")
Back to Journals » Therapeutics and Clinical Risk Management » Volume 11
Profile of pomalidomide and its potential in the treatment of myelofibrosis
Received 17 January 2015
Accepted for publication 25 February 2015
Published 2 April 2015 Volume 2015:11 Pages 549—556
DOI https://doi.org/10.2147/TCRM.S69211
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Professor Garry Walsh
Video presented by Ruben A Mesa and Krisstina L Gowin.
Views: 313
Krisstina L Gowin, Ruben A Mesa
Division of Hematology and Medical Oncology, Mayo Clinic Arizona, Scottsdale, AZ, USA
Abstract: Myelofibrosis, a Philadelphia-negative myeloproliferative neoplasm, is in a new treatment era after the discovery of the JAK2V617F mutation in 2005. JAK inhibitors boast improvements in disease-related symptoms, splenomegaly, and overall survival; however, treatment of myelofibrosis remains a challenge, given the lack of improvement in cytopenias with these agents. Second-generation immunomodulatory agents, such as pomalidomide, have shown efficacy in myelofibrosis-associated anemia within multiple clinical trials. Five major pomalidomide clinical trials have been completed to date, and demonstrate tolerability and efficacy with low-dose pomalidomide (0.5 mg/day) in the treatment of myelofibrosis, and no clinical benefit of elevated dosing regimens (≥2.5 mg/day). Anemia responses ranged from 17% to 36% as per the International Working Group for Myelofibrosis Research and Treatment consensus guidelines, while improvements in splenomegaly were rare, and observed in <1% of most clinical trials. In comparison with earlier immunomodulatory agents, pomalidomide was associated with an improved toxicity profile, with substantially lower rates of myelosuppression and neuropathy. Given the low overall response rate to pomalidomide as a single agent, combination strategies are of particular interest for future studies. Pomalidomide is currently being tested in combination with ruxolitinib, and other novel combinations are likely on the horizon.
Keywords: myelofibrosis, pomalidomide, immunomodulatory, myeloproliferative neoplasm
Myelofibrosis: an introduction
Myelofibrosis is a chronic Philadelphia-negative myeloproliferative neoplasm characterized by clonal proliferation of myeloid hematopoietic cells and intramedullary fibrosis.1 Myelofibrosis can arise de novo, as in primary myelofibrosis, or may evolve from a prior myeloproliferative neoplasm, such as in post essential thrombocytosis myelofibrosis or post polycythemic myelofibrosis. The pathogenesis of myelofibrosis is driven by high levels of profibrogenic and proangiogenic intramedullary cytokines, such as transforming growth factor beta, basic fibroblast growth factor, tumor necrosis factor alpha, and vascular endothelial growth factor.2,3 Clinical manifestations of myelofibrosis include cytopenias, profound splenomegaly, bone pain, night sweats, weight loss, and fatigue.4 Anemia and fatigue can be debilitating in patients afflicted with myelofibrosis.5 The severe anemia manifested in myelofibrosis is driven by multiple mechanisms, including decreased erythropoietic capacity secondary to marrow fibrosis, changes in the intramedullary marrow microenvironment and cytokine expression profile, and sequestration from massive splenomegaly.6 The treatment of anemia in myelofibrosis presents a unique challenge in the management of these patients.
Recommendations for treatment of myelofibrosis are based upon disease risk stratification with calculation of the Dynamic International Prognostic Scoring System (DIPSS) risk score.7 The DIPSS uses five risk factors to predict survival, ie, age older than 65 years, hemoglobin lower than 10 g/dL, leukocytes higher than 25×109/L, circulating blasts >1%, and constitutional symptoms. Risk categories are low (score 0), intermediate-1 (score 1–2), intermediate-2 (score 3–4), and high (score 5–6), with median overall survival decreasing with increased risk. Each variable contributes one point to the overall score, with the exception of anemia, which accounts for a 2-point contribution. This underscores the importance of anemia in prognostication with regard to myelofibrosis and highlights the overall impact of a hemoglobin level <10 g/dL on calculation of risk and subsequent therapeutic recommendations.
Currently, the only treatment modality that offers potential for cure is allogeneic stem cell transplantation.8 For patients with low-risk disease and those who are not transplant-eligible, the therapeutic options are palliative in nature and aim to reduce disease-related symptoms. Classic treatment options include hydroxyurea, erythropoietin, interferon, androgens, immunomodulatory medications (IMiDs), corticosteroids, splenic radiation, and splenectomy.9–18 Agents are often employed in combination, particularly cytoreductive agents and those that aim to improve anemia, such as erythropoietin, corticosteroids, and androgens. Combination therapy with the androgenic agent danazol is a therapeutic strategy that is currently under active clinical investigation (ClinicalTrials.gov NCT01732445).
The therapeutic armamentarium for myelofibrosis has recently expanded after the discovery of the JAK2V617F mutation in 2005 and the subsequent development of JAK inhibitors.19,20 Ruxolitinib, the first JAK inhibitor approved by the US Food and Drug Administration, has led to significant improvements in symptom burden, decreased splenomegaly, and improvement in overall survival; however, its clinical utility is often restricted by dose-limiting anemia.21–23 Responses to ruxolitinib in patients with wild-type JAK are observed, reflecting the underlying activation of the JAK-STAT pathway occurring in the pathogenesis of myeloproliferative neoplasms. Novel agents and combinations are under active investigation, particularly agents that offer improvement in erythropoiesis.24 Multiple studies have investigated the therapeutic potential of IMiDs, such as thalidomide and its subsequent analogs, lenalidomide and pomalidomide, in the treatment of myelofibrosis. In this review, we focus on the history of IMiDs and their clinical application in myelofibrosis, with particular attention to the clinical trials that have evaluated pomalidomide in the treatment of myelofibrosis.
