Back to Journals » Drug Design, Development and Therapy » Volume 15
Pristimerin Protects Against OVX-Mediated Bone Loss by Attenuating Osteoclast Formation and Activity via Inhibition of RANKL-Mediated Activation of NF-κB and ERK Signaling Pathways
Authors Li X, Lin X, Wu Z, Su Y, Liang J, Chen R, Yang X, Hou L, Zhao J, Liu Q, Xu F
Received 24 September 2020
Accepted for publication 2 December 2020
Published 7 January 2021 Volume 2021:15 Pages 61—74
DOI https://doi.org/10.2147/DDDT.S283694
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Jianbo Sun
Xuedong Li,1,2,* Xixi Lin,1,2,* Zuoxing Wu,3 Yuangang Su,1,2 Jiamin Liang,1,2 Runfeng Chen,1,2 Xue Yang,1,2 Lei Hou,4 Jinmin Zhao,1,5 Qian Liu,5 Feng Xu1,2,6
1Guangxi Key Laboratory of Regenerative Medicine, Guangxi Medical University, Nanning, Guangxi 530021, People’s Republic of China; 2Guangxi Collaborative Innovation Center for Biomedicine, Guangxi Medical University, Nanning, Guangxi 530021, People’s Republic of China; 3School of Medicine, Xiamen University, Xiamen, Fujian 361102, People’s Republic of China; 4Department of Cardiology, Tongren Hospital, Shanghai Jiaotong University School of Medicine, Shanghai, People’s Republic of China; 5Research Centre for Regenerative Medicine, Orthopaedic Department, The First Affiliated Hospital of Guangxi Medical University, Nanning, Guangxi 530021, People’s Republic of China; 6Department of Subject Planning Shanghai, Ninth People’s Hospital Shanghai, Jiaotong University School of Medicine, Shanghai 200011, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Feng Xu
Department of Subject Planning Shanghai, Ninth People’s Hospital Shanghai, Jiaotong University School of Medicine, Shanghai, People’s Republic of China
Email [email protected]
Qian Liu
Research Centre for Regenerative Medicine, Orthopaedic Department, The First Affiliated Hospital of Guangxi Medical University, Nanning, Guangxi 530021, People’s Republic of China
Email [email protected]
Introduction: Osteoporosis is an osteolytic bone condition characterized by decreased bone strength and increased bone fragility. It is the result of elevated formation or activity of bone-resorbing osteoclasts. Although current therapeutic agents are efficacious against osteoclast-mediated bone loss, detrimental side effects preclude the long-term use of these agents. Pristimerin (PRI) is a naturally occurring quinone-methide triterpenoid that has been revealed to exert anti-inflammatory and anti-tumor effects via regulating various signaling cascades including NF-κB and MAPK activation.
Methods: The bone marrow macrophages were used to confirm the anti-osteoclastic and anti-resorptive functions of PRI in vitro. An in vivo ovariectomy (OVX) model was applied to verify the function of PRI protecting bone loss.
Results: PRI abolished the early activation of NF-κB and ERK MAPK signal cascades thereby thwarting the downstream expression of c-Fos and NFATc1, which prevented the production of mature osteoclasts. In vivo, PRI protects mice against ovariectomy (OVX)-mediated bone loss by diminishing osteoclast formation and bone resorptive activity.
Conclusion: Our study shows that PRI demonstrates therapeutic potential in the effective treatment against osteoclast-induced osteolytic diseases like osteoporosis.
Keywords: osteoporosis, osteoclast, pristimerin, ERK, NF-κB
Introduction
Bone tissue needs to undergo constant remodeling. The remodeling process is balanced in the alternating work of bone-resorbing and bone-forming.1 Thus, the activities of osteoclasts and osteoblasts are tightly coupled to maintain bone homeostasis. However, the main reason of osteolytic bone diseases including post-menopausal osteoporosis is owing to upgrading osteoclast formation or excessively activating osteoclast bone resorption shifting the balance to net bone loss.2 The pathological manifestations of excessive osteoclast-mediated bone resorption including thinning of bones, decreasing in bone mass and bone mineral density leading to overall exacerbation in bone microstructures and increased risks of bone fractures characterizes post-menopausal osteoporosis.3 Fractures resulting from osteoporotic bone loss is a global health and socioeconomic problem with plentiful bone-associated diseases, and raised mortality and medical costs. Although currently available anti-osteoporotic drugs are effective in alleviating bone loss, the long-term use of such agents are associated with severe side-effects such as ocular, renal and gastrointestinal toxicities, atypical fractures, and osteonecrosis of the jaw.4,5 The deep understanding of key pathways regulating osteoclastogenesis has enabled the discovery of new novel approaches for treating osteolytic diseases with distinctive mechanisms of action.
Multinucleated osteoclasts form through mononuclear precursor cells of the monocyte/macrophage lineage fused under the action of two vital cytokines, receptor activator of nuclear factor-κB (NF-κB) ligand (RANKL) and macrophage-colony stimulating factor (M-CSF).6,7 M-CSF functions can regulate proliferation and survival of monocytic precursor cells, besides upregulate the expression of surface RANK, the homologous receptor for RANKL. In the form of a surface protein or secretory factor osteoblasts and stromal cells produce RANKL, a member of the tumor necrosis factor (TNF) superfamily.8 The interaction between RANKL and RANK initiates a whole train of signaling events via intracellular adaptor proteins and TNF receptor-associated factor (TRAF) proteins. Mitogen-activated protein kinase (MAPK) and NF-κB signaling pathways early activation can lead to robust induction of nuclear factor of activated T-cells 1 (NFATc1), which is a vital osteoclast transcription factor.9 NFATc1 is regarded as defining transcription factor essential to drive osteoclast generation and bone resorption.10 The production of numerous crucial osteoclast genes touched on precursor cell fusion is regulated by NFATc1 transcription for instance, dendritic cell-specific transmembrane protein (Dc-stamp), and proteolytic enzymes involved in osteoclast resorption such as tartrate resistant acid phosphatase (Trap/Acp5), cathepsin K (Ctsk), and matrix metalloproteinase-9 (Mmp-9).11
Pristimerin (PRI) is a natural quinone-methide triterpenoid compound extracted from plants of the Celastraceae and Hippocrateaceae family and exerts multiple pharmacological activities with its anti-inflammatory12 and anti-cancer properties13 the most studied. It has been demonstrated that PRI induces apoptosis via the Akt/NF-κB and ROS/MAPK signal pathways.14 Interestingly, PRI have also been shown to exert protective effects against both arthritic inflammation, and cartilage and bone damage in the joints by modulating expression of RANKL expression and other pro-inflammatory cytokines.15 Given these promising effects, we hypothesized that PRI could potentially exert inhibitory effects against RANKL-induced osteoclastogenesis or bone resorptive function. In this study, we used complementary in vitro cellular and biochemical assays to check up the roles and molecular mechanism of PRI against RANKL-induced osteoclast differentiation, and mature osteoclast resorption function. We further examined potential therapeutic effects of PRI in vivo on bone loss caused by ovariectomy (OVX) in mice. The murine OVX-induced bone loss model is a relatively perfect model that closely mimics the conditions of post-menopausal osteoporosis and often used for investigating the biological effects of pharmacological agents.16
Materials and Methods
Regent and Media
Pristimerin (PRI, purity of 98%) was got from Chengdu Biopurify Phytochemicals Ltd (Chengdu, Sichuan, China). PRI was dissolved in dimethyl sulfoxide (DMSO) to stock concentration of 20 mmol/L and further diluted to desired working concentrations using culture media. Fetal bovine serum (FBS), Gibco Minimum Essential Medium Alpha Modification (α-MEM) and penicillin/streptomycin were obtained from Thermo Fisher Scientific (Scoresby, Victoria, Australia). Complete α-MEM for cultivation contained 10% FBS and 1% penicillin/streptomycin. R&D Systems (Minneapolis, MN, USA) supplied recombinant mouse GST-rRANKL (RANKL) and macrophage-colony stimulating factor (M-CSF). Cell Signaling Technology (Danvers, MA, USA) supplied primary antibodies against p38 (#8690), p-p38 (#4511), p-JNK (#4668), JNK (#9252), p-ERK (#4370), ERK (#4695), and NF-κB p65 (#8242), phospho-NF-κB p65 (#3033), β-actin (#4970), IĸBα (#4814). NFATc1 (#sc-7294) was from Santa Cruz Biotechnology (Dallas, TX, USA). Primary antibodies against c-Fos (#ab134122) were from Abcam (Cambridge, UK). MedChemExpress LLC (Monmouth Junction, NJ, USA) provided cell counting kit-8 (CCK-8). Rhodamine-conjugated phalloidin and 4′,6-diamidino-2-phenylindole (DAPI) were gained from Molecular Probes (Eugene, OR, USA) and Sigma-Aldrich (St Louis, MO, USA) respectively.
