")
Back to Archived Journals » Research and Reports in Endocrine Disorders » Volume 5
Profile of idursulfase for the treatment of Hunter syndrome
Authors Sestito S, Ceravolo F, Grisolia M, Pascale E, Pensabene L, Concolino D
Received 20 March 2015
Accepted for publication 5 May 2015
Published 9 July 2015 Volume 2015:5 Pages 79—90
DOI https://doi.org/10.2147/RRED.S64347
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Professor Mingzhao Xing
Simona Sestito, Ferdinando Ceravolo, Michele Grisolia, Elisa Pascale, Licia Pensabene, Daniela Concolino
Department of Pediatrics, University Magna Graecia of Catanzaro, Catanzaro, Italy
Abstract: Mucopolysaccharidosis type II (MPS II; Hunter syndrome) is a rare X-linked lysosomal storage disorder caused by deficiency of the enzyme iduronate-2-sulfatase (IDS). Enzyme replacement therapy (ERT) with recombinant human IDS, available since 2005, is currently the most appropriate treatment for this progressive, multisystemic, chronic, and life-threatening disease. Efficacy and safety of therapy with idursulfase have been assessed in several clinical trials, and confirmed in many clinical reports. Long-term follow-up of patients receiving ERT has demonstrated the importance of an early onset of treatment with idursulfase, before irreversible pathological changes occur. Intravenously administered idursulfase is not able to cross the blood–brain barrier, so neurological signs and symptoms cannot benefit from ERT, still remaining a major challenge in the treatment of MPS II.
Keywords: MPS II, glycosaminoglycans, enzyme replacement therapy, ERT
Introduction
Mucopolysaccharidosis type II (MPS II; Hunter syndrome; OMIM number 309900), described for the first time by Charles Hunter in 1917,1 is a rare inherited metabolic disorder, caused by deficiency of the lysosomal enzyme iduronate-2-sulfatase (IDS, EC 3.1.6.13), due to a mutation in the gene IDS,2,3 located on chromosome Xq28. IDS is essential for the degradation of the glycosaminoglycans (GAGs) dermatan sulfate (DS) and heparan sulfate, with the deficiency or the absence of its activity leading to progressive accumulation of these GAGs in many cells and tissues. The estimated incidence of the disease is 1.3 in 100,000 male newborns.4 As MPS II is an X-linked recessive condition, it generally affects only males, although few female cases with Hunter syndrome have also been reported.5,6
MPS II is a progressive, multisystemic, chronic, and life-threatening disorder with a wide variability in severity of signs and symptoms, in rate of progression, and in age of onset between patients.7 Although the disease has been traditionally classified into “mild” or “severe”, on the basis of severity of clinical manifestations and the presence or absence of central nervous system (CNS) involvement, MPS II should be considered as a continuum between the two extreme forms of the disease. Patients with MPS II typically appear normal at birth, but because of the progression of the disease, signs and symptoms appear during early childhood,8 usually between 18 months and 4 years of age, depending on the severity of disease.3,7,9 All affected patients experience progressive somatic involvement including facial dysmorphism, short stature due to dysostosis multiplex, reduced joint range of motion and contractures, hepatosplenomegaly, heart involvement (cardiac valve disease and left ventricular hypertrophy), and pulmonary dysfunction (obstructive sleep apnea and restrictive lung disease).3,7,10 In addition, about two-thirds of patients present neurological involvement (severe phenotype), leading to progressive severe mental impairment, progressive neurodegeneration, and death which usually occur during the second decade of life, most often from respiratory and/or cardiac failure.3,7 The patients with attenuated phenotype can also experience some neurological manifestations such as communicating hydrocephalus and carpal tunnel syndrome.7 They usually may live up to the fifth or sixth decade of life or longer,11 presenting with normal intellectual development.7
The limited experiences with hematopoietic stem cell transplantation (HSCT) in patients with Hunter syndrome showed that this approach offered only few clinical benefits and had been associated with a serious risk of morbidity and mortality.12,13 Therefore, HSCT is not at present recommended for patients with MPS II,7 whereas enzyme replacement therapy (ERT) with recombinant IDS (idursulfase, Elaprase®, Shire Human Genetic Therapies, Inc., Cambridge, MA, USA) is currently the most appropriate treatment, used routinely in clinical practice.
In this review of the literature regarding treatment with idursulfase in patients with Hunter syndrome, there was no language nor types of studies restriction in the bibliography search published from August 2006 to January 2015, and the articles evaluated were identified from the Cochrane Central Register of Controlled Trials, PubMed, and EMBASE, using the keywords idursulfase, Hunter syndrome, and MPS II.
Idursulfase
Recombinant IDS (idursulfase) is produced in a continuous human cell line and is a purified form of the lysosomal enzyme IDS. The resulting glycoprotein presents mannose-6-phosphate (M6P) residues on the oligosaccharide chains, and these allow specific binding of IDS to M6P receptors on the cell surface,14 thus enabling the enzyme to enter the cell and to target lysosomes, with subsequent catabolism of accumulated GAGs (Figure 1).10 In addition, purified idursulfase contains complex highly sialylated glycans which lead to a prolongation in the circulating half-life of the enzyme.14
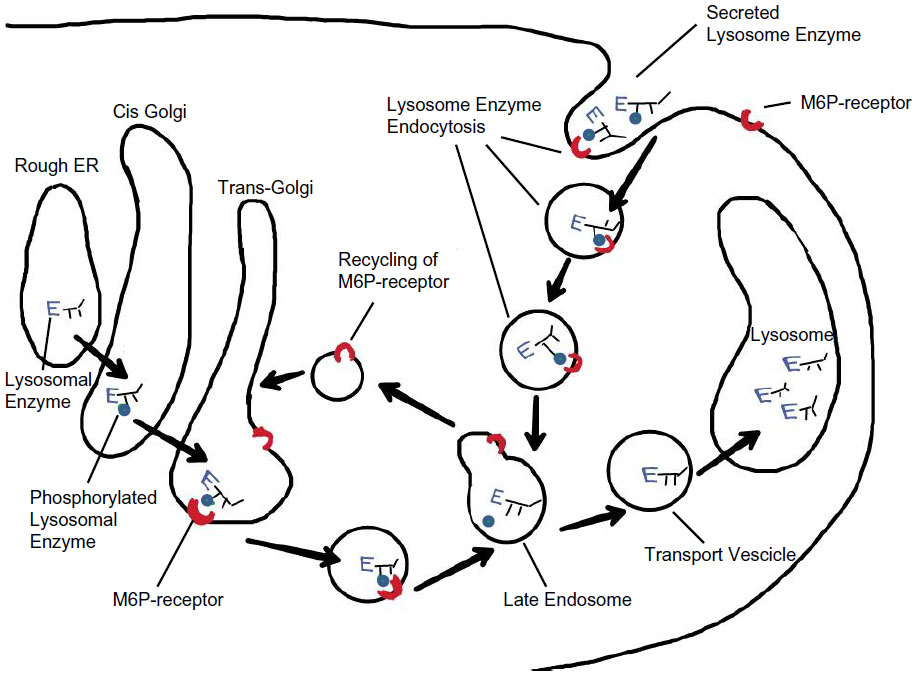 | Figure 1 Lysosomal recombinant iduronate-2-sulfatase transport. |
