")
Back to Archived Journals » Orphan Drugs: Research and Reviews » Volume 4
Profile of aminopyridines for Lambert–Eaton myasthenic syndrome
Authors Maddison P, Keogh M, Sedehizadeh S
Received 26 September 2013
Accepted for publication 6 November 2013
Published 20 January 2014 Volume 2014:4 Pages 11—18
DOI https://doi.org/10.2147/ODRR.S40799
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Paul Maddison,1 Michael J Keogh,2 Saam Sedehizadeh1
1Department of Clinical Neurology, Queen's Medical Centre, Nottingham, UK; 2Institute of Human Genetics, University of Newcastle upon Tyne, International Centre for Life, Newcastle upon Tyne, UK
Abstract: 3,4-diaminopyridine (3,4-DAP) is an effective, symptomatic treatment for Lambert–Eaton myasthenic syndrome (LEMS). Several trials have studied the effects of 3,4-DAP in small numbers of LEMS patients. We systematically reviewed all randomized trials of 3,4-DAP in LEMS to determine the efficacy of this treatment using meta-analysis of clinical and electrophysiological end points. Data from four randomized, placebo-controlled trials revealed that muscle-strength scores increased significantly with 3,4-DAP. Limited meta-analysis performed on two trials using the quantitative myasthenia gravis score indicated that the clinical benefits seen were modest, with an improvement in quantitative myasthenia gravis score of 2.44 points (95% confidence interval: 1.2–3.6). Meta-analysis of the mean change in compound muscle action potential amplitude following 3,4-DAP treatment revealed a significant improvement compared to placebo (1.36 mV, 95% confidence interval: 0.99–1.72). 3,4-DAP is an effective, safe, first-line symptomatic treatment for LEMS, with significant clinical and electrophysiological benefits demonstrated by meta-analysis.
Keywords: Lambert–Eaton myasthenic syndrome, 3,4-diaminopyridine, meta-analysis
Introduction
Lambert–Eaton myasthenic syndrome (LEMS) is an autoimmune, presynaptic disorder of neuromuscular transmission characterized by proximal limb weakness, autonomic disturbance, depressed tendon reflexes, and post-tetanic potentiation.1,2 Approximately 50% of patients with LEMS have an underlying small-cell lung cancer (SCLC).3,4 LEMS is an autoimmune disease mediated by antibodies to P/Q-type voltage-gated calcium channels at the motor nerve terminal.5–7
Given that LEMS is a presynaptic disorder characterized by impaired quantal release of acetylcholine, symptomatic treatment has utilized drugs that increase neurotransmitter release at the neuromuscular junction. Guanidine was first recommended for use in LEMS by Lambert,8 but has not been used in large randomized controlled trials because of the serious side effects of marrow suppression9 and renal failure.10 However, low-dose guanidine (less than 1,000 mg/day) has been used in conjunction with pyridostigmine in one small, open-labeled study that resulted in improved muscle strength and neurophysiological measurements in all nine LEMS patients studied.11 One-third of these study patients had to discontinue guanidine due to persistent gastrointestinal side effects.
The quaternary ammonium compound, 4-aminopyridine (4-AP), was found to increase the release of acetylcholine at the neuromuscular junction,12 and was therefore subsequently used for the symptomatic treatment of two patients with LEMS.13,14 A larger open study of the use of oral 4-AP in four patients with LEMS resulted in clinical and electrophysiological improvement, but one participant suffered a single generalized seizure whilst taking a dose of 120 mg 4-AP per day.15 The possibility of significant central nervous system side effects such as seizures has therefore limited the use of 4-AP, a drug known to cross the blood–brain barrier and result in epileptogenic effects in animals.16
The related aminopyridine 3,4-diaminopyridine (3,4-DAP) has become the mainstay of symptomatic treatment for LEMS in Europe, with the phosphate salt preparation amifampridine recently receiving a license for treatment in LEMS.17 3,4-DAP has been shown in animals to be more potent in improving neuromuscular transmission18 and less convulsant19 than 4-AP. In addition, it has the advantage over 4-AP of crossing the blood–brain barrier less readily,20 resulting in fewer central nervous system side effects. The first use of 3,4-DAP was in three patients with LEMS without lung cancer who all improved clinically and electrophysiologically from intravenous and then oral preparations of 3,4-DAP.21 Follow-up data collected after a mean treatment duration of 5 years demonstrated prolonged clinical benefit with few side effects at daily doses less than 60 mg of 3,4-DAP.22
Since these initial reports, there have been a number of additional trials of 3,4-DAP in LEMS. The objective of this review was to use a systematic approach to examine the efficacy of 3,4-DAP treatment for LEMS by reviewing all data from relevant randomized trials.