Cells Culture and Osteoclast Differentiation Assay
Primary bone marrow macrophages (BMMs) were extracted from the femur and tibia of 6-week-old C57BL/6 mice by marrow lavage. Extracted BMMs grew in complete α-MEM added 30 ng/mL M-CSF at 37°C under the condition of 95% air and 5% CO2. Replenish the culture media every 2 days to clear away non-adherent cells and after 4 days adherent cells were considered BMMs that were suitable for subsequent applications.
To generate mature osteoclasts, BMMs were cultured with complete α-MEM added M-CSF in 96-well plates at a cell number of 8×103 per well and permitted to adhere to the culture plates overnight. Next day BMMs were induced by 50 ng/mL RANKL in the absence or presence of indicated concentrations of PRI (0.3125, 0.625, or 1.25 μM) for 6 days. To further explore which stage of osteoclast differentiation was affected by PRI, cells were induced by RANKL with or without PRI with a concentration of 1.25 μM on day 1–3 (early stage), day 3–5 (middle stage), or day 5–6 (late stage) of osteoclast formation. Cells cultured with RANKL without PRI throughout the 6-day period were used as positive controls. Culture media supplemented with RANKL and PRI were changed every two days. The cells were fixed with 4% paraformaldehyde (PFA) for 30 mins and next tartrate-resistant acid phosphatase (TRAP) staining was implemented. Phase contrast images were captured by light microscope and TRAP-positive cells more than 3 nuclei were recorded as osteoclasts.
Cell Viability/Cytotoxicity Assay
To estimate the viability of BMMs under PRI conditions, the cell counting kit-8 (CCK-8) was used. Briefly, BMMs uniformly planted with a number of 8×103 cells/well in 96-well plates were treated by diverse concentrations of PRI (0.3125, 0.625 and 1.25 μM) for 48 hours. At last, 10 μL CCK-8 was applied to culture cells for further 2 hours and the absorbance at 450 nm was detected using a TriStar2 LB 942 Multimode Microplate Reader (Berthold Technologies Gmbh & Co.KG, Baden-Württemberg, Germany).
Podosomal Actin Belt Staining
To investigate the effects of PRI on osteoclast actin cytoskeleton, BMM-derived osteoclasts were produced as described above under different concentrations of PRI (0, 0.3125, 0.625, or 1.25 μM). After 6 days of osteoclastogenesis, mature multinucleated osteoclasts were fixed in 4% PFA around 15 mins, and then permeated in phosphate-buffered saline (PBS) with 0.1% Triton X-100 about 5 min. After washing with PBS several times, cells were blocked by 3% bovine serum albumin (BSA)-PBS nearly 30 min and next incubated at 4°C with Rhodamine-conjugated phalloidin in 0.2% BSA-PBS for an hour without light. Next, DAPI was used to counterstain the cell nuclei nearly 5 min. Fluorescence images were gained from a Cytation 5 and analysed using associated Gen5 Data Analysis Software (BioTek Instruments Inc, Winooski, VT, USA).
Bone Resorption Assay
To examine anti-resorptive effects of PRI on osteoclasts, BMMs cultured in 6-well plates at a number of 1×105 cells/well were induced by 50 ng/mL RANKL for 3 days until small multinucleated cells were formed. Cells were then dissociated from the wells with trypsin and then equal numbers of pre-osteoclasts were reseeded into 96-well plates coated with bone-mimicking hydroxyapatite matrix. Pre-osteoclasts cultured with 50 ng/mL RANKL and added to various concentrations of PRI (0, 0.3125, 0.625, or 1.25 μM) for further 3 days. After experiment, cells were cleared away using sodium hypochlorite solution and the resorption region visualized under a light microscope. Using ImageJ software (NIH, Bethesda, MD, USA) to quantify bone resorption area under each treatment condition.
RNA Obtainment and Real-Time Quantitative PCR (qPCR)
Osteoclast marker genes expression was assessed by quantitative real-time PCR (qPCR) analysis. Total RNA was collected using TRIzol reagent (Thermo Fisher Scientific) from BMM-derived osteoclasts cultured with 50 ng/mL RANKL without or with PRI (0.625 or 1.25 μM) for 6 days. First strand synthesis was then performed through the RevertAid First Strand cDNA Synthesis Kit (Thermo Fisher Scientific) on the basis of manufacturer’s statement to generate complementary DNAs (cDNA). Real-time qPCR was implemented on a LightCycler® 96 System (Roche, Basel, Switzerland) in reaction mixtures containing FastStart Essential DNA Green Master Mix (Roche), cDNA template and specific primers of interest. The PCR amplification programs were the following: initial denaturation at 95°C for 10 min, followed by 35 cycles at 95°C for 10 sec, 60°C for 15 sec and 72°C for 10 sec. The 2−ΔΔCt method was employed to standardize the expression of objective genes to β-actin housekeeping gene expression. Specific primer sequences against target genes are as follows: Ctsk (Forward: 5ʹ-AGGCGGCTATATGACCACTG-3ʹ; Reverse: 5ʹ-TCTTCAGGGCTTTCTCGTTC-3ʹ), Trap/Acp5 (Forward: 5ʹ-ACGGCTACTTGCGGTTTCA-3ʹ; Reverse: 5ʹ-TCCTTGGGAGGCTGGTCTT-3ʹ), Mmp-9 (Forward: 5ʹ-GAAGGCAAACCCTGTGTGTT-3ʹ; Reverse: 5ʹ-AGAGTACTGCTTGCCCAGGA-3ʹ), Dc-stamp (Forward: 5ʹ-TCTGCTGTATCGGCTCATCTC-3ʹ; Reverse: 5ʹ-ACTCCTTGGGTTCCTTGCTT-3ʹ), and β-actin (Forward: 5ʹ-TCCTCCCTGGAGAAGAGCTA-3ʹ; Reverse: 5ʹ-ATCTCCTTCTGCATCCTGTC-3ʹ).
Protein Obtainment and Western Blotting
To assess the roles of PRI during early RANKL-mediated signaling events, BMMs were cultured in a 6-well plate at 1×106 cells/well overnight, and serum-starved for 3 hours, next added to 1.25 μM PRI for 1 hour, and then cultured with 50 ng/mL RANKL for 5, 10, 20, 30, and 60 min. To evaluate the roles of PRI on late RANKL-stimulated signaling cascades, BMMs cultured in 6-well plates at number of 1×105 cells/well in complete α-MEM were incubated by 50 ng/mL RANKL without or with 1.25 μM PRI 1, 3, and 5 days. Unstimulated BMMs were used as mock controls (time point 0). After the experimental period, total cellular protein was obtained by using RIPA lysis buffer added with 1% (v/v) phosphatase inhibitor, 1% (v/v) Phenylmethylsulfonyl fluoride (PMSF) and 1% (v/v) protease inhibitor. Protein lysates were centrifugated at 15,000 rpm for 10 min and the supernatant containing proteins were gathered and subjected to protein quantification. The same amounts of protein from each treatment condition were separated on 10–12% sodium dodecyl sulfatepolyacrylamide gel electrophoresis (SDS-PAGE) gels and electrotransfer to nitrocellulose membranes. Nitrocellulose membranes were blocked in tris buffered saline with tween (TBST) added with 5% (w/v) skim milk around 1 hour at 37°C and next incubated overnight with specific primary antibodies diluted in TBST containing 1% (w/v) skim-milk at 4°C. TBST was employed to wash away free primary antibodies and the membranes were then incubated with suitable secondary antibodies for imaging by using an Odyssey Sa Infrared Imaging System (LI-COR Biosciences, Lincoln, NE, USA). The ImageJ software was employed to quantify the optical density of protein band intensity based on gray scale image.
Immunofluorescence Staining of NF-κB P65 and NFATc1 Nuclear Translocation
The nuclear localization of NF-κB p65 and NFATc1 in response to RANKL stimulation was examined by confocal fluorescence microscopy. For NF-κB p65 nuclear translocation, BMMs were planted onto microscopy dishes overnight and serum-free medium starved cells for 3 hours, then pre-stimulated without or with 1.25 μM PRI for 1 hour, and next cultured with 50 ng/mL RANKL for 30 min. For NFATc1 nuclear localization, BMMs were cultured with 50 ng/mL RANKL without or with 1.25 μM PRI for 5 days. Afterwards, cells were fixed 15 mins and then permeabilized using 0.1% Triton X-100 in PBS about 5 min. Non-specific immuno-reactivities were blocked with 3% BSA-PBS for 30 min. After extensive washes with PBS, cells were incubated with specific antibodies against NF-κB p65 (sc-8008 PE; Santa Cruz Biotechnology, Santa Cruz, CA) or NFATc1 (sc-7294 AF488; Santa Cruz Biotechnology, Santa Cruz, CA) at 4°C overnight. Cells were then incubated with Alexa Fluor 552 (NF-κB p65) or Alexa Fluor 488 (NFATc1) secondary antibodies for 1 hour in the dark. Cell nuclei were counterstained using DAPI and fluorescence images acquired under a confocal microscopy (Leica, Germany).
