and 7.29 percentage points, respectively, p<0.05), but not in comparison with placebo.
This was the first clinical trial evaluating the combination of tezacaftor/ivacaftor in patients with CF homozygous for F508del and heterozygous F508del/G551D patients. The trial showed that the combination had a satisfactory adverse effect profile, reduced sweat chloride levels and improved lung function as assessed by FEV1. This set the stage for Phase III trials in these two groups of CF patients.
Following on from the Phase-II trial, combination therapy with tezacaftor/ivacaftor has been evaluated in both subsets of CF patients; homozygous for F508del and compound heterozygous for the F508del mutation and a second allele mutation associated with residual CFTR function.30,31 Definition of residual CFTR function and list of mutations is detailed in the supplement to the published article and was based on clinical and lab criteria.30
Five hundred ten patients ≥12 years of age with CF and homozygous for the F508del mutation underwent randomization in this multicenter, double-blind, placebo-controlled, parallel-group randomized trial (EVOLVE).30 Subjects had their FEV1 between 40% and 90% predicted at screening and had stable disease. Of the 510 who underwent randomization 509 received tezacaftor 100 mg od/ivacaftor 150 mg q12hrs in a fixed-dose combination tablet or matched placebo for 24 weeks in a 1:1 ratio. The primary end-point of the study was absolute change in ppFEV1 through week 24. Key secondary end-points were relative change in ppFEV1, number of pulmonary exacerbations, absolute change from baseline in BMI and the absolute change in the respiratory domain score on the Cystic Fibrosis Questionnaire-Revised (CFQ-R).
Of the 510 subjects who underwent randomization, 509 received at least one dose of a trial medication and 475 completed the requisite 24 weeks of the trial. The trial met its primary end-point; significant improvement was noted in favor of tezacaftor/ivacaftor as compared to placebo on the absolute and relative changes in ppFEV1 (4 percentage points and 6.8%, respectively, p<0.001). Improvement in FEV1 was observed at the first assessment point (15 days) and was consistently sustained through to 24 weeks. The risk of pulmonary exacerbation was significantly lower in the tezacaftor/ivacaftor group (p=0.005) as compared to placebo. The risk of pulmonary exacerbations at 24 weeks was 35% in the placebo group as compared to 25% in the tezacaftor/ivacaftor group. No significant differences between the placebo and tezacaftor/ivacaftor groups were noted in the BMI at week-24. At this point, as per study design, testing hierarchy for statistical significance was broken. The CFQ-R respiratory domain scores favored tezacaftor/ivacaftor. Tezacaftor/ivacaftor treatment led to a reduction in sweat chloride levels, with a between group difference of −10.1 mmol/L. The incidence of adverse events was similar in both groups with at least one adverse effect observed in 92.7%. Most of these were either mild (41.8%) or moderate (40.9%) in severity. Serious adverse effects were reported in 12.4% of the tezacaftor/ivacaftor group and 18.2% of the placebo group. Commonly noted adverse effects (>10 incidence in either group) were similar to that observed in the Phase II trial and included infective pulmonary exacerbations, cough, headache, nasopharyngitis, increased sputum production, pyrexia, hemoptysis, oro-pharyngeal pain, and fatigue. Importantly, and in contrast to lumacaftor/ivacaftor, tezacaftor/ivacaftor treatment was not linked with an acute post-dose decline in FEV1 nor increased risk of respiratory events or any derangements in liver function tests. Study results are summarized in Table 4.
 |
Table 4 Clinical trials of tezacaftor and ivacaftor combination therapy in CF |
Two hundred and forty-eight patients ≥12 years of age with CF and heterozygous for the F508del mutation and a residual function CFTR mutation underwent randomization in this multicenter, double-blind, placebo-controlled, two-period, three-intervention crossover trial ( EXPAND).31 The study had two intervention periods of 8 weeks separated by a washout period of 8 weeks. Each patient received two of tezacaftor/ivacaftor combination therapy, ivacaftor monotherapy or placebo. Similar to EVOLVE, patients had their FEV1 between 40% and 90% predicted at screening and had stable disease. Primary end-point of the study was also similar to EVOLVE; absolute change in ppFEV1 from the baseline value to the average of week 4 and week 8 measurement in each intervention period. A key secondary end-point was the absolute change in CFQ-R respiratory domain score. Further secondary end-points included relative change in ppFEV1 and absolute change in sweat chloride concentration.