The pharmacokinetic characteristics of idursulfase were evaluated in several studies in patients with Hunter syndrome. The serum concentration of idursulfase was quantified using an antigen-specific enzyme-linked immunosorbent assay. The pharmacokinetic parameters at the recommended dose regimen (0.5 mg/kg of Elaprase administered weekly as a 3-hour infusion) were determined at week 1 and week 27 in ten patients aged 7.7–27 years. There were no apparent differences in pharmacokinetic parameters between week 1 and week 27. Degradation of idursulfase consists of mechanisms of protein hydrolysis with formation of small peptides or amino acids, so that a possible renal or hepatic impairment does not affect the pharmacokinetics of idursulfase.15
A second recombinant enzyme, idursulfase beta (Hunterase®, Green Cross Corp, Yongin, Korea), has been produced from the Chinese hamster ovary cell line by genetic engineering yielding a glycosylated protein analogous to the native human enzyme.16,17 Its safety and a similar efficacy compared to idursulfase had been demonstrated by a successful clinical trial16 in Korean patients with MPS II older than 6 years, leading to the recent approval of idursulfase beta by the Korean Ministry of Food and Drug Safety. A second clinical trial demonstrated that the safety and efficacy profile of 0.5 mg/kg/week of idursulfase beta infusion in patients younger than 6 years was similar to that previously reported for older Hunter syndrome patients.18
The first clinical trial14 with idursulfase was a randomized, double-blind, placebo-controlled trial conducted in 12 patients with Hunter syndrome in order to assess efficacy and security of treatment with idursulfase in patients with MPS II, and to establish the optimal dose to use in the following Phase II/III pivotal study.19 Another multinational, randomized, double-blind, placebo-controlled trial was performed to evaluate the safety and efficacy of idursulfase 0.5 mg/kg administered weekly compared with placebo. Additionally, the trial evaluated idursulfase 0.5 mg/kg administered every other week (EOW) compared with placebo. The three groups of patients included 96 patients, aged from 5 years to 31 years at baseline, and each of them received a total of 52 infusions of idursulfase, idursulfase alternating weekly with placebo, or placebo.19 Recombinant human IDS (idursulfase, Elaprase®, Shire Human Genetic Therapies, Inc.) was approved for ERT of Hunter syndrome by the US Food and Drug Administration in 2006 and the European Medicines Agency in 2005, and it is today available in the US, European Union, and other countries.
Following registration trials, a long-term (2-year) open-label extension study of weekly infusions of idursulfase was undertaken in all 94 patients who completed the Phase II/III study,20 and since ERT with idursulfase has been approved and commercially distributed, several other clinical studies and many case series have been published in order to demonstrate both safety and efficacy of this therapy for patients with MPS II.
Efficacy
To date, to assess the efficacy of treatment with idursulfase and to monitor the response to ERT, it has been suggested to exclusively evaluate different clinical, laboratory, and instrumental endpoints, listed in Table 1.
 | Table 1 Clinical, laboratory, and instrumental endpoints used to assess efficacy of treatment with idursulfase |
Urinary GAGs excretion
Several evidences confirm the effectiveness of idursulfase in reducing urinary GAGs level. The Phase I/II trial showed a reduction in urinary GAGs within 2 weeks of idursulfase treatment, with GAG levels remaining low for the 48 weeks of the study (P<0.0001).14 Moreover, in the Phase II/III trial, a reduction from baseline in the urinary GAGs excretion was detected in patients treated with either dosing regimens of idursulfase, while no significative changes were reported in patients treated with placebo. After 53 weeks of treatment, urinary GAG levels in the idursulfase groups were significantly different compared to the placebo group (P<0.0001), and the 40.6% of idursulfase-treated patients had normalized urine GAG levels, while no patients in the placebo group experienced this result.19 During the open-label extension study, a progressive decline in urine GAGs level was detected with the mean value decreased to 107±9 μg/mg of creatinine at 16 months, below the upper limit of the normal levels. At month 36, the mean GAG level was even lower, with only 31 out of 94 patients having urine GAG levels above the upper limit of normal, compared to 91 out of 94 patients at baseline.20 Moreover, during a separate 12-month, open-label, clinical study of ten adult Japanese patients with MPS II (Japan Elaprase Treatment [JET] study),21 a rapid reduction in urinary GAGs excretion was detected within the first 3 months of treatment with idursulfase, and urinary GAG levels remained low for the reminder of the study. After 12 months of treatment, a >70% decrease in urinary GAG levels was seen in all nine evaluable patients with normal values experienced by the end of the study.21
Coppa et al22 performed an accurate quantitative and structural characterization of plasmatic and urinary GAGs of a 3-year-old patient affected by the severe form of MPS II, before and during the first 10 months of ERT with idursulfase. Before ERT, DS was characterized as the main (~90%) urinary GAG mainly composed of ~90% of ΔDi4s with minor percentages of monosulfated and disulfated disaccharides, in particular, ΔDi2,4dis. After 10 months of treatment, ~40% of pathological DS was still detected in the urine, showing that ERT, performed by infusion of recombinant IDS at the standard doses, does not totally remove DS from urine (and also plasma) after prolonged treatment, even if it is very effective in eliminating large amounts of this pathological GAG just after the first weeks following the beginning of enzyme infusion.22
Six-minute walk test
The 6-minute walk test (6MWT) consists of measuring the distance (in meters) that the individual is able to go walking in 6 minutes. It is a submaximal exercise test that reflects the integrated function of all systems utilized in day-to-day exercise,23 allowing at the same time an evaluation of cardiac, respiratory, and musculoskeletal functions.