Ovariectomy (OVX)-Induced Osteoporosis Model
The mice model of osteoporosis induced by OVX was applied to determine the in vivo improving efficacy of PRI against post-menopausal bone mass loss. Both of OVX model and subsequent treatment conditions were received approval from Ethics Review Committee of Guangxi Medical University (Guangxi, China). The surgical and experimental programs complied with the orientations set by the Animal Protection Committee of Guangxi Medical University. The Animal Experimental Center of Guangxi Medical University offered twenty-four healthy female C57BL/6 mice (12 weeks old) and were randomly assigned to 3 groups (n = 8 mice per group): Sham-operated group (saline injection), OVX group (saline injection), and OVX + PRI group (5 mg/kg bodyweight). Mice were anaesthetized with tribromoethanol and experienced bilateral ovariectomy (OVX groups) or received sham operation. All mice received intraperitoneal injections of penicillin to minimize post-operative infections. One-week post-surgery, mice received intraperitoneal injections of either PRI in saline (OVX + PRI group) or normal saline (Sham and OVX groups) once every two days for 6-weeks. After treatment period, all mice were euthanized and the tibial bones were removed and dealt with micro-computed tomography (CT) and histological analyses.
Micro-CT Scanning and Histological Analyses
The extracted tibial tissues were fixed in 4% PFA for 24 hours and carried through high-resolution scanning on a Bruker micro-CT system. Scanning was conducted at a current of 142 μA and voltage of 70 kV and the isometric pixel size was 10 μm. Three-dimensional (3D) reconstructions and bone morphometric analyses were carried out using CTAn software. A square region of interest set at 0.5 mm below the growth plate was applied to quantitatively analyze bone parameters which included bone mineral density (BMD, mg/cm2), connectivity density (Conn.Dn, 1/mm3), trabecular number (Tb.N, 1/mm), trabecular separation (Tb.Sp, mm), trabecular thickness (Tb.Th, mm), and percentage bone volume per tissue volume (BV/TV, %).
Histological assessments of fixed tibial bones were carried out following decalcification in 10% EDTA at 4°C for 1 week. Paraffin blocks were employed to embed bone tissues and next section them into 5 μm thick sections. Bone sections were performed staining by hematoxylin and eosin (H&E) and TRAP. Bone histomorphometry was performed in a blinded, unbiased manner, and the amount of TRAP-positive multinucleated osteoclasts was assessed in each group.
Statistical Analyses
Results submitted in this work are indicated as mean ± standard deviation (SD) or representative images from three or more independent experimental repeats. Statistical differences were based on Student’s t-test or analysis of variance (ANOVA) analysis with GraphPad Prism software version 7.0 (GraphPad Software, San Diego, CA, USA). A p-values <0.05 or unless otherwise stated were regarded statistically significant.
Results
PRI Inhibits RANKL-Mediated Osteoclastogenesis in vitro
First of all, we assessed the role of PRI (molecular structure presented in Figure 1A) on BMMs cell proliferation using the CCK-8 assay. As shown in Figure 1B, no toxicity was tested when cells were cultured with up to 1.25 μM PRI for 48 hours. Given that on average, 5–6 days of RANKL stimulation is required to induce osteoclast formation, PRI concentrations of 0.3125, 0.625 and 1.25 μM were selected for downstream investigations. Afterwards, we evaluated the roles of PRI in RANKL stimulating osteoclasts generation process. As demonstrated in Figure 1C, BMMs treated with RANKL only formed large well-spread TRAP-positive multinucleated osteoclasts. On the other hand, a downward trend was showed in the amount of TRAP-positive multinucleated osteoclasts when were cultured with increasing concentration of PRI (Figure 1C-D). Cells treated with 1.25 μM PRI were predominantly TRAP-positive mononuclear cells (Figure 1C).
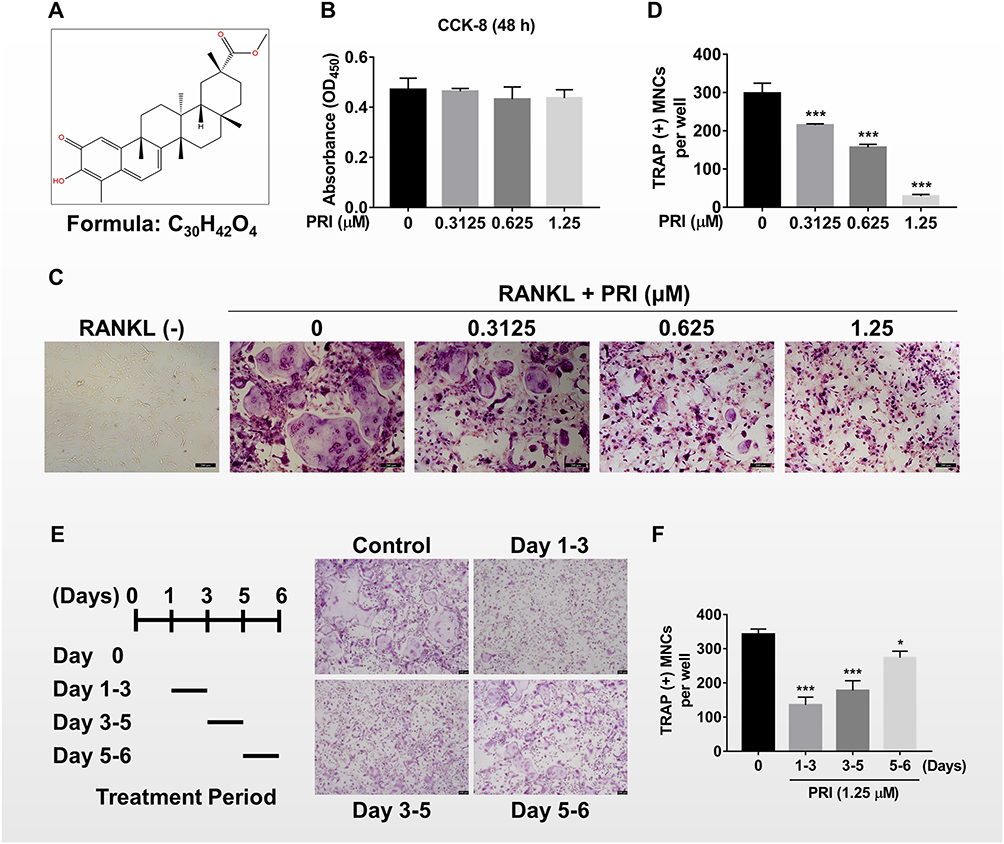 |
Figure 1 PRI inhibits RANKL-induced osteoclast formation in vitro. (A) Chemical structure and molecular formula of Pristimerin (PRI). (B) The cellular cytotoxicity of indicated concentrations of PRI on bone marrow macrophages (BMMs) after 48 hours of treatment as assessed by the CCK-8 assay. (C) Representative TRAP stained phase-contrast images of the dose-dependent effect of PRI against BMM-derived osteoclast formation following stimulation with 50 ng/mL RANKL for 6 days (scale bar = 200 µm). (D) The number of TRAP-positive multinucleated osteoclasts with 3 or more nuclei were counted. (E) Representative TRAP stained phase-contrast images of the time-dependent effect PRI on osteoclast formation. BMMs stimulated with RANKL and treated with 1.25 μM PRI on the indicated days were fixed and stained for TRAP activity (scale bar = 100 μm). (F) The number of TRAP-positive multinucleated osteoclasts with 3 or more nuclei per time period were counted. Bar graphs presented as the mean ± SD of at least three independent experiments; *p < 0.05 and ***p < 0.001 relative to the RANKL-only controls. All experiments are repeated 3 times. |
To further establish the stage of osteoclast formation whereby PRI exerts anti-osteoclastic effects, RANKL-stimulated BMMs were cultured with 1.25 μM PRI for a specified number of days during culture period (Figure 1E). Compared to BMMs stimulated with RANKL alone, a time-dependent effect was noticed when PRI was employed to treat with BMMs and the strongest inhibitory effect was presented when PRI was present on day 1 to day 3 (Figure 1E and F). When cells were exposed to PRI on the last two days of osteoclast formation (days 5 and 6) a modest but the TRAP-positive osteoclasts number was significantly reduced (Figure 1F). When cells were treated with PRI during the middle stages of osteoclast formation (day 3–5), the effect was between early and late treatments (Figure 1E and F). These results suggest that PRI predominantly inhibits the ability of BMM precursor cell to differentiate and fuse in response to RANKL stimulation.