Of the 248 undergoing randomization two patients were later deemed ineligible for the study. Of the remaining 246 subjects, 234 (95%) completed both intervention periods resulting in 481 analyzable periods. This study also met its primary end-point; the least-squares mean difference vs placebo was 4.7 percentage points for ivacaftor monotherapy and 6.8 points for tezacaftor/ivacaftor combination therapy (p<0.001 for both comparisons). The difference between the two treatment arms was statistically significant in favor of tezacaftor/ivacaftor combination therapy (p<0.001). Again improvements in FEV1 were observed at 15 days and were sustained through the study period. Change in CFQ-R respiratory domain score significantly improved with both treatment arms as compared to placebo (11.1 for tezacaftor/ivacaftor and 9.7 for ivacaftor monotherapy vs placebo, p<0.001). The difference between tezacaftor/ivacaftor combination therapy and ivacaftor monotherapy was not significant. Changes in sweat chloride levels were −9.5 mmol/L for tezacaftor/ivacaftor vs placebo, −4.5 mmol/L for ivacaftor vs placebo and −5.1 for tezacaftor/ivacaftor vs ivacaftor monotherapy. The incidence of adverse events was similar across all intervention groups; most events were mild to moderate in severity, similar to the EVOLVE study. Results are summarized in Table 4.
These two Phase III studies demonstrated that tezacaftor/ivacaftor combination therapy improves lung function, as assessed by FEV1, in CF patients homozygous for F508del mutation as well as those heterozygous for F508del and a residual-function mutation. Long-term data will tell whether these improvements noted in both these studies will result in enhanced clinical benefits over time.
Triple combination CFTR modulators (two correctors and one potentiator)
As discussed so far, tezacaftor/ivacaftor combination therapy is efficacious both in F508del homozygotes as well as F508del/RF mutation heterozygotes and is well tolerated with improved side effect profile and less drug-drug interactions. However, like lumacaftor/ivacaftor combination the lung function improvement with tezacaftor/ivacaftor is modest, at best, especially as compared to the improvements seen with ivacaftor in patients with G551D mutations. Indeed, these lung function improvements are comparable to established and inexpensive therapies such as osmotic agents (nebulized hypertonic saline and inhaled mannitol), azithromycin, inhaled tobramycin, and recombinant human DNase.36–42 Dual combination therapy is also not effective in heterozygotes with F508del/minimal function (MF)-CFTR mutations. MF-CFTR mutation refers to mutations that do not make meaningful CFTR protein and are associated with significantly higher rates of pancreatic insufficiency and sweat chloride levels of >86 mmol/L. Therefore, either a more potent corrector or combination therapy addressing most of the CF population is needed.
It has been suggested that the combination of two correctors with different mechanistic actions may have a greater effect on the amount of CFTR protein expressed at the cell surface than either on their own.43 On the basis of this premise next-generation, CFTR correctors have been developed. The hypothesis is a newer-generation corrector in combination with tezacaftor/ivacaftor (ie, a “triple therapy”), will lead to both increased amount and function of F508del CFTR protein. To address this, two next generation correctors (named VX-659 and VX-445) were developed and evaluated in parallel; the results of clinical trials have been recently published.32,33 Both these compounds share a similar structure to each other but have different mechanism of action and structure to tezacaftor. Additive effect to tezacaftor/ivacaftor was described in-vitro. VX-659 was shown to improve processing and trafficking of F508del CFTR protein in a HBE cell-model from cells derived from patients homozygous for the F508del mutation or heterozygous for F508del and a MF mutation allele. The combination of VX-659-tezacaftor, with or without ivacaftor, resulted in significantly higher steady-state levels of mature CFTR protein than either compound alone.32
Twelve patients heterozygous for F508del/MF mutation were randomized to either receive triple therapy (9) or placebo (3) over a period of 14 days to evaluate safety and pharmacokinetics. The adverse effect profile of this triple combination was similar to tezacaftor/ivacaftor, with the most common adverse effects being cough and pulmonary exacerbations. Most adverse effects were mild or moderate and none led to discontinuation of the trial regimen. Triple therapy led to improvement in ppFEV1 as well as sweat chloride levels. VX-445 was evaluated in healthy volunteers in a Phase I study (details not reported). The results of both VX-659 and VX-445 Phase-II studies were designed and reported in parallel.
