Back to Journals » OncoTargets and Therapy » Volume 13
Primary Hepatic Neuroendocrine Tumor Mimicking Ruptured Hepatocellular Carcinoma with AFP Elevation: A Case Report and Literature Review
Authors Huang H, Jin P ![]() , Yang H, Zhang C, Zhang X, Wang J, Yu J, Zhang B
, Yang H, Zhang C, Zhang X, Wang J, Yu J, Zhang B ![]() , Zhang Y, Hu Q
, Zhang Y, Hu Q ![]()
Received 30 October 2019
Accepted for publication 10 January 2020
Published 3 February 2020 Volume 2020:13 Pages 975—979
DOI https://doi.org/10.2147/OTT.S236728
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Sanjay Singh
Hai-feng Huang,1 Piao-piao Jin,2 Han-jin Yang,3 Chun-jun Zhang,1 Xin Zhang,4 Jun-sen Wang,5 Jia-jie Yu,1 Bo Zhang,1 Yun Zhang,6 Qi-da Hu6
1Department of Surgery, Shengzhou People’s Hospital, Shengzhou 312400, People’s Republic of China; 2Health Management Center, First Affiliated Hospital, Zhejiang University School of Medicine, Hangzhou 310003, People’s Republic of China; 3Department of Pathology, First Affiliated Hospital, Zhejiang University School of Medicine, Hangzhou, Zhejiang 310003, People’s Republic of China; 4Department of Radiology, First Affiliated Hospital, Zhejiang University School of Medicine, Hangzhou 310003, People’s Republic of China; 5Department of Pathology, Shengzhou People’s Hospital, Shengzhou 312400, People’s Republic of China; 6Department of Hepatobiliary and Pancreatic Surgery, First Affiliated Hospital, Zhejiang University School of Medicine, Hangzhou 310003, People’s Republic of China
Correspondence: Qi-da Hu
Department of Hepatobiliary and Pancreatic Surgery, First Affiliated Hospital, Zhejiang University School of Medicine, 79 Qingchun Road, Hangzhou 310003, People’s Republic of China
Email [email protected]
Abstract: Liver cancer is a common malignant disease in China, while the primary hepatic neuroendocrine tumor (PHNET) is extremely rare presented with various manifestations. We herein describe an interesting PHNET case, which was clinically diagnosed as hepatocellular carcinoma (HCC) based on strong clinical evidence and the national guideline, but confirmed to be PHNET by pathology. A 42-year-old Chinese male was admitted for persistent upper abdominal pain, and CT scan revealed a huge liver tumor in the left lobe. The tumor presented attributes of tumor rupture, portal vein tumor thrombus, elevated serum AFP level, background hepatitis B virus infection history, and radiological features mimicking typical HCC. After left semi-hepatectomy was performed for curative treatment of the primary “HCC”, the pathology demonstrated the correct diagnosis be poorly differentiated neuroendocrine carcinoma (NEC). The immunohistochemistry assays showed positive neuroendocrine markers of CgA and Syn and negative HCC markers of Hep Par 1 and GPC3, ruling out concurrent HCC. This case and literature review suggest that in spite of rare incidence, PHNET should be considered as a possible diagnosis when lacking a confirmative pathology result, even when sufficient evidence of typical presentation exist to establish the clinical diagnosis of HCC.
Keywords: primary hepatic neuroendocrine tumor, ruptured liver cancer, α-fetoprotein, neuroendocrine carcinoma, misdiagnosis
Introduction
Liver cancer is a common malignancy in Asian countries, especially in China where a high prevalence of hepatitis B virus (HBV) infection is presented.1,2 Among all types of liver cancer, primary hepatocellular carcinoma (HCC) is most frequent. According to the national guideline,3 diagnosis of primary HCC could be clinically established based on radiology appearance, tumor marker level, and history of chronic liver disease. The pathological evidence is not essentially required for diagnosis. After the clinical diagnosis of HCC has been determined, the patient could receive specific healthcare against HCC, including hepatic resection, locoregional therapies like transarterial chemoembolization or ablation, and systemic targeted agents or immunotherapy.4
In comparison to HCC, the primary hepatic neuroendocrine tumor (PHNET), as a rare subset of the neuroendocrine tumor primarily occurred in the digestive system, has a significantly lower incidence.5 There have been frequent reports on PHNET cases, mostly with unremarkable clinical presentations.6,7 Several liver centers also provided cases of HCCs with neuroendocrine differentiation or mixed HCC and PHNET tumors.5,8,9
To date, there has been no report of PHNET that abnormally presents with typical HCC behaviors. We herein describe an interesting and rare PHNET case, which was clinically diagnosed as HCC based on strong clinical evidence and the national guideline, but confirmed to be PHNET by pathology.