Thalidomide: the initial immunomodulatory agent
Thalidomide, the first of the IMiDs, was first introduced in the late 1950s for use as a sedative hypnotic agent.25 However, its use was halted after the discovery of secondary congenital malformations associated with ingestion in pregnant women.26 IMiDs as a class, and thalidomide in particular, were stigmatized and thought to be of little medicinal value until the 1990s, when interest in thalidomide was renewed on the basis of it being an antitumor agent. Thalidomide was found to be an inhibitor of angiogenesis, a pathogenic mechanism known to be important in many malignancies.27 In 1999, thalidomide was found to be active against advanced multiple myeloma, inducing marked and durable responses, including in patients who relapsed after chemotherapy.28 Interestingly, a myeloproliferative-like reaction was observed in patients receiving thalidomide.29
Despite the myeloproliferative reaction observed in some patients treated with thalidomide, the antiangiogenic mechanism implicated in thalidomide therapy resulted in continued optimism for use of this agent in the treatment of myelofibrosis. In 2001, thalidomide was administered to 21 patients with myelofibrosis who were not responsive to standard treatment. Significant improvements in associated anemia and thrombocytopenia were observed with standard doses of thalidomide (>100 mg/day). Despite this, treatment was burdened by a high rate of side effects, which prevented prolonged treatment.30 Subsequently, multiple other small trials observed a response in patients with myelofibrosis who received thalidomide.31–33 In 2003, a prospective Phase II trial of the combination of low-dose thalidomide and prednisone investigated the tolerability and efficacy of a reduced dose of thalidomide (50 mg/day) in the treatment of myelofibrosis.34 An objective clinical response was demonstrated with regard to anemia in the majority (62%) of treated patients, and spleen size decreased in 4/21 patients (19%). Low-dose thalidomide and prednisone therapy was generally tolerable, with 95% of patients enrolled being able to complete 3 months of treatment, indicating that this combination was a promising new regimen for treatment of cytopenias associated with myelofibrosis.34 Later studies corroborated these findings, and confirmed the efficacy of single-agent, low-dose thalidomide; however, low-dose thalidomide was not effective in all patients and responses were often not sustainable. Additionally, although tolerable at reduced doses, thalidomide was associated with a heavy side effect profile, including somnolence, cytopenias, and neuropathy.35–42
Lenalidomide: a potent thalidomide analog
Given the significant side effect profile of thalidomide, pharmacological analogs were developed in an effort to obtain therapeutics with enhanced efficacy and decreased toxicity.43 Thalidomide analogs were found to be associated with a more potent direct antitumor and antiangiogenic effect in vivo.44 Much of the momentum for investigation of lenalidomide, one of the potent thalidomide analogs, was for use in patients with multiple myeloma.45 Interestingly, the biochemical mechanism for antitumor activity with thalidomide analogs is currently unknown. However, thalidomide was recently shown to inhibit cereblon ubiquitin ligase, subsequently altering the ikaros family zinc finger proteins 1 and 3 (IKZF1 and IKZF3), which are necessary and sufficient for the therapeutic effect of lenalidomide in myeloma cell lines.46 Clinical trials in multiple myeloma found lenolidamide to be efficacious and tolerable. The use of lenolidamide continues to be a mainstay of treatment for those afflicted with multiple myeloma.47
In 2006, lenalidomide was tested in myelofibrosis. In that year, two Phase II studies were conducted in a total of 68 patients with myelofibrosis.48 Response rates were 22% for anemia, 33% for splenomegaly, and 50% for thrombocytopenia. Additional treatment effects included resolution of leukoerythroblastosis in four patients, a decrease in medullary fibrosis and angiogenesis in two patients, and del (5) cytogenetic remission accompanied by reduction in JAK2V617F allelic burden in one patient. Grade 3 or greater thrombocytopenia (19%) and neutropenia (31%) were observed. Later, other cases of cytogenetic remissions of myelofibrosis with del (5) and JAK2V617F were discovered after treatment with lenalidomide.49 In 2009, a Phase II study evaluated the efficacy of a combination of lenalidomide and prednisone.50 In this study, 40 patients with myelofibrosis were treated and overall response rates were 30% for anemia and 42% for splenomegaly. Ten patients had a reduction in reticulin fibrosis and eight patients experienced a reduction in their baseline mutant allele burden. Grade 3 or higher toxicity occurred, including neutropenia (58%), anemia (42%), and thrombocytopenia (13%), but was generally well tolerated. A high proportion of patients lost their response after discontinuation of lenalidomide. Although lenalidomide offered promising results, improved tolerability and durability was desired in the treatment of myelofibrosis. Further second-generation IMiDs were created, in the continued hope for better toxicity profiles and enhanced erythropoietic activity.
Pomalidomide: a potentially more tolerable immunomodulatory agent
While both thalidomide and lenalidomide offered improvements in anemia and splenomegaly for patients with myelofibrosis, their therapeutic application was limited by rates of neuropathy and myelosuppression. Pomalidomide, another potent second-generation IMiD, which had shown activity in multiple myeloma, offered new hope for achieving a better toxicity profile and improved erythropoietic activity in myelofibrosis.51,52 Here we discuss the major clinical trial contributions establishing the role of pomalidomide in the treatment of myelofibrosis (see Table 1).