The safety and clinical efficacy of VX-659 in combination therapy was evaluated in a randomized, parallel-track, placebo or active-controlled, double-blind multicenter Phase II trial.32 Three doses of VX-659 (80 mg, 240 mg, and 400 mg od) in combination with tezacaftor 100 mg od/ivacaftor150 mg q12hrs were studied in the F508del/MF mutation CF patients in comparison to triple-placebo. VX-659 (400 mg od) in combination with tezacaftor/ivacaftor was studied in F508del homozygous CF patients; the comparator was placebo/tezacaftor/ivacaftor as tezacaftor/ivacaftor combination therapy has already been shown to be beneficial in this group. VX-651 is a deuterated form of ivacaftor which is dosed once a day as compared to the non-deuterated form which is dosed b.d. VX-659 (400 mg) in combination with tezacaftor/VX-651, the combination leading to once daily dosing of triple therapy, was studied as compared to placebo in F508del/MF mutation CF patients. Treatments were administered over a period of 4 weeks with 4 weeks each of preceding screening/run-in and safety follow-up periods. Stable patients with CF and FEV1 between 40% and 90% predicted at screening were recruited. Primary end-points were safety and improvement in ppFEV1 at day 29. Secondary end-points included absolute change in sweat chloride concentration and the absolute change in CFQ-R respiratory domain score at day 29.
One hundred and seventeen patients were randomized to receive triple therapy or a comparator and 115 completed the 4-week intervention period. In the F508del/MF mutation group 53 received VX-659/tezacaftor/ivacaftor (10 placebo) and 19 received VX-659/tezacaftor/VX-651 (6 placebo). In the F508del homozygous group, 18 received VX-659/tezacaftor/ivacaftor (11 placebo/tezacaftor/ivacaftor). Adverse effect profile was similar to that observed in the Phase Istudy. There were no discontinuations of treatment due to adverse effects in any patient receiving VX-659/tezacaftor/ivacaftor; one subject discontinued in the VX-659/tezacaftor/VX-651 group. The safety profile of VX-659/tezacaftor/VX-651 was noted to be similar to VX-659/tezacaftor/ivacaftor. Bronchoconstriction was not noted in any subject taking VX-659 in combination therapy. VX-659/tezacaftor/ivacaftor led to significant improvements in ppFEV1 from baseline (placebo and placebo/tezacaftor/ivacaftor did not); the range of improvements was from 9.7 (F508del homozygous) to 13.3 percentage points (in F508del/MF heterozygotes). Improvements in FEV1 were observed at day 15 and sustained at day 29. Concomitant improvements were noted in the secondary end-points of sweat chloride levels and CFQ-R respiratory domain score. Sweat chloride level improvements from baseline ranged from 42.2 mmol/L to 51.4 mmol/L. CFQ-R respiratory domain scores improvements ranged from 19.5 to 24.6 points. Improvements in end-points in those receiving VX-659/tezacaftor/VX-651 were similar.
Similar to VX-659, the efficacy of VX-445 was also demonstrated in-vitro and then in a Phase I trial (involving healthy volunteers). VX-445 was evaluated in a similar manner to VX-659 and in the same two genotypes of CF patients and over the same intervention duration of 4-weeks.33 In the F508del/MF genotype 50, 100, or 200 mg once daily VX-445 in combination with tezacaftor 100 mg od/ivacaftor 150 mg q12hrs was compared with triple placebo. In this genotype, the combination of VX-445/tezacaftor/VX-561 was also evaluated. In the F508del homozygous group, VX-445 200 mg o.d in combination with tezacaftor/ivacaftor was compared with placebo/tezacaftor/ivacaftor.
One hundred and twenty-three patients (95 F508del/MF heterozygous and 28 F508del homozygous) were randomized and 119 completed the 4-week study period. The adverse effect profile of VX-445/tezacaftor/ivacaftor was very similar to VX-659/tezacaftor/ivacaftor; 68 of 74 (92%) who received VX-445/tezacaftor/ivacaftor reported an adverse effect with 96% of these being mild or moderate. Serious adverse effects, none considered life-threatening, occurred in 6 subjects; 3 (14%) in the VX-445/tezacaftor/VX-561, 2 (17%) in triple placebo and 1(14%) in the tezacaftor/ivacaftor. The 3 in the VX-445 group and 1 in the tezacaftor/ivacaftor control discontinued treatment.