In Phase I/II trial, the 6MWT distance of patients receiving idursulfase increased an average of 48 m after 48 weeks,14 even if, compared with placebo, there was no statistically significant difference (P=0.013).14 In Phase II/III clinical trial, after 53 weeks of treatment, the increase from baseline in the mean (± standard error) 6MWT distance was significantly greater in patients receiving idursulfase weekly compared with those receiving placebo (+44.3±12.3 m and +7.3±9.5 m, respectively; P=0.0131).19 The idursulfase 0.5 mg/kg EOW group also showed an improvement, but that was not significant compared with placebo.19 In the open-label extension study, the mean increase in 6MWT distance seen in weekly treated patients was maintained with statistical significance after 36 months (P<0.01), although absolute values at the end of the period were not reported.20 The largest increase was seen at 20 months after the start of the initial study – a mean increase of 42±10 m from baseline (P<0.01).20 The JET study21 showed that, by the end of the study, the mean 6MWT distance had increased by 54.5±27.0 m.21 A meta-analysis, performed between the JET study21 and the open-label extension study20 about the variable distance covered in the 6MWT, gave a resulting mean improvement of 43.5 m in the distance walked in 6 minutes.24 Similar findings have been reported in several separate clinical reports.25,26
Forced vital capacity
In the Phase I/II trial, effects of idursulfase treatment on pulmonary function have been assessed by spirometry to measure forced expiratory volume in 1 second (FEV1) and forced vital capacity (FVC).14 In this study, nine out of 12 patients had an increase in FVC after 12 months of idursulfase, but the average increase was small and not statistically significant (FVC =1.03±0.34 L at baseline and 1.10±0.13 L at 12 months; P=0.08). FEV1 did not improve after 12 months (0.82±0.28 L at baseline and 0.84±0.30 L at 12 months; P=0.61).14 The primary endpoint of the Phase II/III study was a two-component composite variable combining percentage of predicted FVC (%FVC) and 6MWT.19 There was a statistically significant difference when comparing placebo and idursulfase 0.5 mg/kg weekly group (treatment difference =18.96±6.47; P=0.0049) or biweekly group (treatment difference =12.86±6.17; P=00416), even if there was no statistical significance comparing the two idursulfase schemes (treatment difference =10.84±7.11; P=0.1329).19 When evaluated individually after 53 weeks, %FVC increased by 3.45% (±1.77%) in patients treated weekly, compared to 0.75% (±1.71%) in those of the placebo group, with this difference approaching statistical significance (P=0.065).19 No difference was seen between idursulfase EOW and placebo. Moreover, a mean 220 mL (±50 mL) increase in absolute FVC was seen in patients on weekly idursulfase, compared to 60 mL (±30 mL) in those on placebo (P=0.0011) and 70 mL (±30 mL) in those on idursulfase EOW (P=0.0176) after 53 weeks.19 During the long-term, open-label extension study, a statistically significant increase in %FVC was seen only at month 16 but not at the end of the study, while absolute FVC showed sustained increase throughout the study, with an improvement of 0.31±0.06 L after 3 years of treatment (25.1%±4%) (P<0.05).20 The JET study reported an increase in %FVC of 15% over baseline with an improvement in absolute FVC of 16.3% over the baseline of 1.4 L.21 A meta-analysis performed using the last two studies showed that there was a mean change of 0.229 L (95% confidence interval 0.028–0.429) in absolute FVC and 10.8% in %FVC.24
Liver and spleen volumes
Changes in liver and spleen size, measured by abdominal magnetic resonance imaging, were considered in many studies as efficacy endpoints of treatment with idursulfase. In the Phase I/II study, both liver and spleen volumes were decreased at 24 weeks (P<0.01) and 48 weeks (P<0.001).14 The Phase II/III trial showed an initial reduction in both liver and spleen volumes in treated patients, already after the first 4 months of therapy. After 53 weeks, the reduction in liver volume was significantly greater both in biweekly and in weekly treated patients compared to placebo (−25.3%±1.6% in the idursulfase weekly group, −24%±1.7% in the idursulfase EOW group, −0.8%±1.6% in the placebo group; P<0.0001 for placebo compared with either idursulfase group). Similar finding was observed for splenic volume after 53 weeks (−25.1%±2.4% in the idursulfase weekly group, −19.8%±3.2% in the idursulfase EOW group, and +7.2%±4.2% in the placebo group; P<0.0001 for placebo compared with either idursulfase group).19 Okuyama et al21 described that, after 12 months of treatment with idursulfase, mean liver volume decreased by −33.2%±4.0% and mean spleen volume decreased by −31.0%±5.5%, and both changes were statistically significant (P=0.002).21 Muenzer et al20 observed that liver and spleen volumes were significantly reduced after 4 months of treatment and that these reductions were sustained throughout the study.20
Joint mobility
Muenzer et al14 did not report the effects of idursulfase treatment on joint mobility because there were difficulties in interpretation due to the heterogeneity of patients and measurements.14 During Phase II/III trial,19 aside from a statistically significant gain in range of motion of the elbow in the weekly idursulfase group compared with placebo (P=0.0476), no other significant differences between treatment groups for any joint range of motion have been observed.19 During the open-label extension study,20 progressive, statistically significant, and clinically relevant improvements in joint range of motion were seen only for the shoulder, both in abduction and in flexion–extension movements (P≤0.005 compared with baseline), whereas no important change in mobility was observed for elbow, wrist, digits, hip, knee, or ankle.20 During JET study,21 following 12 months of treatment, several joints showed increased range of motion (shoulder flexion and abduction, knee flexion, hip flexion, and elbow extension), but most of these changes did not achieve statistical significance.21
Height and linear growth velocity
Muenzer et al20 observed that in patients younger than 12 years, after 36 months of ERT, height had increased by a mean of 11.9±1.0 cm (mean height at baseline =120±1.5 cm), while in the group of patients aged 12–18 years, after the same time, height had increased by a mean of 9.8±1.4 cm (mean height at baseline =128±2.2 cm).20 Schulze-Frenking et al27 focused on the effects of ERT on growth in 18 of the patients previously enrolled in the Phase II/III clinical trial19 and on long-term treatment with idursulfase. They observed that patients who started ERT before 10 years of age (n=9) showed an average growth of 14.6±5.5 cm, remaining with weight and height in the normal curve according to the Centers for Disease Control and Prevention,28 while patients who started ERT after 10 years of age (n=9) had short stature at the beginning of ERT (median 130 cm) and grew on average 8.1±1.6 cm after 3 years, highlighting the importance of an early initiation of ERT.27 Data from Hunter Outcome Survey (HOS) confirmed the positive effects of idursulfase on growth in the study population of 133 patients with both phenotypes of Hunter syndrome aged 8–15 years when they started treatment. In this analysis, both age at start of ERT (8–11 years vs 12–15 years) and type of mutation impacted significantly on growth.29 A recent retrospective analysis of the ERT effects on the linear growth of 32 Korean MPS II patients of different ages and phenotypes showed that in patients who started ERT before 6 years of age, the mean increase in height over the 2-year period of ERT was 11.5±5.8 cm. In the group of patients who started ERT between 6 years and 10 years, during 3 years of ERT, the mean increase in height was 9.4±6.1 cm, while in the group aged 10–20 years at the beginning of ERT, the mean increase in height was 9.5±7.6 cm after the same period. Patients belonging to the last two groups showed a tendency toward growth restoration after initiation of ERT, as well as important growth retardation before treatment initiation, thus showing that age at start of ERT (6–10 years vs 10–20 years) has a significant impact on growth.30