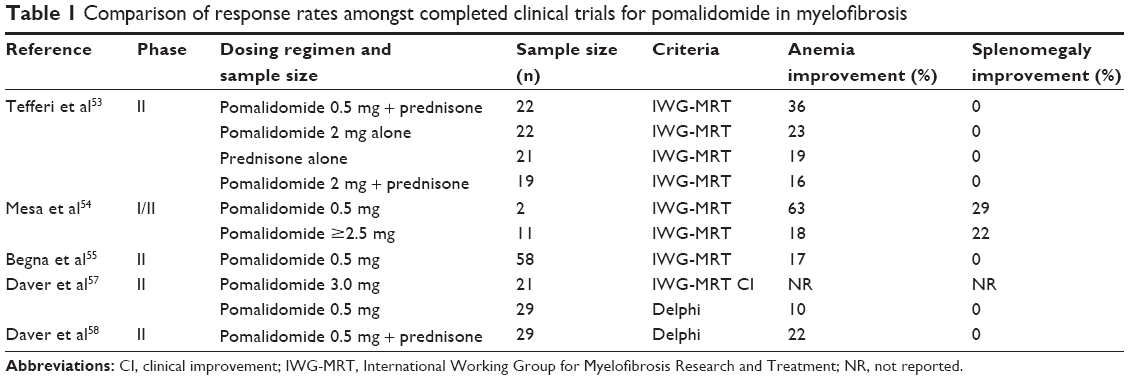 | Table 1 Comparison of response rates amongst completed clinical trials for pomalidomide in myelofibrosis |
In 2009, Tefferi et al reported on a randomized, multicenter, double-blind, adaptive-design Phase II study with four treatment arms, including pomalidomide 2 mg/day plus placebo, pomalidomide 2 mg/day plus prednisone, pomalidomide 0.5 mg/day plus prednisone, and prednisone plus placebo.53 In this study, 84 patients with myelofibrosis-associated anemia were randomly assigned to one of the four treatment arms. The treatment arms were well balanced in terms of patient characteristics, including age, median disease duration, and transfusion dependence. More than 70% of patients in each treatment group were transfusion-dependent. Pomalidomide 2 mg or 0.5 mg (or placebo) was administered daily for up to twelve 28-day treatment cycles, with the option to continue treatment beyond that in the presence of a treatment response and in the absence of unacceptable toxicity. Prednisone or placebo was given in combination with pomalidomide or placebo in a tapering dose schedule during the first three cycles only, with a starting dose of 30 mg/day in the first cycle, 15 mg/day in the second cycle, and 15 mg every other day in the third cycle. Pomalidomide (or placebo) dose reduction by 50% was allowed in the event of adverse events that warranted such action. Dose escalation was not allowed. After a median treatment period of 4.6 (range 0.3–18) months for all patients and 10 (range 6–17.7) months for patients still on study, the response rates were 23% for pomalidomide 2 mg/day plus placebo, 16% for pomalidomide 2 mg/day plus prednisone, 36% for pomalidomide 0.5 mg/day plus prednisone, and 19% prednisone plus placebo; the corresponding anemia response rates per International Working Group for Myelofibrosis Research and Treatment (IWG-MRT) criteria for those who received at least three cycles of treatment (n=62) were 38%, 23%, 40%, and 25%, respectively. The median response duration was 6.5 (range 2.3–16.9) months. No improvement in splenomegaly was observed. Univariate analysis did not show a correlation between response and Lille score, myelofibrosis subtype, platelet count, red blood cell (RBC) transfusion dependence at baseline, age, sex, or presence of JAK2V617F or abnormal cytogenetics. A leukocyte count higher than 10×109/L or a palpable spleen size ≥10 cm correlated with lower response rate; however, only leukocytosis remained significant in multivariable analysis. Grade 3 or higher adverse events included neutropenia (8%), thrombocytopenia (11%), pneumonia/sepsis (11%), and venous thrombosis (4%). This study identified pomalidomide at a dose level of 0.5–2 mg/day with or without a short course of concurrent prednisone to be well tolerated and efficacious in the treatment of myelofibrosis, with the suggestion that low-dose pomalidomide (0.5 mg/day) with prednisone therapy might be as effective and less toxic than higher dosage strategies. Anemia response rates were observed to be as high at 40% in a population that was highly transfusion-dependent, while by comparison prior single-agent thalidomide or lenalidomide studies reported an anemia response rate of 20% or 22%, respectively. The drug was noted to exert minimal myelosuppression when compared with older IMiDs. This study established pomalidomide firmly within the therapeutic armamentarium for myelofibrosis, but further studies were needed to validate the results, assess the impact of pomalidomide on survival, leukemic transformation, bone marrow histology, cytogenetics, and JAK2V617F allelic burden.