PRI Exhibits Anti-Resorption Effect in vitro
The formation of podosomal actin belts and subsequently dense F-actin rings during bone resorption is critical for osteoclast function.17 To determine the influence of PRI about the formation and morphology of the podosomal actin belt, we stained BMM-derived osteoclasts treated in the absence or presence of PRI with Rhodamine-conjugated phalloidin. Consistent with inhibition of osteoclast formation, treatment with PRI also dose-dependently inhibited the actin belt formation (Figure 2A). We also observed significant decrease in the size of podosomal actin belt which correlated with reduced nucleation further indicating that PRI inhibited BMM precursor cell fusion during the early stages of osteoclast formation. This is particularly apparent following treatment with 1.25 μM PRI where cells are predominantly mononuclear in nature with no podosomal actin belts (Figure 2A).
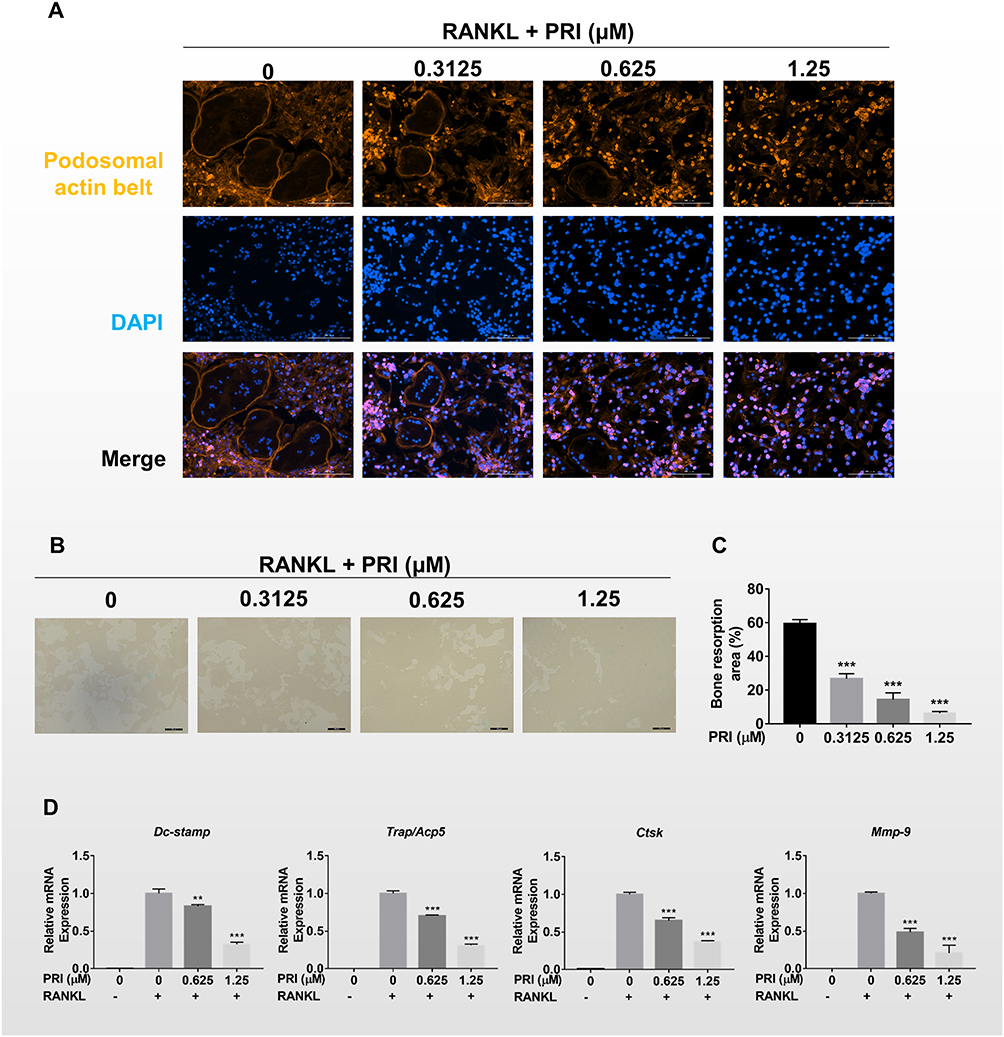 |
Figure 2 PRI impairs podosomal actin belt formation and mature osteoclast bone resorptive activity in vitro. (A) Representative immunofluorescence images of podosomal actin belt (rhodamine-conjugated phalloidin; red) from BMM-derived osteoclasts stimulated with 50 ng/mL RANKL without or with indicated concentrations of PRI. Cell nuclei were counterstained with DAPI (blue). (B) Representative phase-contrast images of the effect of PRI on mature osteoclast bone resorption. Equal numbers of pre-osteoclasts stimulated with 50 ng/mL RANKL for at least 3 days were seeded onto hydroxyapatite-coated OsteoAssay plates and then treated with indicated concentrations of PRI for further 3 days (scale bars = 200 μm). (C) The percentage of bone resorption area relative to total well area at each PRI concentration was quantified using ImageJ. (D) PRI dose-dependently suppressed RANKL-induced upregulation of osteoclast marker gene expression. Real-time PCR was performed on RNA extracted from cells stimulated with 50 ng/mL RANKL without or with indicated concentrations of PRI for 6 days. The mRNA expression levels of Dc-stamp, Trap/Acp5, Ctsk, and Mmp-9 genes were normalized to β-actin using the 2−ΔΔCt method and then displayed as fold change relative to RANKL-only control. Bar graph presented as mean ± SD of at least 3 independent experiment; **p < 0.01 and ***p < 0.001 relative to RANKL-only control. |
Functionally, the only cells that can resorb mineralized bone matrix are osteoclasts. Thus, we test the role of PRI about mature osteoclast bone resorptive function. To this end, pre-cultured osteoclasts around 3 days were planted on bone mimicking hydroxyapatite-coated plates and appointed concentrations of PRI was used or not for further 3 days. In addition to the anti-osteoclastic effect shown earlier, PRI was also found to demonstrate dose-dependent anti-resorptive effects against mature osteoclasts (Figure 2B and C). Together these results further indicate that PRI exerts anti-osteoclastic effects against BMM precursor cell fusion and anti-resorptive effects against bone resorption function of mature osteoclast.
PRI Hampers RANKL-Stimulated Upregulation of Osteoclast Marker Genes
RANKL-stimulated osteoclast formation and subsequent bone resorptive function require the upregulation of osteoclast-specific genes like Dc-stamp, Ctsk, Trap, and Mmp-9 (Figure 2D). These genes encode proteins required for precursor cell fusion (Dc-stamp) or proteolytic enzymes required for bone resorptive function (Ctsk, Trap and Mmp-9). Consistent with cellular roles on osteoclast formation and bone resorption, PRI treatment markedly attenuated these genes expression, which further cemented the dual anti-resorptive and anti-osteoclastic functions of PRI.
PRI Attenuates RANKL-Mediated Activation of NF-κB and ERK Signaling Pathways
The upregulation of osteoclast genes is controlled by the timely activation of several RANKL response signaling pathways including the NF-κB and MAPK pathways. The timely activation of these two signaling cascades is required for efficiently and effectively driving of the differentiation of BMM precursor cells towards the osteoclast lineage. To investigate the effects of PRI against NF-κB and MAPK signaling following RANKL stimulation, Western blot analyses were performed. As demonstrated in Figure 3A and B, within 5 mins of RANKL stimulation and persisting to 30 mins, the NF-κB inhibitory subunit IκBα rapidly degraded via the proteasome. IκBα prevents the nuclear transport of NF-κB subunits by masking the nuclear localization signals (NLS) of NF-κB subunits.18 As IκBα is degraded, it no longer masks the NLS of the NF-κB p65 subunit thus enabling it to be phosphorylated within 5 mins and persisting to 10 mins (Figure 3A and C) and then translocated to the nucleus (Figure 3D) where it can play a transcriptional function on target genes. In contrast, treatment with 1.25 μM PRI completely abolished RANKL-induced IκBα degradation, and NF-κB p65 phosphorylation and nuclear localization (Figure 3A-D). Thus, these data indicate that PRI can completely inhibit RANKL-induced NF-κB signaling pathway activating.