Cardiac disease
Effects of idursulfase on cardiac disease were assessed, in the Phase I/II trial, by echocardiography performed in the 12 patients enrolled. Two out of the six patients with left ventricular hypertrophy at baseline reached the normal limits of left ventricular mass index (LVMI), after 12 months, whereas two out of the six patients with normal LVMI at baseline ended up in the hypertrophic category (final LVMI =105 g/m2 and 122 g/m2) after the same period.14 During JET study, cardiac ejection fraction and valve disease remained mostly stable. A mean decrease in LVMI of −12.4% was observed after 12 months of therapy.21 Brands et al31 studied cardiac abnormalities observed in 24 patients with MPS I, II, and VI (six were MPS II patients), and the effect of ERT upon them. They described a significant reduction in the size of the interventricular septum thickness during diastole (−0.36 Z-score/year; P=0.05; n=5) and LVMI (−0.26 Z-score/year; P=0.032; n=5),31 without significant effects on ejection fraction, valve disease, or other cardiac parameters after treatment with idursulfase. These findings, together with other clinical reports,32,33 suggest that ERT has limited effect on the cardiac valves in MPS II patients which are probably only slightly accessible to ERT, whereas it has some effect on cardiac dimension.31
Skeletal disease
An Italian study evaluated brain and spine abnormalities of 36 patients with MPS II (mean age 12.6 years, range 2.8–32.2 years) using magnetic resonance imaging. Fifteen patients were treated with idursulfase, and after a period of 1.6 years (range 1–2.9 years), cranial bone abnormalities and spinal anomalies remained stable for most patients. Two idursulfase-treated patients experienced a worsening of intervertebral disk abnormalities. The authors concluded that GAG-mediated damages to cranial bone and spine might be difficult to revert once they have occurred.34
Obstructive sleep apnea
The effects of idursulfase on obstructive sleep apnea were evaluated in two studies. Muenzer et al14 reported that, after 12 months of idursulfase, the average number of O2 desaturation events per hour decreased from 19.2 at baseline to 2.4, with six out of the seven patients evaluated for this outcome experiencing a decrease of >78%.14 The JET study21 showed a mean increase in the rate of oxygen desaturation of 3.9±3.5 events per hour, mainly due to the worsening in a single patient, while the other patients had stable oxygen desaturation index value.21
Functional capacity
During the open-label extension study,20 the Child Health Assessment Questionnaire (CHAQ) was used in order to collect data on functional status of patients regarding their ability to perform daily physical activities. CHAQ was administered to all patients aged 12 years or older and to parents or guardians.20 Statistically significant improvements in parent-assessed Disability Index Score (DIS) were seen at months 8, 16, 20, 24, and 30. For the child-assessed DIS, statistically significant improvements were seen at months 20, 24, 30, and 36.20
The effects of treatment with idursulfase are summarized in Table 2.
 | Table 2 Efficacy of treatment with idursulfase |
Safety
In clinical trials, most of the adverse events (AEs) observed were related to the underlying disease rather than ERT with idursulfase.14,19–21 The most commonly reported treatment-related AEs were infusion-related reactions (IRRs) (ie, AEs assessed as drug-related and occurring within 24 hours of the infusion).
Patients enrolled in the Phase I/II trial, belonging to the 0.15 mg/kg dose group, did not experience any IRR, whereas six out of the eight patients in the higher dose groups had IRRs. These IRRs were of moderate-to-severe intensity and were successfully managed by premedication with antihistamines and/or corticosteroids and by extending the infusion time from 1 hour to 3 hours. Only one patient, treated with idursulfase at 0.5 mg/kg and with a history of severe upper airway obstruction requiring tracheostomy, nighttime ventilation, and supplemental oxygen at baseline, experienced three serious and potentially life-threatening episodes of respiratory distress that resolved after an appropriate drug treatment and/or supplemental oxygen.14
The most frequently reported AEs in the Phase II/III trial19 were fever, headache, cough, pharyngitis, upper respiratory tract infection, nasal congestion, nausea, vomiting, abdominal pain, and diarrhea; these AEs were all mild or moderate in severity, and the total number was similar in each group of treated patients. A similar number of patients in each group experienced one or more IRRs during the study, with a maximal incidence between weeks 4 and 12. No patient was withdrawn from the study because of IRR. A total of 49 serious adverse events (SAEs) occurred in 26 patients belonging to all the three treatment groups during the Phase II/III trial. The investigators considered that SAEs that occurred in three patients could be probably or possibly related to the study drug. Two deaths occurred during the study, but neither of them was considered related to blinded study medication by the investigator.19
During the open-label extension study,20 a total of 50 patients (53%) experienced at least one IRR with the incidence being higher in the group originally treated with placebo (74.2%) compared with the groups originally treated with weekly (45.2%) or EOW (40.6%) idursulfase. A total of 27 patients (28.7%) experienced at least one severe or life-threatening AE, and one death occurred, not considered to be related to treatment by the investigators.20
Five out of the ten patients enrolled in the JET study21 experienced a total of eleven drug-related AEs: the most frequent was urticaria followed by erythema. The same number of patients experienced IRR (urticaria, erythema, dyspnea, abdominal pain, vasovagal syncope). Two patients experienced SAEs, one of whom faced death.21
A meta-analysis of two studies19,21 evaluating the AEs related to infusion of idursulfase showed that 63% of patients had at least one IRR with no statistical significance.24
The most frequent adverse drug reactions observed during treatment with idursulfase are listed in Table 3.