In 2010, Mesa et al performed a dose-escalation trial to determine whether higher doses of pomalidomide were effective and well tolerated.54 The trial was a classic 3×3 design that included patients with myelofibrosis and hemoglobin less than 8.0 g/dL or requiring RBC transfusions and/or symptomatic splenomegaly. Pomalidomide was administered at a starting dose of 2.5 mg/day for 21 days on a 28-day cycle. The dose was increased by 0.5 mg/day if no dose-limiting toxicity was observed, including ≥ grade 4 hematologic, ≥ grade 3 febrile neutropenia, or ≥ grade 3 non-hematological toxicity in cycle 1. Cohorts were enrolled until the maximum tolerated dose was determined, as defined by the dose level prior to that resulting in dose-limiting toxicity in more than one in six patients. In the Phase II portion of the study, subjects were treated at the maximum tolerated dose. Nineteen patients with myelofibrosis were enrolled into the trial, with most being RBC transfusion-dependent (58%) and having palpable splenomegaly (84%) and IPSS intermediate-2 risk or greater (78%). The median duration of disease prior to enrollment was 24 (range 1–173) months. Eleven patients received pomalidomide at a dose of >2.5 mg/day, with an anemia response per IWG-MRT criteria in 18% and spleen responses in 22%. Reversal of myelofibrosis-associated thrombocytopenia was noted in some subjects. Responses occurred after a median of 4 (range 2–9) months. The maximum tolerated dose was 3.0 mg/day given on 21 of 28 consecutive days, with myelosuppression being the dose-limiting factor. Most responses occurred after reduction of the dose to 0.5 mg/day, suggesting that higher doses are associated with increasing myelosuppression without increasing efficacy. Non-hematological toxicity was uncommon at any dose, with grade 3 fatigue at 3.0 mg/day observed in one subject. This study reiterated the results of the initial study performed by Teferri et al suggesting that low-dose pomalidomide is well tolerated and efficacious in the treatment of anemia associated with myelofibrosis.
A Phase II clinical trial was conducted in 2011 by Begna et al who evaluated the efficacy of low-dose pomalidomide and predictors of response.55 These investigators enrolled 58 patients with myelofibrosis, all in the high or intermediate-2 risk category, the majority (79%) of whom were RBC transfusion-dependent. Pomalidomide 0.5 mg was administered daily for 28 days to represent one treatment cycle. Dose escalations were allowed in the absence of drug side effects. A total of 49 (84%) patients completed at least three cycles of therapy. Ten patients (17%) achieved an anemia response per IWG-MRT criteria, 90% of whom became RBC transfusion-independent. The median time to response was 2.3 (range 1–4.6) months and the median duration of response was 2.3 (range 5.5–12.8) months. No correlations between anemia response and age, sex, leukocyte count, platelet count, cytogenetic risk, myelofibrosis subtype, or DIPSS were observed. However, anemia responses were significantly associated with the presence of JAK2V617F mutation (P=0.03) and the absence of marked splenomegaly (P=0.04). Pomalidomide therapy also resulted in a >50% increase in the platelet count for 14 (58%) of 24 patients with a baseline platelet count of ≤100×109. Amongst platelet responders, the median platelet count increased from 82×109 (range 32–100) to 160×109 (range 61–332). Grade 3 or 4 adverse events were infrequent (7%), with grade 3/4 neutropenia or thrombocytopenia occurring in 0% and 2% of patients, respectively. No patient discontinued therapy secondary to adverse events. Begna et al demonstrated the safety and therapeutic value of single-agent, low-dose pomalidomide in the treatment of anemia associated with JAK2V617F positive myelofibrosis, particularly in the absence of splenomegaly.
Later in 2012, Begna et al performed another analysis looking at the long-term outcomes of pomalidomide therapy in MF.56 In that analysis, 94 patients in two consecutive clinical trials had an overall anemia response rate of 27%, which increased to 53% in JAK2V617F-positive patients with <10 cm palpable splenomegaly and <5% circulating blasts. The response rate was 0% in mutation-negative patients with either >10 cm splenomegaly or >5% circulating blasts (P=0.0001). The median duration of anemia response was 16 (range 1–32) months. Only one patient (1%) met the IWG-MRT criteria for a spleen response. Of 34 patients with a baseline platelet count of <100×109, 20 (59%) experienced a >50% increase in platelet count, which was predicted by the presence or absence of an anemia response (P=0.03). The majority of patients (89%) had discontinued treatment by 2 years, while eight patients (9%) remained on treatment for a median duration of 34 (range 22–51) months. Interestingly, grade 1 sensory neuropathy developed in four (13%) of 30 patients treated for 12 months or more at a dosage of ≥2 mg/day. To evaluate survival, a comparison patient group with population characteristics matching those of the clinical trial participants were selected, including similar DIPSS risk profiles and hematological study eligibility criteria. Risk-adjusted survival was similar between patients with myelofibrosis who were (n=72) and were not (n=471) exposed to pomalidomide. This analysis underscored the palliative nature of pomalidomide therapy, and highlighted the potential for development of sensory neuropathy with long-term use.