Patients and methods
Trial inclusion criteria
All randomized or quasi-randomized trials involving 3,4-DAP treatment of LEMS were included. Within these trials, all adults and children with a diagnosis of Lambert–Eaton myasthenic syndrome, with or without SCLC were included. Diagnosis of LEMS was based on typical clinical and electrophysiological findings.3,23–25
Outcome measures
The primary outcome measure was a change in the score on a muscle-strength scale, (the quantitative myasthenia gravis [QMG] score)26–28 or, when not available, limb muscle strength measured by myometry. The QMG scoring system ranges from a score of 0 to 39. A score of 0 implies that speech, swallowing, vital capacity, facial muscle strength, external ocular muscles, and all limb muscles are normal.28 Barohn et al tested for interrater reliability of the QMG score and found that, if the QMG score is to be used as a primary efficacy measure, then a treatment must produce more than 2.6 units of change to be of clinical significance.28
The secondary outcome measure was an improvement in the amplitude of the resting compound muscle action potential ([CMAP] mean of all muscles tested).
Search strategy for identification of studies
We searched the Cochrane Neuromuscular Disease Group trials register (April 2011) using “Lambert-Eaton (myasthenic syndrome)” or “LEMS” or “Eaton-Lambert,” and “treatment” as the search terms. We also searched MEDLINE (January 1966 to April 2011) and EMBASE (January 1980 to April 2011), and checked bibliographies and contacted authors to identify additional published or unpublished data. There were no language restrictions. The authors checked the titles and abstracts identified from the literature search. The full text of all potentially relevant studies was obtained for independent assessment by all authors. Two authors (MK and PM) independently assessed trials for inclusion and graded their methodological quality, and disagreements were resolved by discussion. Missing data were obtained from the trial authors whenever possible.
Data analysis
The risk of bias in each identified trial was assessed by considering sequence generation, allocation concealment, and blinding, and addressing incomplete outcome data, selective reporting, or any other forms of bias.29 Results were expressed as mean differences and 95% confidence intervals (CIs) for continuous variable outcomes. Due to the crossover design of some studies,30–32 we pooled data with the generic inverse variance (GIV) method.33 This takes the mean difference between treatment and control, with standard error of the mean (SEM) for the difference. Wherever possible, we used the published SEM; when this was not available, we used the published P-value or original data obtained from the authors to estimate SEM.
For one trial, when no other means of obtaining the variance were possible, we had to assume a known within-subject correlation between the treatment effect in the two periods of the crossover study.30 The subsequent calculated values were then utilized in a GIV analysis.
We undertook a sensitivity analysis on the basis of methodological quality and tested for heterogeneity using a chi-squared test.
Results
To date, there have been four randomized, placebo-controlled trials of 3,4-DAP in a total of 54 patients with LEMS.30–32,34 Characteristics of the studies are shown in Table 1. No trial involved healthy or disease control groups.