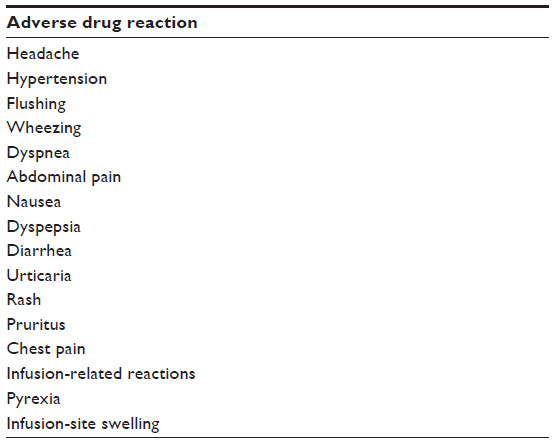 | Table 3 The most frequent adverse drug reactions observed during treatment with idursulfase |
In order to analyze the incidence of IRRs and their timing from the start of ERT in patients with Hunter syndrome, a retrospective analysis was made on 104 patients enrolled in HOS, who began treatment at the time or after the enrollment, and for whom at least 1 year of follow-up was available at October 2009.35 Sixty-five IRRs were observed in 33 patients, all mild or moderate in severity but two, which were reported as SAEs. Nearly all of these initial IRRs occurred during the first 3 months of ERT. Only five patients experienced their first IRR after this period, and only two, after 6 months of ERT.35 Three deaths occurred in this patient population during follow-up, but none was considered to be related to treatment with idursulfase.35
Although even other evidences indicate that the risk of IRR appears to be greatest in the first 6 months of treatment36 and rates of IRRs tend to decline over time, however, in postmarketing reports, patients treated with idursulfase have experienced anaphylactic reactions up to several years after initiating treatment.15
A higher time between infusion and premedication with antihistamines and/or corticosteroids was adopted successfully, in the studies described, in patients who had experienced previous IRR.14,19–21,35 Serrano and Gomez37 performed a short desensitization protocol for a patient with Hunter disease who had experienced urticaria during infusion of idursulfase. After that the patient underwent this desensitization protocol, no further episodes of urticaria or other signs of hypersensitivity occurred.37
Antibodies
Six out of the 12 patients enrolled in Phase I/II trial,14 three in the 0.5 mg/kg, and three in the 1.5 mg/kg group resulted positive for anti-idursulfase IgG antibodies. The development of antibodies did not impact on urinary GAG levels, liver and spleen volumes, and changes in 6MWT and %FVC.14 IgG anti-idursulfase antibodies were detected in 30 patients during Phase II/III trial19 (15 [46.9%] in the idursulfase weekly group, 15 [46.9%] in the EOW group), with the highest prevalence at week 27, when 44.4% of the patients treated were positive. IgM antibodies occurred in two patients, one in each treatment group. About one-third of the patients who developed antibodies were antibody-negative after 1 year of treatment. When antibodies occurred, there was a transient increase in urinary GAGs, whereas changes in distance walked in 6 minutes and FVC measurements did not appear to be affected by antibody status.19 Forty-seven patients (50%) were positive for IgG antibodies at some time during the double-blinded or extension studies. At any time during the extension study,20 22 out of 94 (23.4%) patients had neutralizing antibodies (detected by a test for neutralizing activity), and after 105 weeks, neutralizing antibodies were detected in 19 out of 85 (22.3%) patients, suggesting that tolerization had not occurred. Patients with neutralizing antibodies showed smaller increases in absolute FVC compared with patients without neutralizing antibodies, whereas there was no association with the presence of antibodies and responses on 6MWT, liver and spleen volume, and urine GAG levels.20 Six out of the ten patients (60%) enrolled in the JET study21 were positive for anti-idursulfase IgG antibodies with no differences in urinary GAG levels between seropositive and seronegative patients.21
No IgE antibodies were detected in any of the studies described above.14,19–21 Recently, Kim et al38 identified specific IgE antibodies against Elaprase® in six out of 34 patients with MPS II treated with idursulfase. All these six patients experienced immediate-type allergic reactions during the infusion (three patients [8.8%] demonstrated recurrent anaphylactic reactions, while urticaria was shown in four of the patients studied [11.8%], even if one patient with urticaria refused to undergo the tests proposed). The authors concluded that this immediate-type hypersensitivity is mediated by anti-idursulfase IgE antibodies, which might be produced by the de novo synthesis.38
The results of a post hoc analysis of data from the Phase II/III study and extension study were presented by Barbier et al,39 in order to evaluate the association between anti-idursulfase antibody status and the efficacy and safety of idursulfase, administered at the dose of 0.5 mg/kg weekly for 105 weeks, in an attenuated, treatment-naïve population of patients aged 5 years and older. Thirty-two out of the 63 patients analyzed (51%) developed at least a single sample that was positive for anti-idursulfase antibodies without a decrease in 6MWT distance, a decrease in %FVC response, or an increase in liver and/or spleen volume. Antibody positivity was associated with higher urinary GAG levels (P<0.001 at study week 105), although even patients positive for anti-idursulfase antibodies showed large and statistically significant decreases in urinary GAG levels from baseline. The authors found that IRR rates were higher in patients who would develop antibodies over the course of idursulfase treatment, and interestingly, this was true even before the antibodies were detected, when the IRR rates decreased, likely due to preventive measures.39
Current treatment with idursulfase
Idursulfase is administered by weekly intravenous infusions over 3 hours at a dose of 0.5 mg/kg diluted in an appropriate volume of saline: the duration of infusion can be shortened gradually to 1 hour if there are no IRRs.15 A premedication with antihistamines and/or corticosteroids can be considered for those patients who have experienced previous IRRs during the infusions.
Home therapy with idursulfase
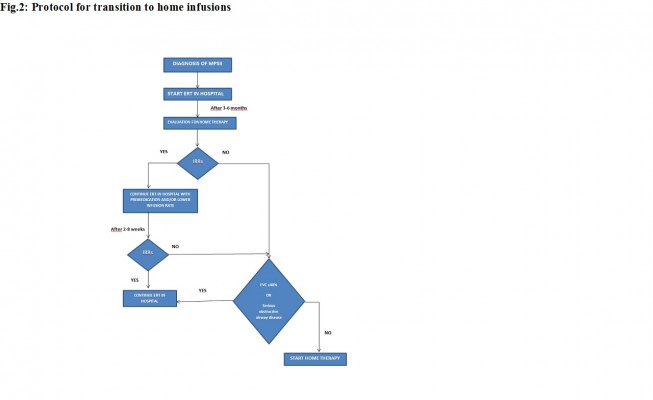
A change to the European product license for idursulfase in March 2010 led to the availability of home treatment with idursulfase for selected patients with MPS II.40 Home therapy with idursulfase has been associated with an increase in patient compliance compared to hospital management and with an improvement in quality of life of both patients and their families.41–44 In order to make home therapy with idursulfase safe and to standardize the timing of beginning of home treatment, protocols have been developed to manage transition to home infusions (Figure 2). Before entering the homecare service, patients must have received ERT in hospital for 3–6 months; if previous IRRs occurred, they must be under control with premedication, and they must not have had an IRR in the 2–8 weeks before homecare is approved, and premedication must be given. If a patient has significant respiratory disease (%FVC, 40% or less; or evidence of serious obstructive airway disease), homecare may not be suitable.45
 | Figure 2 Protocol for transition to home infusions. |
Idursulfase treatment in early childhood
The exclusion of children below 5 years of age from the Phase II/III clinical trial, mainly because of the difficulty in performing meaningful evaluations (eg, 6MWT and spirometry), represents a challenge in the management of very young patients with ERT. To date, only limited data on the effect of ERT in children under 5 years of age are available.19,20