Significant improvements in ppFEV1 as compared to baseline, and similar to VX-659, were observed and ranged from 11 percentage points (F508del homozygous group) to 7.9 percentage points (F508del/MF heterozygous). Improvements in sweat chloride levels as well as CFQ-R respiratory domain were similar to that observed with VX-659; sweat chloride level improvements ranged from 33.2 to 39.6 mmol/L and that of CFQ-R respiratory domain from 15.4 to 25.7 points.
The results of these Phase II trials are summarized in Table 5.
 |
Table 5 Clinical trials of next-generation corrector, tezacaftor and ivacaftor combination therapy in CF |
Conclusion
The development of CFTR modulator therapies targeting defective protein function and expression marked a paradigm change in the management of CF. Ivacaftor, a potentiator, leads to a marked and sustained improvement and is now considered the standard of care in CF patients with the appropriate genotype. However, only a small proportion of CF patients with specific mutations are eligible for treatment with ivacaftor. Lumacaftor/ivacaftor combination is currently accepted as standard of care for F50del homozygous patients in many parts of the world but it is not approved for routine prescription in England on cost effectiveness grounds [44]. However, it is available for patients with advanced disease from Vertex on compassionate grounds.
Phase III studies clearly demonstrated that tezacaftor/ivacaftor combination therapy is effective in F508del homozygotes as well as F508del/RF mutation heterozygotes. This treatment is well tolerated with an acceptable side effect profile which is comparable to placebo. Unlike lumacaftor/ivacaftor, there is no early bronchoconstriction noted and the drug–drug interactions are less of a concern. Lung function improvements seen in F508del homozygotes were similar in both dual combination therapies.
The improvements reported recently with triple therapy regimes represent a major advance compared to dual therapy. Addition of a next generation corrector (VX-659 or VX-445) to tezacaftor/ivacaftor was demonstrated to further improve ppFEV1 by around 10 percentage points. The adverse effect profile of these triple combinations is acceptable and is similar to tezacaftor/ivacaftor combination.
Triple therapy promises an effective CFTR modulator therapy for more than 90% of the CF patients. At present, we do not know whether improvements in lung function will be sustained and whether this in turn would lead to reduction in exacerbations and improve other CF outcomes. The results of ongoing Phase III studies are eagerly awaited and will address the above queries. There are also a number of other potential corrector/potentiator/amplifier combination therapies in various stages of clinical development which might benefit patients in the future.1
Disclosure
Dr. Shiferaw has received support from Vertex Pharmaceuticals to attend Cystic Fibrosis specific conferences. The authors report no other conflicts of interest in this work.
References
1. Cystic Fibrosis Foundation. Available from: https://www.cff.org/. Accessed July 26, 2019.
2. Riordan JR, Rommens JM, Kerem B, et al. Identification of the cystic fibrosis gene: cloning and characterization of complementary DNA. Science. 1989;245(4922):1066–1073.
3. Riordan JR. CFTR function and prospects for therapy. Annu Rev Biochem. 2008;77:701–726. doi:10.1146/annurev.biochem.75.103004.142532
4. Anderson MP, Gregory RJ, Thomson S, et al. Demonstration that CFTR is a chloride channel by alteration of anion selectivity. Science. 1991;253:202–205.
5. Smith JJ, Welsh MJ. cAMP stimulates bicarbonate secretion across normal, but not cystic fibrosis airway epithelia. J Clin Invest. 1992;89:1148–1153. doi:10.1172/JCI115696
6. Stutts MJ, Canessa CM, Olsen JC, et al. CFTR as a cAMP-dependent regulator of sodium channels. Science. 1995;269:847–850.
7. Andersen DH. Cystic fibrosis of the pancreas and its relation to celiac disease. Am J Dis Child. 1938;56:344. doi:10.1001/archpedi.1938.01980140114013
8. Cystic Fibrosis Foundation. Patient registry: annual data report; 2017. Available from: https://www.cff.org/Research/Researcher-Resources/Patient-Registry/2017-Patient-Registry-Annual-Data-Report.pdf.