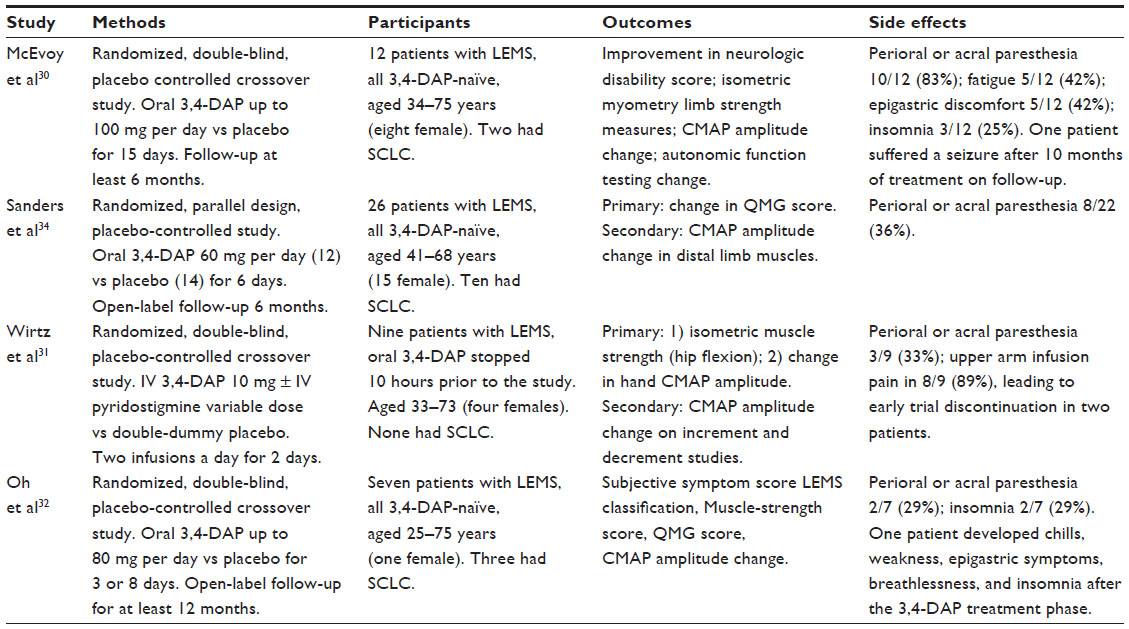 | Table 1 Characteristics of studies |
Trial details
The first 3,4-DAP trial to be published was a crossover trial of 12 participants that compared the effect of maximum-dose oral 3,4-DAP (100 mg/day) for 6 days with placebo, using a muscle-strength score and electrophysiological testing at 3 and 6 days.30 The authors found a significant improvement in isometric muscle strength and a parallel increase in resting CMAP amplitudes following 3,4-DAP treatment in all participants compared with placebo.
A second trial with a parallel-group design compared oral 3,4-DAP (60 mg/day) with placebo (oral lactose capsules) in 26 participants (12 received 3,4-DAP, 14 placebo).34 A QMG muscle – strength score and electrophysiological measurements were taken on days 5 and 6. The authors demonstrated a significant improvement in both the QMG muscle strength score and summated median CMAP amplitude after 6 days in patients taking 3,4-DAP compared to placebo.
The third trial reported was a placebo-controlled, double-dummy, double-blind, randomized crossover study of nine patients with LEMS, in which the study group compared 10 mg of intravenous 3,4-DAP with placebo infusion, intravenous pyridostigmine (varying doses), and a combination of 3,4-DAP and pyridostigmine.31 Isometric muscle strength of hip flexion, measured by dynamometry, and resting CMAP amplitude from the hypothenar eminence, were recorded every 20 minutes between 10 and 170 minutes post-infusion. The authors found that both CMAP amplitude and isometric muscle strength increased significantly during treatment with 3,4-DAP compared with placebo. The addition of pyridostigmine to 3,4-DAP did not confer any additional benefit, and treatment with pyridostigmine alone did not improve CMAP amplitude and isometric muscle strength compared with placebo.
The fourth, and latest, trial published was a randomized, double-blind, placebo-controlled, crossover study of oral 3,4-DAP in seven patients with LEMS.32 The treatment protocol varied, with the first group of three cases treated with an initial daily dose of 15 mg, increasing to 80 mg per day by the end of the 8-day period. A second group was treated with 30 mg per day, increasing to 75 mg/day over a 3-day study period, due to time constraints. End points included a subjective symptoms score, LEMS classification, Medical Research Council summated muscle-strength score, QMG score, and abductor digiti quinti (ADQ) CMAP amplitude and repetitive nerve stimulation. All four clinical scores improved significantly with 3,4-DAP compared with placebo, as did resting ADQ CMAP amplitudes after 6 to 16 days.
Methodological quality of the trials
Sequence generation was acceptable in all four trials. Participants were assigned treatment or placebo by either random allocation table31,34 or random number table.30,32
In two studies, insufficient information was given about the process of allocation concealment to determine whether this was adequate.30,31 Participant blinding was intended in all four trials, although only two32,34 clearly stated that the active drug was identical in appearance to the placebo preparation. The details of the compound used for the placebo in one trial were not stated.30 Three studies recorded the effectiveness of blinding in terms of the side effects noted when taking active drug (3,4-DAP) rather than placebo (Table 1).31,32,34 The methods by which observer blinding was achieved was only infrequently outlined.