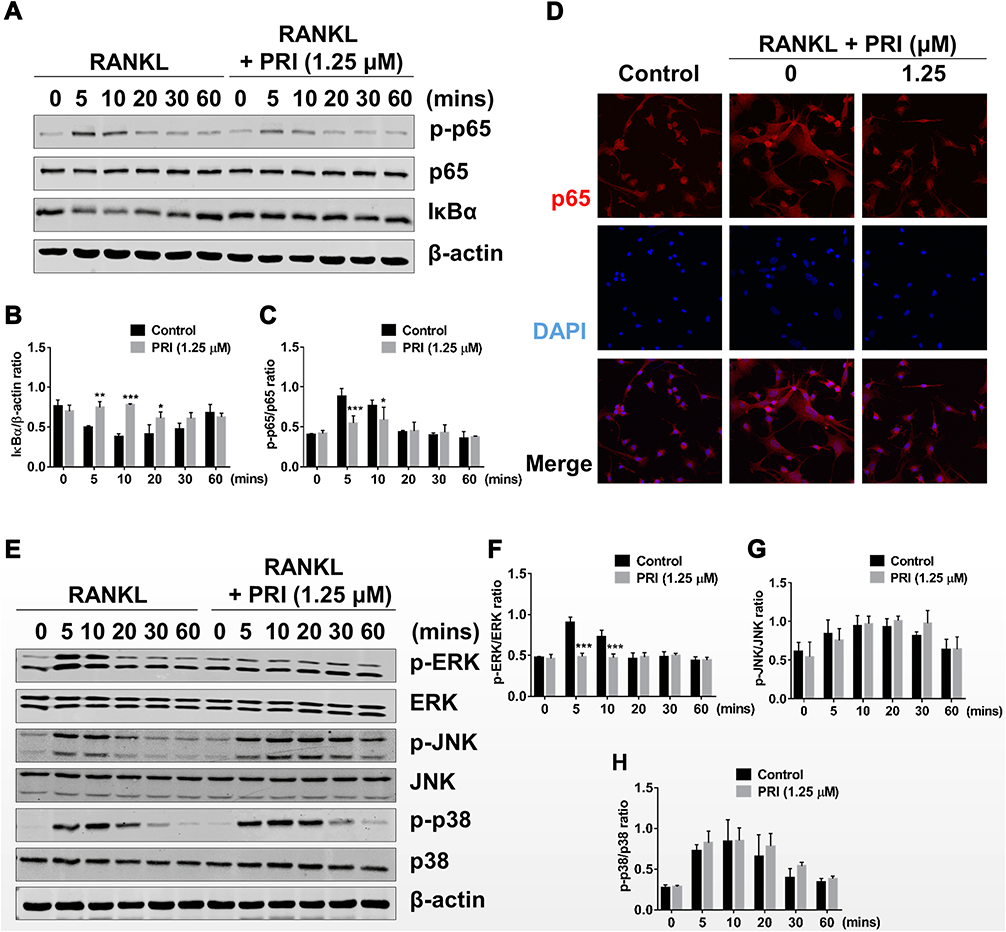 |
Figure 3 PRI abolishes RANKL-induced activation of NF-κB and ERK MAPK signaling pathways. (A) Representative immunoblot images of the effect of PRI on the early activation of NF-κB signaling in response to RANKL stimulation. Total cellular proteins were extracted from BMM pretreated with 1.25 μM PRI for 1 hour and then stimulated with 50 ng/mL RANKL for indicated times. Protein expression and phosphorylation status of p65 and IκBα were assessed using specific antibodies. β-actin was used as internal loading control. (B and C) Densitometric quantitation of protein expression of IκBα relative to β-actin, and p-p65 relative to p65, using ImageJ software. (D) Immunofluorescence images of p65 nuclear translocation following RANKL stimulation without or with 1.25 μM PRI treatment (Magnification = 40×). Cell nuclei were counterstained with DAPI. (E) Representative immunoblot images of the effect of PRI on the early activation of MAPK signaling in response to RANKL stimulation. Protein expression and phosphorylation status of ERK, JNK, and p38 were assessed using specific antibodies. β-actin was used as internal loading control. (F-H) Densitometric quantitation of protein expression of p-ERK relative to ERK, p-JNK relative to JNK, and p-p38 relative to p38 by using ImageJ software. Bar graphs presented as mean ± SD of at least 3 independent experiments; *p < 0.05, **p < 0.01, and ***p < 0.001 relative to RANKL-only control. |
The MAPK signaling pathways activating consisting of three members, ERK, JNK, and p38, is also necessary for driving osteoclast differentiation. Stimulation of BMM precursor cells with RANKL rapidly stimulates the activation phosphorylation of all three members of MAPK signaling cascade (Figure 3E-H). The ERK, JNK, and p38 phosphorylation activation occurs within 5 mins of RANKL stimulation. For ERK phosphorylation lasted for 10 mins before returning to basic levels. For JNK and p38, phosphorylation persisted for 20 mins before returning to basic levels. Nevertheless, PRI treatment only inhibited the activation phosphorylation of ERK not that of JNK or p38 (Figure 3E-H). Surprisingly, PRI treatment sustained the activation of JNK compared to RANKL stimulation alone (Figure 3E and G). Together, these results suggested that PRI can inhibit RANKL-stimulated ERK activation but at the same time potentiates RANKL-induced JNK activation.
PRI Inhibits RANKL-Induced Induction and Activation of NFATc1
The timely activation of NF-κB and MAPK signaling cascades is crucial for the efficient downstream induction of nuclear transcription factors such as c-Fos and NFATc1 (Figure 4A-C), with the latter being a direct downstream transcriptional target of c-Fos.10,19 The most important distal transcription factor, NFATc1, transcription controls many osteoclast marker genes that drive osteoclast generation, fusion and resorption.20 Consistent with ablated NF-κB and ERK signaling, the downstream expression of c-Fos and NFATc1 on day 3 was obviously hampered by PRI treatment (Figure 4A-C). Furthermore, the nuclear translocation of NFATc1 was significantly blocked following PRI treatment further attesting to impediment of NFATc1 transcriptional activity (Figure 4D). Collectively, the ablation of early NF-κB and ERK signaling by PRI effectively hampered c-Fos and NFATc1 proteins induction and transcriptional activities thereby contributing to the anti-osteoclastic effects of PRI.
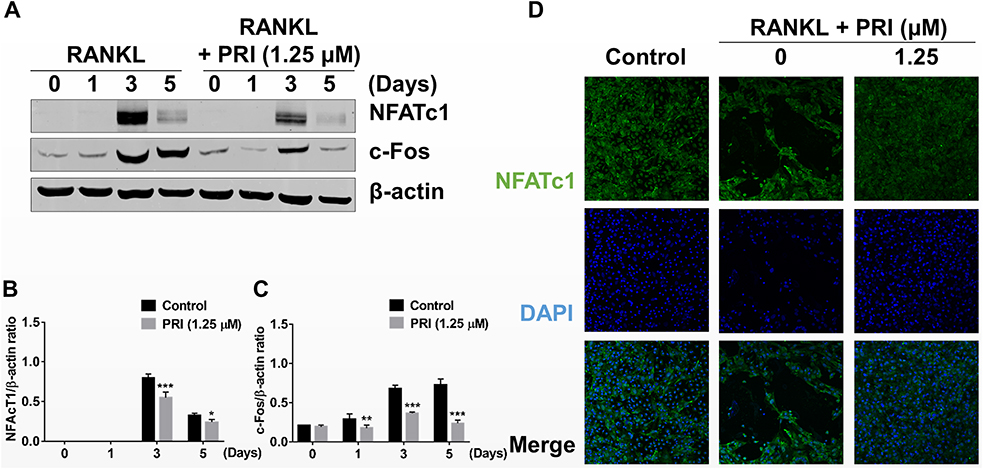 |
Figure 4 PRI thwarted RANKL-induced induction of c-Fos and NFATc1. (A) Representative immunoblot images of the effect of PRI on the downstream induction of c-Fos and NFATc1 transcription factors in response to RANKL stimulation. Total cellular proteins extracted from BMM-derived osteoclasts co-treated with 50 ng/mL RANKL and 1.25 μM PRI for 0, 1, 3 and 4 days were subjected to Western blot analyses using specific antibodies against c-Fos and NFATc1. β-actin was used as internal loading control. (B and C) Densitometric quantitation of protein expression of NFATc1 relative to β-actin, and c-Fos relative to β-actin by using ImageJ software. (D) Immunofluorescence images of NFATc1 nuclear translocation following RANKL stimulation without or with 1.25 μM PRI treatment (Magnification = 20×). Cell nuclei were counterstained with DAPI. Bar graphs presented as mean ± SD of at least 3 independent experiments; *p < 0.05, **p < 0.01, and ***p < 0.001 relative to RANKL-only control group. |
PRI Protects Mice Against Ovariectomy (OVX)-Mediated Bone Loss in vivo
We next assessed whether the encouraging in vitro anti-resorptive and anti-osteoclastic roles of PRI translates to protective effects against osteoclast-mediated bone loss caused by OVX in mice. One week after bilateral OVX or sham operation, mice were treated with intraperitoneal injections of normal saline or 5 mg/kg PRI for further 6 weeks. Compared with Sham-operated mice, three-dimensional reconstructions of high-resolution micro-CT images of excised tibial bone revealed extensive bone loss in normal saline-treated OVX mice (Figure 5A). Morphometric analysis presented significant reductions in bone volume (BV/TV; Figure 5B), trabecular number (Tb.N; Figure 5C), bone mineral density (BMD; Figure 5F) and connectivity density (Conn.Dn; Figure 5G), and increasing trend in trabecular spacing (Tb.Sp; Figure 5D), bone changes that were consistent with osteoporotic phenotype. Treatment with PRI on the other side, protected mice against OVX-induced bone loss, improving morphometric parameters of BV/TV, Tb.N, Tb.Sp, BMD and Conn.Dn (Figure 5A-G). No difference in trabecular thickness (Tb.Th; Figure 5E) was observed in both groups.