9. JS E. Cystic Fibrosis. Lancet. 2016;383:2519–2531.
10. Available from: www.CFTR2.org. Accessed July 26, 2019.
11. Sosnay PR, Siklosi KR, Van Goor F, et al. Defining the disease liability of variants in the cystic fibrosis transmembrane conductance regulator gene. Nat Genet. 2013;45(10):1160–1167. doi:10.1038/ng.2745
12. Zielenski J. Genotype and phenotype in cystic fibrosis. Respiration. 2000;67(2):
13. Oates GR, Schechter MS. Socioeconomic status and health outcomes: cystic fibrosis as a model. Expert Rev Respir Med. 2016;10(9):967–977. doi:10.1080/17476348.2016.1196140
14. Rafeeq MM, Murad HAS. Cystic fbrosis: current therapeutic targets and future approaches. J Transl Med. 2017;15(1):84. doi:10.1186/s12967-017-1193-9
15. Ramsey BW, Davies J, McElvaney NG, et al. A CFTR potentiator in patients with cystic fibrosis and the G551D mutation. N Engl J Med. 2011;365(18):1663–1672. doi:10.1056/NEJMoa1105185
16. Davies JC, Wainwright CE, Canny GJ, et al. Efficacy and safety of ivacaftor in patients aged 6 to 11 years with cystic fibrosis with a G551D mutation. Am J Respir Crit Care Med. 2013;187(11):1219–1225. doi:10.1164/rccm.201301-0153OC
17. McKone EF, Borowitz D, Drevinek P, et al. Long-term safety and efficacy of ivacaftor in patients with cystic fibrosis who have the Gly551Asp-CFTR mutation: a phase 3, open-label extension study (PERSIST). Lancet Respir Med. 2014;2(11):902–910. doi:10.1016/S2213-2600(14)70218-8
18. Bessonova L, Volkova N, Higgins M, et al. Data from the US and UK cystic fibrosis registries support disease modification by CFTR modulation with ivacaftor. Thorax. 2018;73(8):731–740. doi:10.1136/thoraxjnl-2017-210394
19. Faruqi S, Shiferaw D, Morice AH. Effect of ivacaftor on objective and subjective measures of cough in patients with cystic fibrosis. Open Respir Med J. 2016;10:105–108. doi:10.2174/1874306401610010105
20. Zeybel GL, Pearson JP, Krishnan A, et al. Ivacaftor and symptoms of extra-oesophageal reflux in patients with cystic fibrosis and G551D mutation. J Cyst Fibros. 2017;16(1):124–131. doi:10.1016/j.jcf.2016.07.004
21. Van Goor F, Yu H, Burton B, Hoffman BJ. Effect of ivacaftor on CFTR forms with missense mutations associated with defects in protein processing or function. J Cyst Fibros. 2014;13(1):29–36. doi:10.1016/j.jcf.2013.06.008
22. Flume PA, Kiou TG, Borowitz DS, et al. Ivacaftor in subjects with cystic fibrosis who are homozygous for the F508D- CFTR mutation. Chest. 2012;142:718–724. doi:10.1378/chest.11-2672
23. Van Goor F, Hadida S, Grootenhuis PD, et al. Correction of the F508D-cftr protein processing defect in vitro by the investigational drug vx-809. Proc Natl Acad Sci U S A. 2011;108:18843–18848. doi:10.1073/pnas.1105787108
24. Clancy JP, Rowe SM, Accurso FJ, et al. Results of a phase IIa study of VX-809, an investigational CFTR corrector compound, in subjects with cystic fibrosis homozygous for the F508D-CFTR mutation. Thorax. 2012;67:12–18. doi:10.1136/thoraxjnl-2011-200393
25. Wainwright CE, Elborn JS, Ramsey BW, et al. Lumacaftor-ivacaftor in patients with cystic fibrosis homozygous for phe508del cftr. N Engl J Med. 2015;373:220–231. doi:10.1056/NEJMoa1409547
26. Konstan MW, McKone EF, Moss RB, et al. Assessment of safety and efficacy of long-term treatment with combination lumacaftor and ivacaftor therapy in patients with cystic fibrosis homozygous for the F508D-cftr mutation (PROGRESS): A phase 3, extension study. Lancet Respir Med. 2017;5(2):107–118. doi:10.1016/S2213-2600(16)30427-1