While the Phase I/II study by Mesa et al was underway, Daver et al initiated another Phase II study of single-agent pomalidomide in patients with myelofibrosis and significant anemia.57 This study was undertaken in two parts, one evaluating the toxicity and efficacy of pomalidomide at a dose of 3.0 mg/day at the 21 days on/7 days off schedule, and the other at a dose of pomalidomide 0.5 mg/day continuously. Response was per IWG-MRT criteria for anemia response however, unlike prior clinical trials of pomalidomide in myelofibrosis, the attainment of transfusion independency was assessed per Delphi consensus criteria. All 21 patients enrolled to receive single-agent pomalidomide at a dose of 3.0 mg/day were intermediate-2 DIPSS or higher. Over a median follow-up of 3 (range 0.4–5.5) months, 12 patients discontinued therapy because of drug-related toxicity (n=6, 29%), comorbidities (n=1, 10%), progression to acute myeloid leukemia (n=1, 5%), or patient preference (n=3, 14%). Due to poor tolerance and a high early dropout rate, further enrollment was suspended. Twenty-nine patients were enrolled to receive 0.5 mg/day continuously. Mean hemoglobin at the time of study was 9.0 g/dL and ten patients were RBC transfusion-dependent per Delphi criteria. The median follow-up was 23.6 (range 1.2–28.5) months. Three of the 29 patients (10%) met the response criteria for clinical improvement in hemoglobin per IWG-MRT criteria, none of whom were transfusion-dependent at initiation of therapy. The median time to response was 1.6 (range 3.8–19.5) months. Of the ten patients who were RBC transfusion-dependent at the time of enrollment, two (20%) became transfusion-independent. Univariate analysis did not correlate response with age, hemoglobin, leukocyte count, platelet count, circulating blasts, presence of JAK2V617F mutation, presence of constitutional symptoms, prior therapy, transfusion dependence, splenomegaly, risk score, cytogenetics, or subtype of myelofibrosis. No responses were observed in splenomegaly. One patient was observed to have a 100% increase in platelet count. No significant change in JAK2V617F allelic burden occurred in patients on therapy. Treatment-related toxicities in patients who received pomalidomide 0.5 mg/day included grade 1 neuropathy in one patient and grade 3 neutropenia in one patient. The results of this study are in concordance with prior studies showing that low-dose pomalidomide is well tolerated and has an erythropoietic effect in patients with myelofibrosis. However, although pomalidomide may represent an effective and well tolerated regimen for selected patients with myelofibrosis and anemia, overall response rates remained low. Combination therapeutic techniques using pomalidomide with other agents that could potential improve response rates as well as symptomatic splenomegaly became an active area of investigation.
Daver et al evaluated combination therapy with pomalidomide and prednisone for the treatment of myelofibrosis in a Phase II trial in 2014.58 This study evaluated 29 patients, 18 of whom were transfusion-dependent, who received pomalidomide 0.5 mg daily in continuous 28-day cycles, with prednisone administered on a taper schedule for three cycles. Prednisone was administered at a dose of 30 mg/day for the first cycle, 15 mg/day for the second cycle, and 15 mg every other day for the third cycle. Pomalidomide was continued for at least 6 months if tolerated. Eighteen patients (62%) were transfusion-dependent at baseline, and four (22%) achieved transfusion independence as defined by Delphi consensus criteria. The median time to transfusion independence was 92 (range 21–130) days. Of the 29 patients enrolled, five remained on study at the time of response analysis. Two patients achieved a documented response, including clinical improvement in spleen size per IWG-MRT criteria in one patient and transfusion independence per Delphi consensus criteria in the other patient. The remaining 24 patients discontinued therapy due to lack of response (n=18), progressive disease (n=2), loss of response (n=1), grade 3 toxicity (n=1), and patient preference (n=2). The most common toxicities involved grade 1 gastrointestinal disturbance in five patients and grade 3/4 fatigue in one patient.
A Phase III study of pomalidomide in myeloproliferative neoplasm-associated myelofibrosis with RBC transfusion dependence was recently completed.59 In this study, 252 patients with myelofibrosis were randomly assigned to continuous treatment with pomalidomide 0.5 mg/day or placebo. Interestingly, anemia response rates in the cohorts were similar (16% per Delphi criteria), as were response durations. In contrast, platelet response rates were significantly different between cohorts (22% for pomalidomide versus 0% for placebo). The authors concluded that responses to pomalidomide and placebo differed, but were not distinguished by Delphi response criteria, and additional research designs are needed to study the efficacy of pomalidomide in myelofibrosis.
In general, pomalidomide and other IMiDs offer therapeutic value in improving erythropoiesis, with differing side effect profiles and spleen responses when applied at a low dose with concurrent steroid therapy (Table 2). Pomalidomide has emerged as the IMiD with the least treatment-related toxicity; however, no true analysis comparing IMiD agents has been performed, so data comparing IMiD medications should be interpreted with caution.
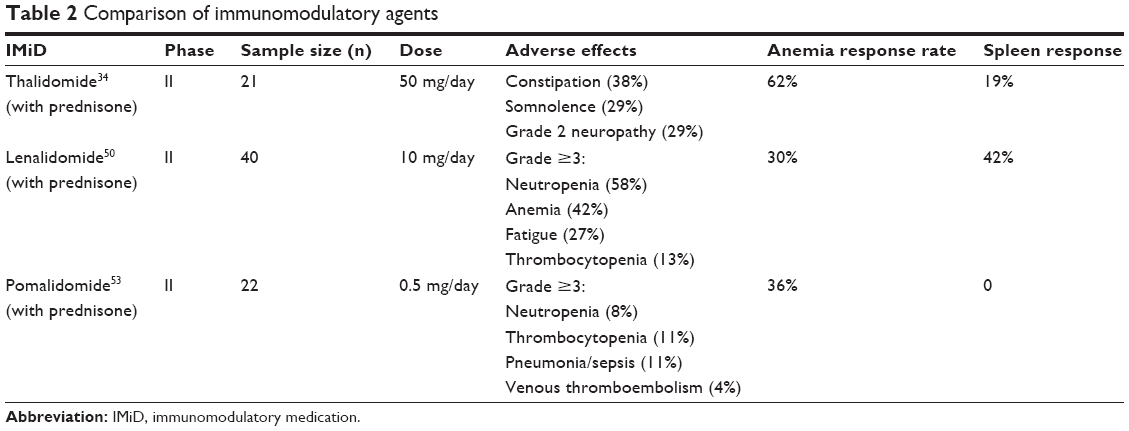 | Table 2 Comparison of immunomodulatory agents |
Despite the multiple clinical trials completed to date investigating pomalidomide in the treatment of myelofibrosis (Table 1), no clear consensus on efficacy has emerged from the data. This can be attributed to differences in study design, including dosing strategies, sample size, and response criteria utilized. In particular, Phase III analysis results were underwhelming and less impressive than prior Phase II analysis. The difference may be secondary to the differential definitions of transfusion dependence, being more stringent in Phase III analysis and including the most symptomatic of patients, potentially hiding the benefit. Additionally, the eligibility was stricter in Phase III analysis, potentially missing benefit of pomalidomide for patients with milder anemia.