Although clearly described clinical and electrophysiological outcome measures were listed in all trials, only two studies differentiated between primary and secondary outcomes.31,34
The only trial of the four that was parallel in design, and not crossover, detailed full baseline characteristics for the LEMS participants receiving either 3,4-DAP or placebo. There were no significant differences between the groups in terms of participant age, sex, presence of SCLC, CMAP amplitudes, or QMG scores.34
Incomplete outcome data were well explained and accounted for in all studies. Follow-up of patients was completed for the short duration of the three trials. Three of the trials reported on an extended period of follow-up, but the long-term effect of treatment was not a planned end point. Extended follow-up of 12 to 21 months by McEvoy et al30 showed sustained benefit in favor of 3,4-DAP. Almost all (22 of 25) participants studied by Sanders et al34 gained sustained benefit from 3,4-DAP treatment over a 6-month follow-up period. Oh et al32 described patients’ choice for long-term treatment at the cessation of the trial, and gave a subjective or objective account of their progress.
Pooled meta-analysis of end point results 3,4-DAP versus placebo
Primary outcome measure: the score on a muscle-strength scale (QMG score or limb muscle strength measured by myometry)
All trials reported a significant improvement in either muscle-strength score or myometric limb measurement following treatment. However, a meta-analysis of the results from all four studies was not possible because of marked differences between these trials regarding primary outcome measures. Sanders et al34 and Oh et al32 both used the QMG score as a primary outcome. The trial by McEvoy et al30 used a different muscle-strength score from the QMG score and the dynamometry isometric muscle strength reported by Wirtz et al.31 The scoring system and isometric limb measurements used by McEvoy et al30 or Wirtz et al31 were not detailed enough for calculation of an equivalent QMG score. We failed to obtain individual participant data from the authors of one study.30
We were able to perform a meta-analysis on QMG scores based on the data provided in the papers by Sanders et al34 and Oh et al32 Sanders et al’s34 was the first trial to use the QMG muscle score and showed that there was a 2.25-point improvement between the placebo and 3,4-DAP treated group. In the study by Oh et al,32 QMG score improved in four of the six patients treated with 3,4-DAP, with a mean improvement of 2.76 points. Meta-analysis showed that QMG score improved with 3,4-DAP treatment compared to baseline QMG values, with a mean overall improvement of 2.44 points (95% CI: 1.22–3.6 points) (GIV analysis, Figure 1).
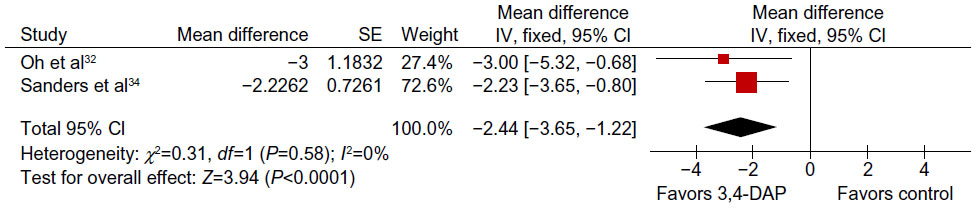 | Figure 1 Meta-analysis of the change in quantitative myasthenia gravis score with 3,4-DAP treatment using generic inverse variance method. |
Secondary outcome measure: improvement in the amplitude of the resting CMAP(s) (mean of all muscles tested)
All trials recorded changes in the amplitude of resting CMAPs after active treatment or placebo. Resting CMAP values in both one arm and one leg muscle, obtained before and after treatment with 3,4-DAP, were available for each participant in one trial.30 Oh et al32 also recorded, before, during, and after 3,4-DAP treatment, ADQ CMAP values in their results section. An averaged CMAP obtained from one foot and two hand muscles was used in the trial by Sanders et al,34 and the averaged change in CMAP amplitude was given for the participant cohort by Wirtz et al.31 The original CMAP data was subsequently provided by Sanders et al34 and Wirtz et al31 in order to be included in this meta-analysis.
All trials recorded significant improvement in resting CMAP amplitudes following 3,4-DAP treatment compared with placebo. We were able to compare overall treatment effect by using an averaged (or hand muscle) CMAP amplitude response. As three trials were a crossover design30–32 and the other used a parallel protocol,34 it was necessary to employ a GIV analysis. We assumed a known correlation between the individual responses in the two treatment periods (within-patient treatment effects) of the crossover trial by McEvoy et al.30 This made it possible to deduce the correct SEM difference. This was then used in the meta-analysis to pool the results of this crossover trial30 (where only the mean responses and their standard errors were available for the two treatment periods) with those of the parallel group trial34 and the remaining two crossover trials,31,32 with known standard mean differences and standard error values. These values were then used in a GIV analysis to assess the overall effect of treatment. Meta-analysis of the CMAP secondary end point showed a significant overall benefit in CMAP amplitude after treatment with 3,4-DAP. The overall mean improvement on GIV analysis was 1.36 mV (95% CI: 0.99–1.72), in favor of the treatment (Figure 2).