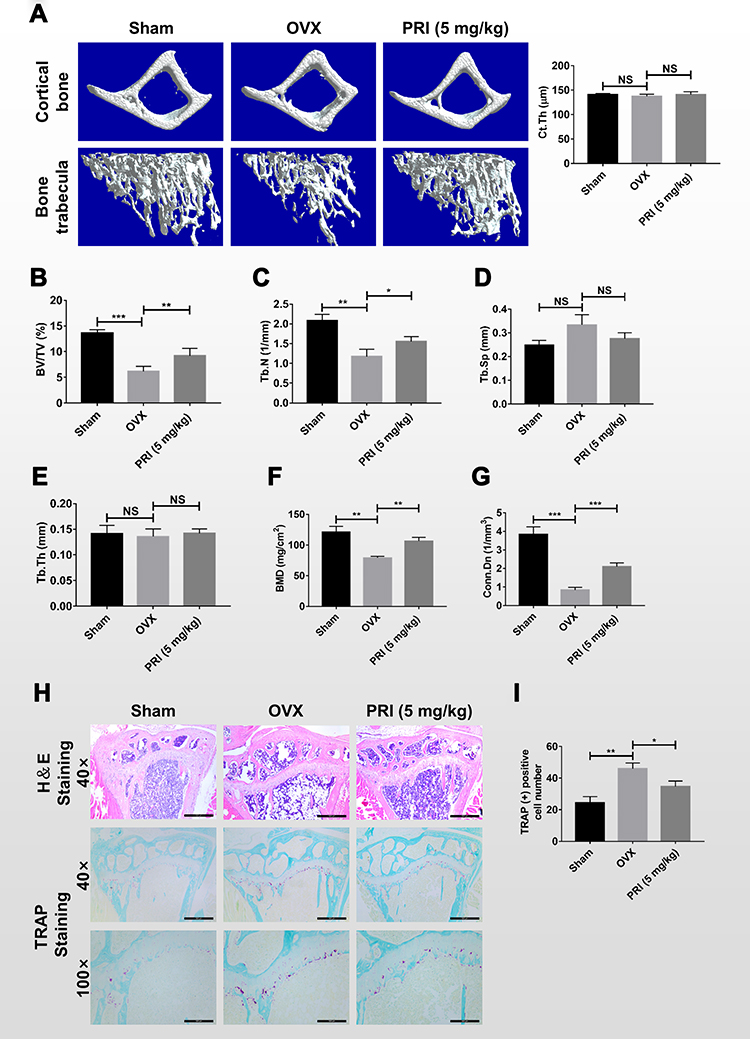 |
Figure 5 PRI protects against OVX-induced bone loss in mice in vivo. (A) Representative 3D reconstructions of micro-CT scans of tibial trabecular and cortical bone (cross-section) from mice in Sham (normal saline), OVX (normal saline), and OVX + PRI (5 mg/kg bodyweight) groups. (B-G) Morphometric analysis of bone parameters including percentage bone volume to tissue volume (BV/TV, %), trabecular bone number (Tb.N, mm−1), trabecular separation (Tb.S, mm), trabecular bone thickness (Tb.Th, mm), bone mineral density (BMD, mg/cm2) and connectivity density (Conn.Dn, 1/mm3). (H) Representative H&E and TRAP stained histological sections of tibial bone from control and experimental groups. (I) Quantitative histomorphometric analysis of the number of TRAP-positive cells in each group. Bar graph presented as mean ± SD from 8 samples per group; *p < 0.05, **p < 0.01, and ***p < 0.001 relative to control group. |
H&E histological assessments provided further evidence for PRI treatment in protecting mice against the deleterious effects of OVX-induced bone loss (Figure 5H). Additionally, TRAP stained sections (Figure 5H) demonstrated significant differences in the number of TRAP-positive osteoclasts in each group (Figure 5I). Together these results clearly suggest that the in vitro anti-resorptive and anti-osteoclastic effects of PRI can be recapitulated to protect mice from bone loss caused by OVX in vivo.
Discussion
Bone metabolism is balanced by the mutual activities of osteoclasts bone resorption and osteoblasts bone formation. The synergistic interplay between these two bone cells ensures the structural integrity of the skeletal tissues remain in optimal condition. However, this disorder of balance leads to excessive osteoclast bone resorption underlies metabolic osteolytic conditions including post-menopausal osteoporosis.21 Current pharmacological interventions that offer some therapeutic benefits against osteoclast-mediated bone loss include bisphosphonates, calcium supplements, estrogen replacements and selective estrogen receptor modulators, and anti-RANKL antibody.22 However, undesirable and in some cases severe side effects exist including cardiovascular events, nephrotoxicity, osteonecrosis of the jaw, atypical fractures, and malignant tumor formation,4,5,23 limit their long-term use. Hence, the identification of new effective and safer alternatives is urgently needed for the treatment and management of osteolytic diseases, for instance osteoporosis. Therefore, this study showed that pristimerin (PRI) inhibits osteoclast generation and resorption and prevents OVX-induced bone loss partly by attenuating the inhibition of RANKL-stimulated activation of NF-κB and ERK MAPK signaling pathways.
PRI is a natural triterpenoid compound extracted from plants of the Celastraceae and Hippocrateaceae family. PRI has attracted much attention over the years for its chemopreventive and chemotherapeutic properties,24 anti-inflammatory, anti-oxidant, anti-arthritic, anti-malarial, anti-viral, anti-bacterial activities,12,15,25,26 and anti-tumour effects against breast cancer, lung cancer, prostate cancer, cervical cancer, gliomas, leukemias, and multiple myeloma tumors.14,27,28 Based on the results presented in this study, we now add anti-osteoclastic and anti-resorptive properties to the list of biological effects exerted by PRI. Mononuclear cells responding to RANKL fused as multinucleated osteoclasts. The combination of RANKL and its cognate receptor RANK on monocytic precursor cells recruits the adaptor protein TRAF6 and rapidly motivates serial signaling events which drives precursor cell differentiation and fusion into multinucleated giant cells and subsequent bone resorption.29 The NF-κB and MAPK (consisting of family members ERK, JNK, and p38) signaling pathways are two of the earliest pathways initiated in answer to RANKL stimulation.
TRAF6 stimulates NF-κB and MAPK by recruiting and activating TAK1. Activated TAK1 then activates IκB kinase (IKK) complex which successively phosphorylates and mediates IκBα ubiquitin-dependent proteasomal degradation. The degradation of IκBα releases p65/p50 NF-κB heterodimer subunits from NF-κB heterodimer/IκBα complex, enabling the p65/p50 subunits to be phosphorylated and then translocated to the nucleus for initiation of transcriptional activity.30 The importance of NF-κB proteins have been demonstrated in double-knockout mice which exhibits critical osteopetrotic phenotype owing to impaired osteoclast formation.31 Our biochemical analyses provide evidence for an inhibitory function of PRI on NF-κB activation in response to RANKL stimulation. PRI treatment completely abolished the degradation of IκBα and consequently hindered p65 phosphorylation, nuclear translocation, and NF-κB transcriptional activity. The suppression function of PRI on NF-κB signaling is consistent with previous reports in murine macrophages,26 and various cancer cells including myeloma, fibrosarcoma, osteosarcoma, esophageal cancer, ovarian carcinoma, pancreatic cancer, and leukemia cells.15,32–35 Interestingly, the study by Lu and colleagues suggests that PRI inhibits two key steps in the NF-κB signaling cascade: TAK1 activation of IKK and IKK-dependent phosphorylation of IκBα, the latter of which is in line with our observations.