Moving forward, the rational next step in applying this important therapeutic agent to the myelofibrosis patient population is with combination therapy. Investigation of combination therapy with the JAK inhibitor ruxolitinib and pomalidomide is underway, with initial feasibility and toxicity data available (ClinicalTrials.gov NCT01644110).60 At the time of this interim analysis, six patients with myelofibrosis were enrolled. The median time on treatment was 4.3 (range 1–9.4) months. Ruxolitinib was maintained at 10 mg twice daily in five patients, and escalated to 20 mg twice daily in one patient until cycle 6 and administered concomitantly with pomalidomide 0.5 mg daily. No grade 4 toxicity was observed. Notably, anemia was worsened in five patients and one patient developed neuropathy. In general, the combination was well tolerated and the observed toxicities were within the expected range according to the reported toxicity profiles for the respective single agents. The trial has continued recruitment and we await efficacy data, with the primary study endpoint being response rate after 12 treatment cycles according to IWG-MRT criteria.
Conclusion
A new era of therapy has evolved in the treatment of myelofibrosis. Since the discovery of the JAK2V617F mutation in 2005 and the subsequent development of therapeutic JAK inhibitors, treatment for primary, post essential thrombocytosis, and post polycythemic myelofibrosis has been revolutionized. The advent of ruxolitinib has led to improvements in symptom burden, splenomegaly, and even overall survival.21,22 Despite the important contribution of ruxolitinib to patients afflicted with myelofibrosis, JAK inhibitor therapy remains undermined by significant treatment-related anemia and thrombocytopenia, which is often dose-limiting.61 The newest of the immunomodulatory agents, pomalidomide, has emerged as an asset in the treatment armamentarium for anemia associated with myelofibrosis. Its use is associated with tolerable toxicity; however, modest response rates and little effect on splenomegaly are observed. The future of treatment for myelofibrosis may lie in combination-based approaches wherein pomalidomide may play an integral role in stimulating erythropoiesis. Currently, investigation is underway evaluating pomalidomide in combination with ruxolitinib for the treatment of myelofibrosis with associated anemia. More novel combination therapy trials with pomalidomide are likely to be on the horizon for patients afflicted with myelofibrosis and anemia.
Author contributions
Both authors made substantial contributions to conception and design; took part in drafting the article or revising it critically for important intellectual content; gave final approval of the version to be published.
Disclosure
RAM has received research support from Incyte, Gilead, CTI, Celgene, Genentech, and Promedior, and has received honoraria for providing consultancy services to Novartis. The authors report no other conflicts of interest in this work.
References
Tefferi A. Pathogenesis of myelofibrosis with myeloid metaplasia. J Clin Oncol. 2005;23(33):8520–8530. | ||
Schmitt A, Jouault H, Guichard J, Wendling F, Drouin A, Cramer EM. Pathologic interaction between megakaryocytes and polymorphonuclear leukocytes in myelofibrosis. Blood. 2000;96(4):1342–1347. | ||
Xu M, Bruno E, Chao J, et al. Constitutive mobilization of CD34+ cells into the peripheral blood in idiopathic myelofibrosis may be due to the action of a number of proteases. Blood. 2005;105(11):4508–4515. | ||
Tefferi A. Primary myelofibrosis: 2014 update on diagnosis, risk-stratification, and management. Am J Hematol. 2014;89(9):915–925. | ||
Mesa RA, Niblack J, Wadleigh M, et al. The burden of fatigue and quality of life in myeloproliferative disorders (MPDs): an international Internet-based survey of 1179 MPD patients. Cancer. 2007;109(1):68–76. | ||
Birgegard G. Does anything work for anaemia in myelofibrosis? Best Pract Res Clin Haematol. 2014;27(2):175–185. | ||
Cervantes F, Dupriez B, Pereira A, et al. New prognostic scoring system for primary myelofibrosis based on a study of the International Working Group for Myelofibrosis Research and Treatment. Blood. 2009;113(13):2895–2901. | ||
Kroger N, Holler E, Kobbe G, et al. Allogeneic stem cell transplantation after reduced-intensity conditioning in patients with myelofibrosis: a prospective, multicenter study of the Chronic Leukemia Working Party of the European Group for Blood and Marrow Transplantation. Blood. 2009;114(26):5264–5270. | ||