In the first published case series of six Spanish children with Hunter syndrome under 5 years of age, idursulfase demonstrated a similar benefit and safety profiles to that seen in older children. With regard to safety, only mild IRRs were noted. IgA and IgG antibodies were reported in one of the three patients tested, and no IgE antibodies were detected. A median decrease in urinary GAG levels of 66% and a small reduction in spleen and liver volume after 12 months of therapy in a limited number of patients were taken as parameters of effectiveness.46 The same results in terms of safety and effectiveness were reported by Tomanin et al in 27 patients, clustered into three groups according to the age at start of ERT: ≤5 years, >5 years and ≤12 years, and >12 years. No differences between the younger and the older populations were registered except for urinary GAGs excretion, significantly reduced only in the younger groups.47 A retrospective study using data from the HOS evaluated the safety and effectiveness of idursulfase in 124 patients younger than 6 years and compared these findings with those seen in 289 older patients. IRRs were the most common drug-associated AE observed in these two subpopulations. The incidence of all IRRs (26.6% vs 18.8% of patients) and SAEs (12.9% vs 20.2%) seems to be higher in the <6-year-old subpopulation than in the >6-year-old group. IgG antibodies were detected in 38 out of 71 (53.5%) patients whose plasma was assayed, while no IgE antibodies were detected in any patient. Reduction in urinary GAG levels was similar in the two subgroups (63% vs 66%). Liver size reduction was also reported, but its evaluation was conducted by palpation, which might be influenced by a certain degree of subjectivity.48 Safety profile of idursulfase was confirmed in a 53-week, open-label, safety trial in children aged 1.4–7.5 years (28 patients). The most common AEs reported (57%) were IRRs such as in the trial conducted in older patients. With regard to effectiveness, at week 53, the mean decrease from baseline in urinary GAGs was 402.4 (±162.1) μg/mg creatinine (range −750.3 μg/mg to −104.3 μg/mg), representing a mean decrease of 54.4% (range −11.7% to −81.8%). At the same time, liver and spleen volume, calculated with index of liver and spleen size,49 had a mean decrease of 17.4% (range −47.8% to +48.4%) and 20.6% (range −55.1% to +37.2%), respectively, compared with baseline.50 Furthermore, no new safety concerns were seen in eight patients under 1 year of age, and no IRRs were reported after a treatment duration ranging from 6 weeks to 5 years. In terms of efficacy, the authors evidenced that early ERT has resulted in somatic, urinary GAGs, and splenomegaly improvements, although two out of the eight patients only received ERT for a short period of time before HSCT.51 Recently, two case studies reported about the use of idursulfase in younger brothers of an affected patient. Tajima et al published a case study of two Japanese brothers affected by MPS II, in which the older brother initiated treatment with idursulfase at 3.0 years of age, while the younger sibling initiated treatment at 4 months. After 32 months of ERT, the younger brother remained free from most of the somatic features that had already appeared in his brother at the same age.52 Tylki-Szymanska et al published a case study of twin boys, one of whom had MPS II, compared to an older affected sister. After 3 years of treatment, the only somatic sign was a mild deformity of one vertebra, while the sister at the age of 3 showed mild coarse facial features, decreased range of motion in elbow, hip, and ankle joints, slight hepatomegaly, and a small umbilical hernia.53
Overall, the safety and effectiveness of idursulfase were similar in patients aged <6 years to those observed in older patients. There are still conflicting data regarding the benefits on joint mobility,47 growth,27,54 or other symptoms50,51 but, albeit with limited data, most authors’ opinion is that treatment of patients with MPS should occur early to obtain better long-term outcomes.55
Idursulfase in treatment of patients with severe phenotype of MPS II
Approximately two-thirds of patients with MPS II have severe phenotype characterized by early onset of signs and symptoms and progressive cognitive impairment. Neither Phase II/III trial19 nor its extension20 enrolled patients with severe MPS II; therefore, there are no clinical trial data available about the effects of idursulfase treatment on the disease progression in these patients. In a recent consensus report,56 a panel of experts described their experience with the use of idursulfase in 66 Hunter syndrome patients with the severe phenotype. After at least 1 year of ERT, 50 of these patients experienced at least one type of somatic improvements (reduction in frequency of respiratory infections or in the coarseness of facial features, improvement in joint range of motion or in sleep apnea). Nearly all of the patients experienced a reduction in liver volume (61 out of 66).46 No result of the effect of ERT on cognitive function was found in any of the patients, as expected, since idursulfase does not cross the blood–brain barrier.57 In 61 out of 66 cases, physicians and families found sufficient benefit to continue ERT.56 Current US and European guidelines suggest initiating a 6- to 18-month trial of ERT in severe MPS II patients to assess the response (through certain evaluations useful in this population, like joint range of motion, liver and spleen size, coarseness of facial features, macroglossia, frequency of respiratory infections, improvement in quality of life) before stopping or continuing therapy.40,56 Recently, Lampe et al58 reported a retrospective case series providing details on their experiences with treating severe MPS II patients with ERT for more than 2 years, coming from five international centers. All the 22 patients described experienced somatic improvements on ERT. Eighteen patients (82%) had improvements in five to seven signs and symptoms, and all of the patients experienced improvements in at least four of the somatic signs and symptoms evaluated. Cognitive disease progressed in 17/22 (77%) patients and stabilized in 3/22 (14%), and slight improvements were reported in 2/22 (9%) patients due to an increased ability of these patients to interact with their environment consequent to improvements in somatic symptoms. The authors concluded that improvements in somatic signs and symptoms may be experienced by severe patients and can help improve the quality of patients’ and caregivers’ lives.58
Intrathecal administration of idursulfase
A major challenge for research is to develop well-tolerated therapies able to cross the blood–brain barrier, so as to produce positive effects on neurological involvement of patients with Hunter syndrome. Several animal studies demonstrated the feasibility and tolerability of intrathecal administration of idursulfase in monkey models,59 and that idursulfase delivered via the intrathecal route distributes throughout the CNS, penetrates brain tissue, and promotes clearance of lysosomal storage material,60 with deposition in the lysosomal compartment of target cells, the site of pathological GAGs accumulation.61 These findings have supported ongoing clinical trials which are currently investigating intrathecal ERT in patients with MPS II (for example, US National Institutes of Health ClinicalTrials.gov identifiers NCT00920647, NCT02055118, and NCT01506141).
Conclusion
ERT with idursulfase is, actually, the most appropriate available therapy for patients with MPS II. Results coming from clinical studies and clinical practice confirm its efficacy and safety in the treatment of this multisystemic, progressive disorder.
Long-term follow-up of patients receiving ERT have demonstrated that many disease manifestations do not generally improve if irreversible pathological changes have already occurred, so that early diagnosis of the disease and timely initiation of ERT may heavily modify the natural history of MPS II, slowing or preventing the development of some of these irreversible manifestations.
The lack of a specific biomarker useful to assess treatment efficacy led to the evaluation of different tools for monitoring patients’ response to ERT during clinical trials. Probably, more validated clinical outcome parameters able to assess the effects of idursulfase on disease burden are needed, particularly in patients with CNS involvement and in children younger than 5 years, who are not able to perform respiratory or walking tests.
CNS disease remains a major challenge, since intravenously administered idursulfase is not able to cross the blood–brain barrier, and experience of intrathecal administration of ERT is limited to clinical trials. There is great hope that intrathecal administration of idursulfase will be available and will enter clinical practice together with intravenously administered ERT, so that also the neurological signs and symptoms of MPS II can be treated.
In our opinion, an early start of ERT, the validation of more appropriate clinical efficacy tools, and the availability of intrathecal-administered therapy can be useful to reach the full potential of idursulfase in treatment of MPS II.
Acknowledgment
The authors thank Prof Pietro Strisciuglio for the review of the manuscript.
Disclosure
The authors report there are no potential conflicts of interest, real or perceived.