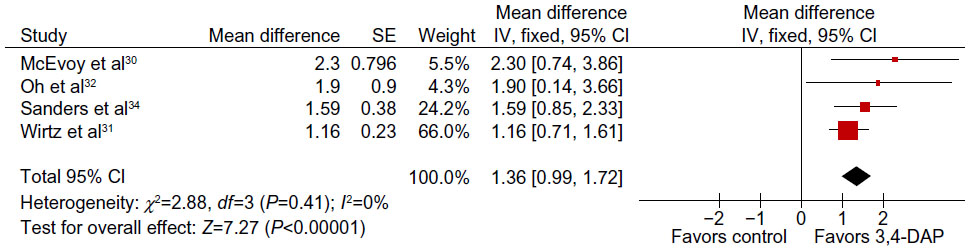 | Figure 2 Meta-analysis of the change in mean compound muscle action potential amplitude (mV) with 3,4-DAP treatment using generic inverse variance method. |
The number of participants with SCLC included in the trials was small (only 15 patients in three of the four trials).30,32,34 In the analyses of the effects of 3,4-DAP treatment in these three trials, the numbers of patients with an associated SCLC (n=15) was too small to enable statistically meaningful subgroup analysis.
Side effects
Major adverse events from 3,4-DAP were seldom encountered in these clinical trials.30–32,34 Mild, common side effects encountered included brief perioral tingling and acral paresthesias, insomnia, and epigastric discomfort (Table 1). One patient suffered from a seizure after 10 months of 3,4-DAP treatment.30 This occurred soon after her daily dose of 3,4-DAP was increased from 90 mg to 100 mg, and her pyridostigmine dose doubled to 240 mg a day, and never recurred when the daily dose of 3,4-DAP was reduced to 40 mg. Almost all patients receiving intravenous 3,4-DAP in one study reported upper limb pain on the side of the infusions, leading to trial withdrawal in two patients.31 Monitoring of routine blood tests, electrocardiography, and electroencephalography did not reveal any abnormalities during 3,4-DAP treatment.
Discussion
3,4-DAP has been shown to be a well-tolerated and effective treatment for patients with LEMS in a meta-analysis of the results from four randomized controlled trials.30–32,34 All primary end point measures of isometric muscle strength,31 neurological disability score.30 and QMG score32,34 improved significantly following the administration of 3,4-DAP. However, due to the heterogeneity of these primary end point scores, we were unable to perform an overall meta-analysis for these data. The apparent improvement in QMG score in two trials, in a limited meta-analysis,32,34 must be placed into the context of the reliability of the QMG score as a primary efficacy measurement in clinical trials as outlined by Barohn et al.28 The QMG score in these validation studies was principally designed for patients with myasthenia gravis, and the score changes judged to represent a meaningful clinical improvement were not calculated in patients with LEMS. The other two primary end point clinical scores have never been validated in further studies. The use of a uniform primary outcome clinical measure such as QMG, ideally validated for LEMS, in future trials would enable a more definitive treatment effect to be established with meta-analysis.
Meta-analysis demonstrated a significant improvement in the secondary end point of mean CMAP amplitude following treatment with 3,4-DAP, although statistical assumptions had to be employed in the analysis. This was because one of the four trials had a crossover design and we did not have access to individual patient data from this study to help determine within-patient treatment effects in the two crossover periods.30 However, original data from the other three trials could be used directly in the analysis, and the overall results revealed a significant overall improvement of CMAP to be determined on GIV testing. Therefore, change in mean CMAP amplitude following treatment seems to be an ideal, objective, and reproducible secondary end point for trials of treatment in LEMS.