Tefferi A, Silverstein MN. Recombinant human erythropoietin therapy in patients with myelofibrosis with myeloid metaplasia. Br J Haematol. 1994;86(4):893. | ||
Tefferi A, Elliot MA, Yoon SY, et al. Clinical and bone marrow effects of interferon alfa therapy in myelofibrosis with myeloid metaplasia. Blood. 2001;97(6):1896. | ||
Hasselbalch HC, Clausen NT, Jensen BA. Successful treatment of anemia in idiopathic myelofibrosis with recombinant human erythropoietin. Am J Hematol. 2002;70(2):92–99. | ||
Martinez-Trillos A, Gaya A, Maffioli M, et al. Efficacy and tolerability of hydroxyurea in the treatment of the hyperproliferative manifestations of myelofibrosis: results in 40 patients. Ann Hematol. 2010;89(12):1233–1237. | ||
Cervantes F, Mesa R, Barosi G. New and old treatment modalities in primary myelofibrosis. Cancer J. 2007;13(6):377–383. | ||
Singh PP, Timucin T, Nagorney DM, Tefferi A. Splenectomy for massive splenomegaly associated with myelofibrosis: outcomes from 63 patients at Mayo Clinic. ASH Annual Meeting Abstracts. 2012;120(21):2848. | ||
Pardanani A, Brown P, Neben-Wittich M, Tobin R, Tefferi A. Effective management of accelerated phase myelofibrosis with low-dose splenic radiotherapy. Am J Hematol. 2010;85(9):715–716. | ||
Mesa RA. How I treat symptomatic splenomegaly in patients with myelofibrosis. Blood. 2009;113(22):5394–5400. | ||
Barosi G, Elliott M, Canepa L, et al. Thalidomide in myelofibrosis with myeloid metaplasia: a pooled-analysis of individual patient data from five studies. Leuk Lymphoma. 2002;43(12):2301–2307. | ||
Kiladjian JJ, Cassinat B, Chevret S, et al. Pegylated interferon-alfa-2a induces complete hematologic and molecular responses with low toxicity in polycythemia vera. Blood. 2008;112(8):3065–3072. | ||
Levine RL, Wadleigh M, Cools J, et al. Activating mutation in the tyrosine kinase JAK2 in polycythemia vera, essential thrombocythemia, and myeloid metaplasia with myelofibrosis. Cancer Cell. 2005;7(4):387–397. | ||
James C, Ugo V, Le Couedic JP, et al. A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature. 2005;434(7037):1144–1148. | ||
Santos FPS, Verstovsek S. Breakthroughs in myeloproliferative neoplasms. Hematology. 2012;17 Suppl 1:S55–S58. | ||
Harrison C, Kiladjian JJ, Al-Ali HK, et al. JAK inhibition with ruxolitinib versus best available therapy for myelofibrosis. N Engl J Med. 2012;366(9):787–798. | ||
Cervantes F, Vannucchi AM, Kiladjian JJ, et al. Three-year efficacy, safety, and survival findings from COMFORT-II, a phase 3 study comparing ruxolitinib with best available therapy for myelofibrosis. Blood. 2013;122(25):4047–4053. | ||
Gowin K, Mesa R. Emerging therapies for the treatment of chronic Philadelphia chromosome-negative myeloproliferative neoplasm-associated myelofibrosis. Expert Opin Investig Drugs. 2013;22(12):1603–1611. | ||
Azima H, Arthurs D. Control study of thalidomide (Kevadon), a new hypnotic agent. Am J Psychiatry. 1961;118:554–555. | ||
Somers GS. Thalidomide and congenital abnormalities. Lancet. 1962;1(7235):912–913. | ||
D’Amato RJ, Loughnan MS, Flynn E, Folkman J. Thalidomide is an inhibitor of angiogenesis. Proc Natl Acad Sci U S A. 1994;91(9):4082–4085. | ||
Singhal S, Mehta J, Desikan R, et al. Antitumor activity of thalidomide in refractory multiple myeloma. N Engl J Med. 1999;341(21):1565–1571. | ||
Tefferi A. Serious myeloproliferative reactions associated with the use of thalidomide in myelofibrosis with myeloid metaplasia. Blood. 2000;96(12):4007. | ||
Barosi G, Grossi A, Comotti B, Musto P, Gamba G, Marchetti M. Safety and efficacy of thalidomide in patients with myelofibrosis with myeloid metaplasia. Br J Haematol. 2001;114(1):78–83. | ||
Canepa L, Ballerini F, Varaldo R, et al. Thalidomide in agnogenic and secondary myelofibrosis. Br J Haematol. 2001;115(2):313–315. | ||
Pozzato G, Zorat F, Nascimben F, Comar C, Kikic F, Festini G. Thalidomide therapy in compensated and decompensated myelofibrosis with myeloid metaplasia. Haematologica. 2001;86(7):772–773. | ||
Elliott MA, Mesa RA, Li CY, et al. Thalidomide treatment in myelofibrosis with myeloid metaplasia. Br J Haematol. 2002;117(2):288–296. | ||
Mesa RA, Steensma DP, Pardanani A, et al. A phase 2 trial of combination low-dose thalidomide and prednisone for the treatment of myelofibrosis with myeloid metaplasia. Blood. 2003;101(7):2534–2541. | ||
Marchetti M, Barosi G, Balestri F, et al. Low-dose thalidomide ameliorates cytopenias and splenomegaly in myelofibrosis with myeloid metaplasia: a phase II trial. J Clin Oncol. 2004;22(3):424–431. | ||
Mesa RA, Elliott MA, Schroeder G, Tefferi A. Durable responses to thalidomide-based drug therapy for myelofibrosis with myeloid metaplasia. Mayo Clin Proc. 2004;79(7):883–889. | ||