TAK1 also functions as the upstream MAPK kinase kinase (MAP3K) that activates all three downstream MAPK pathways including JNK, p38, and ERK.36 Cooperatively, activation of three MAPK signaling pathways is important in the efficient stimulation of osteoclastogenesis. Monocytic precursors derived from JNK1 or ERK1 knockout mice presents with reduced capacity to differentiate towards the osteoclast lineage.37,38 Additionally, ERK is also important for osteoclast survival and in the maintenance of osteoclast cell polarity in the process of bone resorption.39 Besides, p38 signaling is mainly participated in osteoclast generation and not activity.40 Our present work discovered that PRI significantly restrained the activation of ERK signaling but had no influence in p38 activation. The inhibitory effect of PRI on ERK has previously been reported and is associated with pro-apoptotic effect of PRI.41 Interestingly, PRI treatment was found to sustain the activation of RANKL-induced activation of JNK. PRI has previously been displayed can induce cell apoptosis and autophagy via the elevation in ROS generation which subsequently activated the ASK1/JNK signaling pathway in human breast cancer and in chronic myeloid leukemia cells.42 The sustained activation of phosphorylation of JNK could be put down to the activation of the ROS/ASK1/JNK pathway but further investigation will need to be carried out.
The early activation of NF-κB and MAPK signals coordinates the induction of c-Fos, an important component of the AP-1 transcription factor complex, and NFATc1, the main transcription factor in osteoclast differentiation.9 The loss of c-Fos or NFATc1 leads to complete loss of osteoclast formation, which causes severe osteopetrosis in mice.10 Over-expression of NFATc1 is capable of rescuing the osteoclast defect in NF-κB-deficient and c-Fos-deficient mice indicating that NFATc1 is the most distal transcription factor required for osteoclast generation process.43 NFATc1 induction and transcriptional activity is not only markedly induced by early RANKL signaling cascades but also sustained by the course of osteoclast generation process via the co-stimulatory Ca2+ oscillation-mediated self-sustaining auto-amplification system.44 Activated NFATc1 translocates to the nucleus and cooperates with other transcription factors to induce the production of relevant osteoclast-specific genes, for example Dc-stamp, Trap/Acp5, Mmp-9, and Ctsk.11 In agreement with the suppression role of PRI on NF-κB and ERK signaling activation, the downstream expression of c-Fos and NFATc1, and the nuclear localization of NFATc1 was markedly thwarted. Furthermore, quantitative gene expression analysis attested to the inhibition of NFATc1 transcription with the gene expression of Trap/Acp5, Dc-stamp Mmp-9, and Ctsk significantly downregulated following PRI treatment.
Together our data suggest that PRI exerts its anti-osteoclastic effects via the restraint of RANKL-mediated activation of ERK MAPK and NF-κB signaling pathways which hence thwarted the downstream activation transcription factors c-Fos and NFATc1, the latter of which is a key driver in the transcriptional upregulation of osteoclast genes participated in differentiation, fusion, and bone resorption. Additionally, these promising in vitro was successfully translated into in vivo benefits, with PRI administration protecting mice against the deleterious bone loss effects of estrogen-deficiency following OVX. Morphometric analyses of bone parameters show marked improvements in bone volume and trabecular bone architecture. Histological assessment revealed reductions in osteoclast numbers and activity following PRI administration. Thus collectively, our research data provide convincing evidence for the potential application of PRI as a therapeutic agent against osteoclast-mediated osteolytic conditions.
Abbreviations
PRI, pristimerin; NF-κB, nuclear factor-κB; M-CSF, macrophage-colony stimulating factor; RANKL, receptor activator of nuclear factor-κB ligand; FBS, fetal bovine serum; TNF, tumor necrosis factor; MAPK, mitogen-activated protein kinase; NFATc1, nuclear factor of activated T-cells 1; Dc-stamp, dendritic cell-specific transmembrane protein; Trap/Acp5, tartrate resistant acid phosphatase; Ctsk, cathepsin K; Mmp-9, matrix metalloproteinase-9; α-MEM, Minimum Essential Medium Alpha Modification; CCK-8, cell counting kit-8; DAPI, 4′,6-diamidino-2-phenylindole; BMMs, bone marrow macrophages; PFA, paraformaldehyde; PBS, phosphate-buffered saline; BSA, bovine serum albumin; PCR, polymerase chain reaction; cDNA, complementary DNAs; TBST, tris buffered saline with tween; SDS-PAGE, sodium dodecyl sulfatepolyacrylamide gel electrophoresis; c-Fos, protooncogene; JNK, c-Jun N-terminal kinase; ERK, extracellular signal-regulated kinase; OVX, ovariectomy; BMD, bone mineral density; Conn.Dn, connectivity density; Tb.N, trabecular number; Tb.Sp, trabecular separation; Tb.Th, trabecular thickness; BV/TV, percentage bone volume per tissue volume.
Data Sharing Statement
All data generated or analysed during this study are included in this published article.The data that support the findings of this study are available from the corresponding author upon reasonable request.
Ethics Statement
This work fully considered and protected the rights and interests of the study objects. The use of animals in experiments was approved by the ethics Committee of Guangxi medical university (SYXK 2020-0004). All experimental procedures were performed in conformity with institutional guidelines for the care and use of laboratory animals Guangxi medical university, China. This project meets the criteria of the Ethical Review Committee.
Acknowledgments
This work is supported by the Natural Science Foundation of China (No.81770254, No.81970236) and Natural Science Foundation of Guangxi Province (2018GXNSFAA050092).
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest for this work.
References
1. Su N, Yang J, Xie Y, et al. Bone function, dysfunction and its role in diseases including critical illness. Int J Biol Sci. 2019;15(4):776–787. doi:10.7150/ijbs.27063
2. Cao X. Targeting osteoclast-osteoblast communication. Nat Med. 2011;17(11):1344–1346. doi:10.1038/nm.2499
3. Compston JE, McClung MR, Leslie WD. Osteoporosis. Lancet (London, England). 2019;393(10169):364–376. doi:10.1016/S0140-6736(18)32112-3
4. Milat F, Ebeling PR. Osteoporosis treatment: a missed opportunity. Med J Aust. 2016;205(4):185–190. doi:10.5694/mja16.00568
5. Reid IR. Efficacy, effectiveness and side effects of medications used to prevent fractures. J Intern Med. 2015;277(6):690–706. doi:10.1111/joim.12339
6. Feng X. RANKing intracellular signaling in osteoclasts. IUBMB Life. 2005;57(6):389–395. doi:10.1080/15216540500137669
7. Chen R, Liu G, Sun X, et al. Chitosan derived nitrogen-doped carbon dots suppress osteoclastic osteolysis via downregulating ROS. Nanoscale. 2020;12(30):16229–16244. doi:10.1039/D0NR02848G
8. Nakashima T, Takayanagi H. New regulation mechanisms of osteoclast differentiation. Ann N Y Acad Sci. 2011;1240:E13–18. doi:10.1111/j.1749-6632.2011.06373.x
9. Wagner EF. Bone development and inflammatory disease is regulated by AP-1 (Fos/Jun). Ann Rheum Dis. 2010;69(Suppl 1):i86–88. doi:10.1136/ard.2009.119396
10. Takayanagi H, Kim S, Koga T, et al. Induction and activation of the transcription factor NFATc1 (NFAT2) integrate RANKL signaling in terminal differentiation of osteoclasts. Dev Cell. 2002;3(6):889–901. doi:10.1016/S1534-5807(02)00369-6
11. Fretz JA, Shevde NK, Singh S, Darnay BG, Pike JW. Receptor activator of nuclear factor-kappaB ligand-induced nuclear factor of activated T cells (C1) autoregulates its own expression in osteoclasts and mediates the up-regulation of tartrate-resistant acid phosphatase. Mol Endocrinol. 2008;22(3):737–750. doi:10.1210/me.2007-0333
12. Sun Y, Gao LL, Tang MY, Feng BM, Pei YH, Yasukawa K. Triterpenoids from Euphorbia maculata and Their Anti-Inflammatory Effects. Molecules (Basel, Switzerland). 2018;23:9. doi:10.3390/molecules23092112
13. Buffa Filho W, Corsino J, da Bolzani SV, Furlan M, Pereira AM, Franca SC. Quantitative determination for cytotoxic Friedo-nor-oleanane derivatives from five morphological types of Maytenus ilicifolia (Celastraceae) by reverse-phase high-performance liquid chromatography. Phytochem Anal. 2002;13(2):75–78. doi:10.1002/pca.626
14. Li JJ, Yan YY, Sun HM, et al. Anti-cancer effects of pristimerin and the mechanisms: a critical review. Front Pharmacol. 2019;10:746. doi:10.3389/fphar.2019.00746
15. Tong L, Nanjundaiah SM, Venkatesha SH, Astry B, Yu H, Moudgil KD. Pristimerin, a naturally occurring triterpenoid, protects against autoimmune arthritis by modulating the cellular and soluble immune mediators of inflammation and tissue damage. Clin Immunol. 2014;155(2):220–230. doi:10.1016/j.clim.2014.09.014
16. Xiao Y, Li K, Wang Z, et al. Pectolinarigenin prevents bone loss in ovariectomized mice and inhibits RANKL-induced osteoclastogenesis via blocking activation of MAPK and NFATc1 signaling. J Cell Physiol. 2019;234(8):13959–13968. doi:10.1002/jcp.28079
17. Jurdic P, Saltel F, Chabadel A, Destaing O. Podosome and sealing zone: specificity of the osteoclast model. Eur J Cell Biol. 2006;85(3–4):195–202. doi:10.1016/j.ejcb.2005.09.008
18. Jacobs MD, Harrison SC. Structure of an IkappaBalpha/NF-kappaB complex. Cell. 1998;95(6):749–758. doi:10.1016/S0092-8674(00)81698-0
19. Arai A, Mizoguchi T, Harada S, et al. Fos plays an essential role in the upregulation of RANK expression in osteoclast precursors within the bone microenvironment. J Cell Sci. 2012;125(Pt 12):2910–2917. doi:10.1242/jcs.099986
20. Balkan W, Martinez AF, Fernandez I, Rodriguez MA, Pang M, Troen BR. Identification of NFAT binding sites that mediate stimulation of cathepsin K promoter activity by RANK ligand. Gene. 2009;446(2):90–98. doi:10.1016/j.gene.2009.06.013
21. D’Amelio P, Grimaldi A, Di Bella S, et al. Estrogen deficiency increases osteoclastogenesis up-regulating T cells activity: a key mechanism in osteoporosis. Bone. 2008;43(1):92–100. doi:10.1016/j.bone.2008.02.017.