Complete follow-up data in three trials indicated a sustained ongoing clinical and electrophysiological benefit from oral 3,4-DAP, with no additional adverse events.30,32,34 Due to the occurrence of seizures in three other LEMS patients treated with 3,4-DAP outside the confines of these four trials, usually at a daily dose of 100 mg, it has been recommended that the daily oral dose of 3,4-DAP should not exceed 80 mg.34 This would seem to be a reasonable recommendation, given that most patients in the four reported randomized trials received doses of oral (or intravenous equivalent) 3,4-DAP lower than 100 mg per day, with clinical and electrophysiological improvement. No cardiac abnormalities were encountered in the trials at these doses of 3,4-DAP, as would be expected, considering the only case reported in detail of cardiac side effects (supraventricular tachyarrhythmia) in a patient taking 3,4-DAP for LEMS occurred in a patient given an erroneously high daily dose of 360 mg.35
The importance in establishing a treatment effect of 3,4-DAP in LEMS by meta-analysis has arisen recently, partly due to the release of new, licensed products for this disorder and the costs associated with this.36 Orphan drug legislation, originating in the United States, was designed to encourage the development of medicines that would benefit “orphan” diseases. The European Union followed this with similar legislation in 2000, and LEMS was designated one of these orphan conditions.37,38 As a result, BioMarin Pharmaceutical Inc (San Rafael, CA, USA) have been able to produce a licensed phosphate salt version of 3,4-DAP for LEMS, amifampridine, based on existing evidence of efficacy from unlicensed preparations. An ongoing patient registry, with adverse effect reporting, will be in place for amifampridine, as required by the Medicines and Healthcare Products Regulatory Agency (UK) and the European Medicines Agency, and should provide standardized clinical safeguards.36 The significant price increase associated with the licensed product in Europe (patient costs per year up to £44,200; €52,000; US $71,300) will lead to health care providers seeking robust evidence of the efficacy of 3,4-DAP in LEMS.39 As yet, US Food and Drug Administration approval for 3,4-DAP has not been granted in the United States.
Results of this meta-analysis of data from four trials of 3,4-DAP in patients with LEMS have shown a significant benefit in muscle-strength score (QMG) and resting CMAP amplitude in distal limb muscles following treatment compared with placebo. However, it is not clear whether the improvement in the muscle strength score (QMG, analyzed in two trials for meta-analysis) represents a meaningful clinical improvement, based on previous studies using this QMG score in patients with myasthenia gravis.28 It can be expected that 3,4-DAP will remain the drug of choice for symptomatic treatment of LEMS. In patients not adequately controlled on symptomatic treatment, long-term oral immunosuppression with prednisolone and azathioprine is usually considered, with treatment regimens based on previous trial data in myasthenia gravis (prednisolone increasing initially from 10 mg on alternate days, by 10 mg every other day, up to 1.5 mg/kg (maximum 100 mg) on alternate days: azathioprine 50 mg per day, increasing by 25 mg per day to a total dose of 2.5 mg/kg per day) showing beneficial effects from combined therapy.40 Retrospective, observational follow-up data on 47 non-paraneoplastic LEMS patients in one center demonstrated that combination therapy of prednisolone and azathioprine was required in almost all patients (approximately 90%), with 43% achieving sustained clinical remission within the first 3 years of treatment.41 Further studies of 3,4-DAP and other treatments in LEMS, such as established and newer immunosuppressive agents,42 should utilize a primary end point muscle-strength score, validated in patients with LEMS.
Acknowledgments
We are grateful for the statistical input kindly provided by Dr AV Swan. Statistical calculations of the effects of 3,4-DAP in LEMS, and associated graphs of the meta-analyses, have previously been reported in a review of all randomized controlled trials of treatments for LEMS.43 This paper is in part based on a Cochrane Review published in The Cochrane Library 2010, Issue 2 (see http://www.thecochranelibrary.com for information).43 Cochrane Reviews are regularly updated as new evidence emerges and in response to feedback, and The Cochrane Library should be consulted for the most recent version of the review.
Disclosure
Dr Paul Maddison received a single honorarium for consultancy work from BioMarin. The authors report no other conflicts of interest in this work.