27. Rowe SM, McColley SA, Rietschel E, et al. Lumacaftor/ivacaftor treatment of patients with cystic fibrosis heterozygous for F508D-cftr. Ann Am Thorac Soc. 2017;14(2):213–219. doi:10.1513/AnnalsATS.201609-689OC
28. Van Goor F, Grootenhuis P, Hadida S, et al. Nonclinical profile of the CFTR corrector VX-661. Pediatr Pulmonol. 2016;51(S45):S194–S485. (abstract from conference proceedings [number 217]. doi:10.1002/ppul.23576)
29. Donaldson SH, Pilewski JM, Griese M, et al. Tezacaftor/Ivacaftor in Subjects with Cystic Fibrosis and F508del/F508del-CFTR or F508del/G551D-CFTR. Am J Respir Crit Care Med. 2018;197(2):214–224. doi:10.1164/rccm.201704-0717OC
30. Taylor-Cousar JL, Munck A, McKone EF, et al. Tezacaftor-ivacaftor in patients with cystic fibrosis homozygous for Phe508del. N Engl J Med. 2017;377(21):2013–2023. doi:10.1056/NEJMoa1709846
31. Rowe SM, Daines C, Ringshausen FC, et al. Tezacaftor-ivacaftor in residual-function heterozygotes with cystic fibrosis. N Engl J Med. 2017;377(21):2024–2035. doi:10.1056/NEJMoa1709847
32. Davies JC, Moskowitz SM, Brown C, et al. VX-659-tezacaftor-ivacaftor in patients with cystic fibrosis and one or two Phe508del alleles. N Engl J Med. 2018;379(17):1599–1611. doi:10.1056/NEJMoa1807119
33. Keating D, Marigowda G, Burr L, et al. VX-445-tezacaftor-ivacaftor in patients with cystic fibrosis and one or two Phe508del alleles. N Engl J Med. 2018;379(17):1612–1620. doi:10.1056/NEJMoa1807120
34. Schneider EK. Cytochrome P450 3A4 induction: lumacaftor versus ivacaftor potentially resulting in significantly reduced plasma concentration of ivacaftor. Drug Metab Lett. 2018;12(1):71–77. doi:10.2174/1872312812666180328105259
35. Garg V, Shen J, Li C, et al. Pharmacokinetic and drug-drug interaction profiles of the combination of tezacaftor/ivacaftor. Clin Transl Sci. 2019;12(3):267–275. doi:10.1111/cts.12610
36. Mayer ML. umacaftor-ivacaftor (Orkambi) for cystic fibrosis: behind the ‘breakthrough’. BMJ Evidence Based Med. 2016;21:83–86. doi:10.1136/ebmed-2015-110325
37. Mogayzel PJ
38. Cystic fibrosis: diagnosis and management: NICE guideline [NG78]; October, 2017. Available from: https://www.nice.org.uk/guidance/ng78. Accesssed 26 July, 2019.
39. Fuchs HJ, Borowitz DS, Christiansen DH, et al. Effect of aerosolized recombinant human DNase on exacerbations of respiratory symptoms and on pulmonary function in patients with cystic fibrosis. The pulmozyme study group. N Engl J Med. 1994;331:637–642. doi:10.1056/NEJM199407073310103
40. Yang C, Montgomery M. Dornase alfa for cystic fibrosis. Cochrane Database Syst Rev. 2018;6(9):CD001127.
41. Elkins MR, Robinson M, Rose BR, et al. A controlled trial of long-term inhaled hypertonic saline in patients with cystic fibrosis. N Engl J Med. 2006;354:229–240. doi:10.1056/NEJMoa043900
42. Nevitt SJ, Thornton J, Murray CS, et al. Inhaled mannitol for cystic fibrosis. Cochrane Database Syst Rev. 2018;9(2):CD008649.
43. Grootenhuis P, Van Goor F, Hadida S, et al. Discovery and biological profile of next-generation CFTR correctors. Pediatr Pulmonol. 2016;51(S45):S194–S485. (abstract from conference proceedings [number 188]. doi:10.1002/ppul.23576)
44. Lumacaftor–ivacaftor for treating cystic fibrosis homozygous for the F508del mutation. Technology appraisal guidance [TA398]; July 27, 2016. Available from: https://www.nice.org.uk/guidance/ta398. Accessed 26 July, 2019.
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.