References
Hunter C. A rare disease in two brothers. Proc R Soc Med. 1917;10:104–106. | |
Bach G, Eisenberg F Jr, Cantz M, Neufeld EF. The defect in the Hunter syndrome: deficiency of sulfoiduronate sulfatase. Proc Natl Acad Sci U S A. 1973;70(7):2134–2138. | |
Neufeld EF, Muenzer J. The mucopolysaccharidoses. In: Scriver CR, Beaudet AL, Sly WS, Valle D, editors. The Metabolic and Molecular Bases of Inherited Disease. New York: McGraw-Hill; 2001:3421–3452. | |
Beck M, Wijburg FA, Gal A. Clinical utility gene card for: mucopolysaccharidosis type II. Eur J Hum Genet. 2012;20(1):1. | |
Tuschl K, Gal A, Paschke E, Kircher S, Bodamer OA. Mucopolysaccharidosis type II in females: case report and review of literature. Pediatr Neurol. 2005;32:270–272. | |
Pinto LL, Vieira TA, Giugliani R, Schwartz IV. Expression of the disease on female carriers of X-linked lysosomal disorders: a brief review. Orphanet J Rare Dis. 2010;5:14. | |
Martin R, Beck M, Eng C, et al. Recognition and diagnosis of mucopolysaccharidosis II (Hunter syndrome). Pediatrics. 2008;121: e377–e386. | |
Wraith JE, Beck M, Giugliani R, Clarke J, Martin R, Muenzer J. Initial report from the Hunter outcome survey. Genet Med. 2008;10:508–516. | |
Schwartz IV, Ribeiro MG, Mota JG, et al. A clinical study of 77 patients with mucopolysaccharidosis type II. Acta Paediatr Suppl. 2007;96(455):63–70. | |
Wraith JE, Scarpa M, Beck M, et al. Mucopolysaccharidosis type II (Hunter syndrome): a clinical review and recommendations for treatment in the era of enzyme replacement therapy. Eur J Pediatr. 2008;167:267–277. | |
Jones SA, Almássy Z, Beck M, et al; HOS Investigators. Mortality and cause of death in mucopolysaccharidosis type II – a historical review based on data from the Hunter outcome survey (HOS). J Inherit Metab Dis. 2009;32:534–543. | |
Vellodi A, Young E, Cooper A, Lidchi V, Winchester B, Wraith JE. Long-term follow-up following bone marrow transplantation for Hunter disease. J Inherit Metab Dis. 1999;22(5):638–648. | |
Guffon N, Bertrand Y, Forest I, Fouilhoux A, Froissart R. Bone marrow transplantation in children with Hunter syndrome: outcome after 7 to 17 years. J Pediatr. 2009;154(5):733–737. | |
Muenzer J, Gucsavas-Calikoglu M, McCandless SE, Schuetz TJ, Kimura A. A phase I/II clinical trial of enzyme replacement therapy in mucopolysaccharidosis II (Hunter syndrome). Mol Genet Metab. 2007;90(3):329–337. | |
Elaprase® (idursulfase) Solution for Intravenous Infusion [Prescribing Information] Shire Human Genetic Therapies; 2011. | |
Sohn YB, Cho SY, Park SW, et al. Phase I/II clinical trial of enzyme replacement therapy with idursulfase beta in patients with mucopolysaccharidosis II (Hunter syndrome). Orphanet J Rare Dis. 2013; 18(8):42. | |
Chung YK, Sohn YB, Sohn JM, et al. A biochemical and physicochemical comparison of two recombinant enzymes used for enzyme replacement therapies of Hunter syndrome. Glycoconj J. 2014;31(4):309–315. | |
Sohn YB, Cho SY, Lee J, Kwun Y, Huh R, Jin DK. Safety and efficacy of enzyme replacement therapy with idursulfase beta in children aged younger than 6 years with Hunter syndrome. Mol Genet Metab. 2015;114(2):156–160. | |
Muenzer J, Wraith JE, Beck M, et al. A phase II/III clinical study of enzyme replacement therapy with idursulfase in mucopolysaccharidosis II (Hunter syndrome). Genet Med. 2006;8(8):465–473. | |
Muenzer J, Beck M, Eng CM, et al. Long-term, open-labeled extension study of idursulfase in the treatment of Hunter syndrome. Genet Med. 2011;13(2):95–101. | |
Okuyama T, Tanaka A, Suzuki Y, et al. Japan Elaprase Treatment (JET) Study: idursulfase enzyme replacement therapy in adult patients with attenuated Hunter syndrome (mucopolysaccharidosis II, MPS II). Mol Genet Metab. 2010;99(1):18–25. | |
Coppa GV, Buzzega D, Zampini L, et al. Plasmatic and urinary glycosaminoglycans characterization in mucopolysaccharidosis II patient treated with enzyme-replacement therapy with idursulfase. JIMD Rep. 2012;4:79–90. | |
Glamuzina E, Fettes E, Bainbridge K, et al. Treatment of mucopolysaccharidosis type II (Hunter syndrome) with idursulfase: the relevance of clinical trial end points. J Inherit Metab Dis. 2011;34(3):749–754. | |
Alegra T, Eizerik DP, de Cerqueira CC, Pereira TV, Dornelles AD, Schwartz IV. Efficacy and safety of idursulfase therapy in patients with mucopolysaccharidosis type II with and without comparison to placebo: systematic review and meta-analysis. Cad Saude Publica. 2013;29(Suppl 1):S45–S58. | |
Tchan MC, Devine KT, Sillence DO. Three adult siblings with mucopolysaccharidosis type II (hunter syndrome): a report on clinical heterogeneity and 12 months of therapy with idursulfase. JIMD Rep. 2011;1:57–64. | |
Westhoff M, Litterst P. Successful noninvasive ventilation and enzyme replacement therapy in an adult patient with morbus hunter. JIMD Rep. 2012;5:77–82. | |
Schulze-Frenking G, Jones SA, Roberts J, Beck M, Wraith JE. Effects of enzyme replacement therapy on growth in patients with mucopolysaccharidosis type II. J Inherit Metab Dis. 2011;34(1):203–208. | |
Kuczmarski RJ, Ogden CL, Guo SS, et al. 2000 CDC growth charts for the United States: methods and development. Vital Health Stat. 2002;246:1–190. | |
Jones SA, Parini R, Harmatz P, et al; HOS Natural History Working Group on behalf of HOS Investigators. The effect of idursulfase on growth in patients with Hunter syndrome: data from the Hunter outcome survey (HOS). Mol Genet Metab. 2013;109(1):41–48. | |
Cho SY, Huh R, Chang MS, et al. Impact of enzyme replacement therapy on linear growth in Korean patients with mucopolysaccharidosis type II (Hunter syndrome). J Korean Med Sci. 2014;29(2):254–260. | |
Brands MM, Frohn-Mulder IM, Hagemans ML, et al. Mucopolysaccharidosis: cardiologic features and effects of enzyme-replacement therapy in 24 children with MPS I, II and VI. J Inherit Metab Dis. 2013;36(2):227–234. | |