References
Lambert EH, Eaton LM, Rooke ED. Defect of neuromuscular conduction associated with malignant neoplasms. Am J Physiol. 1956;187:612–613. | |
Lambert EH, Elmqvist D. Quantal components of end-plate potentials in the myasthenic syndrome. Ann N Y Acad Sci. 1971;183:183–199. | |
O’Neill JH, Murray NMF, Newsom-Davis J. The Lambert-Eaton myasthenic syndrome. A review of 50 cases. Brain. 1988;111: 577–596. | |
Wirtz PW, van Dijk JG, van Doorn PA, et al. The epidemiology of the Lambert-Eaton myasthenic syndrome in The Netherlands. Neurology. 2004;63:397–398. | |
Lang B, Newsom-Davis J, Wray D, Vincent A, Murray N. Autoimmune aetiology for myasthenic (Eaton-Lambert) syndrome. Lancet. 1981;2(8240):224–226. | |
Lennon VA, Kryzer TJ, Griesmann GE, et al. Calcium-channel antibodies in the Lambert-Eaton syndrome and other paraneoplastic syndromes. N Engl J Med. 1995;332:1467–1474. | |
Motomura M, Johnston I, Lang B, Vincent A, Newsom-Davis J. An improved diagnostic assay for Lambert-Eaton myasthenic syndrome. J Neurol Neurosurg Psychiatry. 1995;58:85–87. | |
Lambert EH. Defects of neuromuscular transmission in syndromes other than myasthenia gravis. Ann N Y Acad Sci. 1966;135:367–384. | |
Oh SJ, Kim KW. Guanidine hydrochloride in the Eaton-Lambert syndrome. Electrophysiologic improvement. Neurology. 1973;23: 1084–1090. | |
Blumhardt LD, Joekes AM, Marshall J, Philalithis PE. Guanidine treatment and impaired renal function in the Eaton-Lambert syndrome. Br Med J. 1977;1(6066):946–947. | |
Oh SJ, Kim DS, Head TC, Claussen GC. Low-dose guanidine and pyridostigmine: relatively safe and effective long-term symptomatic therapy in Lambert-Eaton myasthenic syndrome. Muscle Nerve. 1997;20:1146–1152. | |
Lundh H. Effects of 4-aminopyridine on neuromuscular transmission. Brain Res. 1978;153:307–318. | |
Lundh H, Nilsson O, Rosén I. 4-Aminopyridine – a new drug tested in the treatment of Eaton-Lambert syndrome. J Neurol Neurosurg Psychiatry. 1977;40:1109–1112. | |
Agoston S, van Weerden T, Westra P, Broekert A. Effects of 4-aminopyridine in Eaton Lambert Syndrome. Br J Anaesth. 1978;50: 383–385. | |
Murray NM, Newsom-Davis J. Treatment with oral 4-aminopyridine in disorders of neuromuscular transmission. Neurology. 1981;31: 265–271. | |
Lemeignan M. Pharmacological approach to the study of convulsive action mechanism of amino-4 pyridine. Therapie. 1971;26:927–940. French. | |
European Medicines Agency. Summary of Product Characteristics. Firdapse® (amifampridine). Date of Preparation of Text Jan 2010. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/001032/WC500069915.pdf. Accessed December 9, 2013. | |
Molgó J, Lundh H, Thesleff S. Potency of 3,4-diaminopyridine and 4-aminopyridine on mammalian neuromuscular transmission and the effect of pH changes. Eur J Pharmacol. 1980;61:25–34. | |
Lechat P, Deysson G, Lemeignan M, Adolphe M. [Comparison of acute toxicity of some aminopyridines in vivo (mice) and in vitro (tissue culture)]. Ann Pharm Fr. 1968;26:345–349. French. | |
Lemeignan M, Millart H, Letteron N, et al. The ability of 4-aminopyridine and 3,4-diaminopyridine to cross the blood-brain barrier can account for their difference in toxicity. In: Lechat P, Thesleff S, Bowman WC, editors. Aminopyridines and Similarly Acting Drugs: Effects on Nerves, Muscles And Synapses. Advances in the Biosciences. Oxford: Pergamon Press; 1982;35:222–229. | |
Lundh H, Nilsson O, Rosén I. Novel drug of choice in Eaton-Lambert syndrome. J Neurol Neurosurg Psychiatry. 1983;46:684–685. | |
Lundh H, Nilsson O, Rosén I, Johansson S. Practical aspects of 3,4-diaminopyridine treatment of the Lambert-Eaton myasthenic syndrome. Acta Neurol Scand. 1993;88:136–140. | |
AAEM Quality Assurance Committee, American Association of Electrodiagnostic Medicine. Practice parameter for repetitive nerve stimulation and single fiber EMG evaluation of adults with suspected myasthenia gravis or Lambert-Eaton myasthenic syndrome: summary statement. Muscle Nerve. 2001;24:1236–1238. | |