Silver RT. Myelofibrosis: thalidomide finds a new disease. Mayo Clin Proc. 2004;79(7):857–858. | ||
Strupp C, Germing U, Scherer A, et al. Thalidomide for the treatment of idiopathic myelofibrosis. Eur J Haematol. 2004;72(1):52–57. | ||
Abgrall J-F, Guibaud I, Bastie J-N, et al. Thalidomide versus placebo in myeloid metaplasia with myelofibrosis: a prospective, randomized, double-blind, multicenter study. Haematologica. 2006;91(8):1027–1032. | ||
Thomas DA, Giles FJ, Albitar M, et al. Thalidomide therapy for myelofibrosis with myeloid metaplasia. Cancer. 2006;106(9):1974–1984. | ||
Berrebi A, Feldberg E, Spivak I, Shvidel L. Mini-dose of thalidomide for treatment of primary myelofibrosis. Report of a case with complete reversal of bone marrow fibrosis and splenomegaly. Haematologica. 2007;92(2):e15–e16. | ||
Weinkove R, Reilly JT, McMullin MF, Curtin NJ, Radia D, Harrison CN. Low-dose thalidomide in myelofibrosis. Haematologica. 2008;93(7):1100–1101. | ||
Koh KR, Janz M, Mapara MY, et al. Immunomodulatory derivative of thalidomide (IMiD CC-4047) induces a shift in lineage commitment by suppressing erythropoiesis and promoting myelopoiesis. Blood. 2005;105(10):3833–3840. | ||
Lentzsch S, LeBlanc R, Podar K, et al. Immunomodulatory analogs of thalidomide inhibit growth of Hs Sultan cells and angiogenesis in vivo. Leukemia. 2003;17(1):41–44. | ||
Chamberlain PP, Lopez-Girona A, Miller K, et al. Structure of the human Cereblon-DDB1-lenalidomide complex reveals basis for responsiveness to thalidomide analogs. Nat Struct Mol Biol. 2014;21(9):803–809. | ||
Anderson KC, Prince HM. Lenalidomide and thalidomide: an evolving paradigm for the management of multiple myeloma. Semin Hematol. 2005;42(4 Suppl 4):S1–S2. | ||
Blade J, Rosinol L, Fernandez de Larrea C. How I treat relapsed myeloma. Blood. January 13, 2015. [Epub ahead of print]. | ||
Tefferi A, Cortes J, Verstovsek S, et al. Lenalidomide therapy in myelofibrosis with myeloid metaplasia. Blood. 2006;108(4):1158–1164. | ||
Tefferi A, Lasho TL, Mesa RA, Pardanani A, Ketterling RP, Hanson CA. Lenalidomide therapy in del(5)(q31)-associated myelofibrosis: cytogenetic and JAK2V617F molecular remissions. Leukemia. 2007;21(8):1827–1828. | ||
Quintas-Cardama A, Kantarjian HM, Manshouri T, et al. Lenalidomide plus prednisone results in durable clinical, histopathologic, and molecular responses in patients with myelofibrosis. J Clin Oncol. 2009;27(28):4760–4766. | ||
Streetly MJ, Gyertson K, Daniel Y, Zeldis JB, Kazmi M, Schey SA. Alternate day pomalidomide retains anti-myeloma effect with reduced adverse events and evidence of in vivo immunomodulation. Br J Haematol. 2008;141(1):41–51. | ||
Lacy MQ, Rajkumar SV. Pomalidomide: a new IMiD with remarkable activity in both multiple myeloma and myelofibrosis. Am J Hematol. 2010;85(2):95–96. | ||
Tefferi A, Verstovsek S, Barosi G, et al. Pomalidomide is active in the treatment of anemia associated with myelofibrosis. J Clin Oncol. 2009;27(27):4563–4569. | ||
Mesa RA, Pardanani AD, Hussein K, et al. Phase 1/-2 study of pomalidomide in myelofibrosis. Am J Hematol. 2010;85(2):129–130. | ||
Begna KH, Mesa RA, Pardanani A, et al. A phase-2 trial of low-dose pomalidomide in myelofibrosis. Leukemia. 2011;25(2):301–304. | ||
Begna KH, Pardanani A, Mesa R, et al. Long-term outcome of pomalidomide therapy in myelofibrosis. Am J Hematol. 2012;87(1):66–68. | ||
Daver N, Shastri A, Kadia T, et al. Modest activity of pomalidomide in patients with myelofibrosis and significant anemia. Leuk Res. 2013;37(11):1440–1444. | ||
Daver N, Shastri A, Kadia T, et al. Phase II study of pomalidomide in combination with prednisone in patients with myelofibrosis and significant anemia. Leuk Res. 2014;38(9):1126–1129. | ||
Tefferi A, Passamonti F, Barbui G, et al. Phase 3 study of pomalidomide in myeloproliferative neoplasm (MPN)-associated myelofibrosis with RBC-transfusion-dependence. Presented at the American Society of Hematology 55th Annual Meeting, December 7–10, 2013, New Orleans, LA, USA. | ||
Stefelmann F, Reiter A, et al. A multicenter Phase-Ib/II study of ruxolitinib/pomalidomide combination therapy in patients with primary and secondary myelofibrosis: safety data from the MPNSG-0212 trial. Presented at the American Society of Hematology 56th Annual Meeting, December 6–9, 2014, San Francisco, CA, USA. | ||
Verstovsek S. Ruxolitinib: an oral Janus kinase 1 and Janus kinase 2 inhibitor in the management of myelofibrosis. Postgrad Med. 2013;125(1):128–135. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.