Sato Y, Fujiwara M, Kobayashi H, Ida H. Massive accumulation of glycosaminoglycans in the aortic valve of a patient with Hunter syndrome during enzyme replacement therapy. Pediatr Cardiol. 2013;34(8):2077–2079. | |
Fesslová V, Corti P, Sersale G, et al. The natural course and the impact of therapies of cardiac involvement in the mucopolysaccharidoses. Cardiol Young. 2009;19(2):170–178. | |
Manara R, Priante E, Grimaldi M, et al. Brain and spine MRI features of Hunter disease: frequency, natural evolution and response to therapy. J Inherit Metab Dis. 2011;34(3):763–780. | |
Burton BK, Whiteman DA; HOS Investigators. Incidence and timing of infusion-related reactions in patients with mucopolysaccharidosis type II (Hunter syndrome) on idursulfase therapy in the real-world setting: a perspective from the Hunter outcome survey (HOS). Mol Genet Metab. 2011;103(2):113–120. | |
Miebach E. Management of infusion-related reactions to enzyme replacement therapy in a cohort of patients with mucopolysaccharidosis disorders. Int J Clin Pharmacol Ther. 2009;47(Suppl 1):S100–S106. | |
Serrano CD, Gomez JF. Successful desensitization to idursulfase in a patient with type II mucopolysaccharidosis (Hunter syndrome). J Investig Allergol Clin Immunol. 2011;21(7):571–572. | |
Kim J, Park MR, Kim DS, et al. IgE-mediated anaphylaxis and allergic reactions to idursulfase in patients with Hunter syndrome. Allergy. 2013;68(6):796–802. | |
Barbier AJ, Bielefeld B, Whiteman DA, Natarajan M, Pano A, Amato DA. The relationship between anti-idursulfase antibody status and safety and efficacy outcomes in attenuated mucopolysaccharidosis II patients aged 5 years and older treated with intravenous idursulfase. Mol Genet Metab. 2013;110(3):303–310. | |
Scarpa M, Almássy Z, Beck M, et al; Hunter Syndrome European Expert Council. Mucopolysaccharidosis type II: European recommendations for the diagnosis and multidisciplinary management of a rare disease. Orphanet J Rare Dis. 2011;6:72. | |
Burton BK, Wiesman C, Paras A, Kim K, Katz R. Home infusion therapy is safe and enhances compliance in patients with mucopolysaccharidoses. Mol Genet Metab. 2009;97(3):234–236. | |
Milligan A, Hughes D, Goodwin S, Richfield L, Mehta A. Intravenous enzyme replacement therapy: better in home or hospital? Br J Nurs. 2006;15(6):330–333. | |
Hughes DA, Mlilligan A, Mehta A. Home therapy for lysosomal storage disorders. Br J Nurs. 2007;16(22):1386–1389. | |
Ceravolo F, Mascaro I, Sestito S, et al. Home treatment in paediatric patients with Hunter syndrome: the first Italian experience. Ital J Pediatr. 2013;39:53. | |
Bagewadi S, Roberts J, Mercer J, Jones S, Stephenson J, Wraith JE. Home treatment with Elaprase and Naglazyme is safe in patients with mucopolysaccharidoses types II and VI, respectively. J Inherit Metab Dis. 2008;31(6):733–737. | |
Alcalde-Martín C, Muro-Tudelilla JM, Cancho-Candela R, et al. First experience of enzyme replacement therapy with idursulfase in Spanish patients with Hunter syndrome under 5 years of age: case observations from the Hunter outcome survey (HOS). Eur J Med Genet. 2010;53(6):371–377. | |
Tomanin R, Zanetti A, D’Avanzo F, et al. Clinical efficacy of enzyme replacement therapy in paediatric Hunter patients, an independent study of 3.5 years. Orphanet J Rare Dis. 2014;9(1):129. | |
Muenzer J, Beck M, Giugliani R, et al. Idursulfase treatment of Hunter syndrome in children younger than 6 years: results from the Hunter outcome survey. Genet Med. 2011;13(2):102–109. | |
Dittrich M, Milde S, Dinkel E, Baumann W, Weitzel D. Sonographic biometry of liver and spleen size in childhood. Pediatr Radiol. 1983;13:206–211. | |
Giugliani R, Hwu WL, Tylki-Szymanska A, Whiteman DA, Pano A. A multicenter, open-label study evaluating safety and clinical outcomes in children (1.4–7.5 years) with Hunter syndrome receiving idursulfase enzyme replacement therapy. Genet Med. 2014;16(6):435–441. | |
Lampe C, Atherton A, Burton BK, et al. Enzyme replacement Therapy in mucopolysaccharidosis II patients under 1 year of age. JIMD Rep. 2014;14:99–113. | |
Tajima G, Sakura N, Kosuga M, Okuyama T, Kobayashi M. Effects of idursulfase enzyme replacement therapy for mucopolysaccharidosis type II when started in early infancy: comparison in two siblings. Mol Genet Metab. 2013;108:172–177. | |
Tylki-Szymanska A, Jurecka A, Zuber Z, Rozdzynska A, Marucha J, Czartoryska B. Enzyme replacement therapy for mucopolysaccharidosis II from 3 months of age: a 3-year follow-up. Acta Paediatr. 2012;101:e42–e47. | |
Żuber Z, Różdżyńska-Świątkowska A, Jurecka A, Tylki-Szymańska A. The effect of recombinant human iduronate-2-sulfatase (Idursulfase) on growth in young patients with mucopolysaccharidosis type II. PLoS One. 2014;9(1):e85074. | |
Muenzer J. Early initiation of enzyme replacement therapy for the mucopolysaccharidoses. Mol Genet Metab. 2014;111(2):63–72. | |
Muenzer J, Bodamer O, Burton B, et al. The role of enzyme replacement therapy in severe Hunter syndrome – an expert panel consensus. Eur J Pediatr. 2012;171(1):181–188. | |
Boado RJ, Hui EK, Lu JZ, Sumbria RK, Pardridge WM. Blood-brain barrier molecular Trojan horse enables imaging of brain uptake of radioiodinated recombinant protein in the rhesus monkey. Bioconjug Chem. 2013;24(10):1741–1749. | |
Lampe C, Bosserhoff AK, Burton BK, et al. Long-term experience with enzyme replacement therapy (ERT) in MPS II patients with a severe phenotype: an international case series. J Inherit Metab Dis. 2014;37(5):823–829. | |
Felice BR, Wright TL, Boyd RB, et al. Safety evaluation of chronic intrathecal administration of idursulfase-IT in cynomolgus monkeys. Toxicol Pathol. 2011;39(5):879–892. | |
Dickson PI. Novel treatments and future perspectives: outcomes of intrathecal drug delivery. Int J Clin Pharmacol Ther. 2009;47(Suppl 1): S124–S127. | |
Calias P, Papisov M, Pan J, et al. CNS penetration of intrathecal-lumbar idursulfase in the monkey, dog and mouse: implications for neurological outcomes of lysosomal storage disorder. PLoS One. 2012;7(1): e30341. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.