AAEM Quality Assurance Committee, American Association of Electrodiagnostic Medicine. Literature review of the usefulness of repetitive nerve stimulation and single fiber EMG in the electrodiagnostic evaluation of patients with suspected myasthenia gravis or Lambert-Eaton myasthenic syndrome. Muscle Nerve. 2001;24:1239–1247. | |
Oh SJ, Kurokawa K, Claussen GC, Ryan HF Jr. Electrophysiological diagnostic criteria of Lambert-Eaton myasthenic syndrome. Muscle Nerve. 2005;32:515–520. | |
Besinger UA, Toyka KV, Hömberg M, Heininger K, Hohlfeld R, Fateh-Moghadam A. Myasthenia gravis: long-term correlation of binding and bungarotoxin blocking antibodies against acetylcholine receptors with changes in disease severity. Neurology. 1983;33:1316–1321. | |
Tindall RS, Rollins JA, Phillips JT, Greenlee RG, Wells L, Belendiuk G. Preliminary results of a double-blind, randomized, placebo-controlled trial of cyclosporine in myasthenia gravis. N Engl J Med. 1987;316: 719–724. | |
Barohn RJ, McIntire D, Herbelin L, Wolfe GI, Nations S, Bryan WW. Reliability testing of the quantitative myasthenia gravis score. Ann N Y Acad Sci. 1998;841:769–772. | |
Higgins JPT, Altman DG. Chapter 8: Assessing risk of bias in included studies. In: Higgins JPT, Green S, editors. Cochrane Handbook for Systematic Reviews of Interventions Version 5.0.2. Chichester: John Wiley & Sons; 2009. Chapter 8. | |
McEvoy KM, Windebank AJ, Daube JR, Low PA. 3,4-Diaminopyridine in the treatment of Lambert-Eaton myasthenic syndrome. N Engl J Med. 1989;321:1567–1571. | |
Wirtz PW, Verschuuren JJ, van Dijk JG, et al. Efficacy of 3,4-diaminopyridine and pyridostigmine in the treatment of Lambert-Eaton myasthenic syndrome: a randomized, double-blind, placebo-controlled, cross-over study. Clin Pharmacol Ther. 2009;86:44–48. | |
Oh SJ, Claussen GG, Hatanaka Y, Morgan MB. 3,4-diaminopyridine is more effective than placebo in a randomized, double-blind, cross-over drug study in LEMS. Muscle Nerve. 2009;40:795–800. | |
Deeks JJ, Higgins JPT, Altman DG. Chapter 9: Analysing data and undertaking meta-analyses. In: Higgins JPT, Green S, editors. Cochrane Handbook for Systematic Reviews of Interventions Version 5.0.2. Chichester: John Wiley & Sons; 2009. Chapter 9. | |
Sanders DB, Massey JM, Sanders LL, Edwards LJ. A randomized trial of 3,4-diaminopyridine in Lambert-Eaton myasthenic syndrome. Neurology. 2000;54:603–607. | |
Boerma CE, Rommes JH, van Leeuwen RB, Bakker J. Cardiac arrest following an iatrogenic 3,4-diaminopyridine intoxication in a patient with Lambert-Eaton myasthenic syndrome. J Toxicol Clin Toxicol. 1995;33:249–251. | |
Hawkes N, Cohen D. What makes an orphan drug? BMJ. 2010;341:c6459. | |
Cheung RY, Cohen JC, Illingworth P. Orphan drug policies: implications for the United States, Canada, and developing countries. Health Law J. 2004;12:183–200. | |
European Medicines Agency. Orphan medicinal product designation. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Brochure/2011/03/WC500104234.pdf. Accessed December 9, 2013. | |
Ferner RE, Hughes DA. The problem of orphan drugs. BMJ. 2010;341:c6456. | |
Palace J, Newsom-Davis J, Lecky B. A randomized double-blind trial of prednisolone alone or with azathioprine in myasthenia gravis. Myasthenia Gravis Study Group. Neurology. 1998;50:1778–1783. | |
Maddison P, Lang B, Mills K, Newsom-Davis J. Long term outcome in Lambert-Eaton myasthenic syndrome without lung cancer. J Neurol Neurosurg Psychiatry. 2001;70:212–217. | |
Maddison P, McConville J, Farrugia ME, et al. The use of rituximab in myasthenia gravis and Lambert-Eaton myasthenic syndrome. J Neurol Neurosurg Psychiatry. 2011;82(6):671–673. | |
Keogh M, Sedehizadeh S, Maddison P. Treatment for Lambert-Eaton myasthenic syndrome. Cochrane Database Syst Rev. 2011;2:CD003279. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.