22. Brommage R. New targets and emergent therapies for osteoporosis. Handb Exp Pharmacol. 2020;262:451–473. doi:10.1007/164_2019_329.
23. Zhang J, Saag KG, Curtis JR. Long-term safety concerns of antiresorptive therapy. Rheum Dis Clin North Am. 2011;37(3):387–400, vi. doi:10.1016/j.rdc.2011.08.001
24. Niampoka C, Suttisri R, Bavovada R, Takayama H, Aimi N. Potentially cytotoxic triterpenoids from the root bark of Siphonodon celastrineus Griff. Arch Pharm Res. 2005;28(5):546–549. doi:10.1007/BF02977756
25. Costa PM, Ferreira PM, Bolzani Vda S, et al. Antiproliferative activity of pristimerin isolated from Maytenus ilicifolia (Celastraceae) in human HL-60 cells. Toxicol in Vitro. 2008;22(4):854–863. doi:10.1016/j.tiv.2008.01.003
26. Dirsch VM, Kiemer AK, Wagner H, Vollmar AM. The triterpenoid quinonemethide pristimerin inhibits induction of inducible nitric oxide synthase in murine macrophages. Eur J Pharmacol. 1997;336(2–3):211–217. doi:10.1016/S0014-2999(97)01245-4
27. Huang S, He P, Peng X, Li J, Xu D, Tang Y. Pristimerin inhibits prostate cancer bone metastasis by targeting PC-3 stem cell characteristics and VEGF-induced vasculogenesis of BM-EPCs. Cell Physiol Biochem. 2015;37(1):253–268. doi:10.1159/000430350
28. Yan YY, Bai JP, Xie Y, Yu JZ, Ma CG. The triterpenoid pristimerin induces U87 glioma cell apoptosis through reactive oxygen species-mediated mitochondrial dysfunction. Oncol Lett. 2013;5(1):242–248. doi:10.3892/ol.2012.982
29. Kobayashi N, Kadono Y, Naito A, et al. Segregation of TRAF6-mediated signaling pathways clarifies its role in osteoclastogenesis. EMBO J. 2001;20(6):1271–1280. doi:10.1093/emboj/20.6.1271
30. Ruocco MG, Maeda S, Park JM, et al. I{kappa}B kinase (IKK){beta}, but not IKK{alpha}, is a critical mediator of osteoclast survival and is required for inflammation-induced bone loss. J Exp Med. 2005;201(10):1677–1687. doi:10.1084/jem.20042081
31. Iotsova V, Caamano J, Loy J, Yang Y, Lewin A, Bravo R. Osteopetrosis in mice lacking NF-kappaB1 and NF-kappaB2. Nat Med. 1997;3(11):1285–1289. doi:10.1038/nm1197-1285
32. Tiedemann RE, Schmidt J, Keats JJ, et al. Identification of a potent natural triterpenoid inhibitor of proteosome chymotrypsin-like activity and NF-kappaB with antimyeloma activity in vitro and in vivo. Blood. 2009;113(17):4027–4037. doi:10.1182/blood-2008-09-179796
33. Wang Y, Zhou Y, Zhou H, et al. Pristimerin causes G1 arrest, induces apoptosis, and enhances the chemosensitivity to gemcitabine in pancreatic cancer cells. PLoS One. 2012;7(8):e43826. doi:10.1371/journal.pone.0043826
34. Gao X, Liu Y, Deeb D, Arbab AS, Gautam SC. Anticancer activity of pristimerin in ovarian carcinoma cells is mediated through the inhibition of prosurvival Akt/NF-kappaB/mTOR signaling. J Exp Ther Oncol. 2014;10(4):275–283.
35. Tu Y, Tan F, Zhou J, Pan J. Pristimerin targeting NF-kappaB pathway inhibits proliferation, migration, and invasion in esophageal squamous cell carcinoma cells. Cell Biochem Funct. 2018;36(4):228–240. doi:10.1002/cbf.3335
36. Mizukami J, Takaesu G, Akatsuka H, et al. Receptor activator of NF-kappaB ligand (RANKL) activates TAK1 mitogen-activated protein kinase kinase kinase through a signaling complex containing RANK, TAB2, and TRAF6. Mol Cell Biol. 2002;22(4):992–1000. doi:10.1128/MCB.22.4.992-1000.2002
37. He Y, Staser K, Rhodes SD, et al. Erk1 positively regulates osteoclast differentiation and bone resorptive activity. PLoS One. 2011;6(9):e24780. doi:10.1371/journal.pone.0024780
38. David JP, Sabapathy K, Hoffmann O, Idarraga MH, Wagner EF. JNK1 modulates osteoclastogenesis through both c-Jun phosphorylation-dependent and -independent mechanisms. J Cell Sci. 2002;115(Pt 22):4317–4325. doi:10.1242/jcs.00082
39. Nakamura H, Hirata A, Tsuji T, Yamamoto T. Role of osteoclast extracellular signal-regulated kinase (ERK) in cell survival and maintenance of cell polarity. J Bone Mineral Res. 2003;18(7):1198–1205. doi:10.1359/jbmr.2003.18.7.1198
40. Lee K, Chung YH, Ahn H, Kim H, Rho J, Jeong D. Selective regulation of MAPK signaling mediates RANKL-dependent osteoclast differentiation. Int J Biol Sci. 2016;12(2):235–245. doi:10.7150/ijbs.13814
41. Hayashi D, Shirai T, Terauchi R, et al. Pristimerin inhibits the proliferation of HT1080 fibrosarcoma cells by inducing apoptosis. Oncol Lett. 2020;19(4):2963–2970.
42. Zhao Q, Liu Y, Zhong J, et al. Pristimerin induces apoptosis and autophagy via activation of ROS/ASK1/JNK pathway in human breast cancer in vitro and in vivo. Cell Death Discovery. 2019;5:125. doi:10.1038/s41420-019-0208-0
43. Matsuo K, Galson DL, Zhao C, et al. Nuclear factor of activated T-cells (NFAT) rescues osteoclastogenesis in precursors lacking c-Fos. J Biol Chem. 2004;279(25):26475–26480. doi:10.1074/jbc.M313973200
44. Park YJ, Yoo SA, Kim M, Kim WU. The role of calcium-calcineurin-NFAT signaling pathway in health and autoimmune diseases. Front Immunol. 2020;11:195. doi:10.3389/fimmu.2020.00195
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.