Case Presentation
A 42-year-old Chinese male was admitted to our hospital for persistent upper abdominal pain. The patient had an abrupt episode of subxiphoid pain, which lasted for half an hour and eventually relieved. He recalled a maximum pain scale as 4/10 according to the Wong-Baker Faces Pain Rating Scale system. He immediately received an emergent CT scan to show a huge liver tumor (14.0 cm × 9.7 cm × 7.7 cm) in the left lobe, with involvement of left branch of the portal vein and left hepatic artery (Figure 1). The CT also indicated concurrent hemorrhage from the liver tumor, causing a small amount of free blood collection in the pelvic cavity. The hemoglobin level was 13.4 g/dL, but the α-fetoprotein (AFP) and neuron specific enolase (NSE) notably elevated to 384 ng/mL and 151 ng/mL, respectively, while the other tumor markers were in normal range. Routine contrasted MRI scan ruled out intrahepatic spread of the liver cancer, and supported the initial diagnosis of HCC. Considering that the patient had a long-term history of chronic hepatitis B infection, we established the initial diagnosis of ruptured HCC.
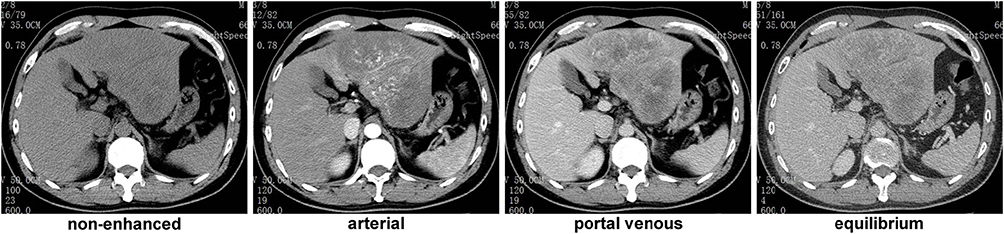 |
Figure 1 CT scan results 1 month before the surgery. |
The patient refused surgical treatment or transarterial chemoembolization at first, and received proton pump inhibitor (PPI), terlipressin, hepatoprotective agents, antiviral therapy, and other best supportive care. Further blood test results revealed a high viral load of 1.32 × 104 copies per mL and Pugh-Child class A. Unfortunately, his hemoglobin and platelet levels gradually decreased to 12.6 g/dL and 84,000/μL, respectively, without any abnormal change of the vital signs. The AFP and NSE levels reached 1016 ng/mL and 172 ng/mL, respectively. The upper abdominal pain persistently existed due to untreated liver disease. One month later, the patient had a second CT scan to find the tumor size increased rapidly to 17.1 cm × 11.2 cm × 8.9 cm within the past month (Figure 2). He then decided to receive surgical treatment.
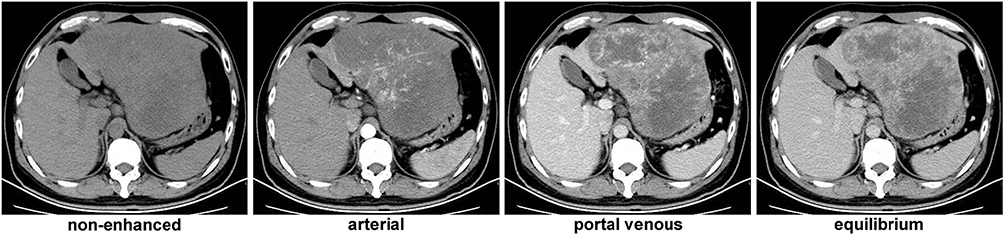 |
Figure 2 CT scan results 3 days before the surgery. |
An open left semi-hepatectomy with regional lymph node dissection was successfully performed. Gross dissection of the resected sample showed a huge tumor with left portal vein tumor thrombus (PVTT), and confirmed hemorrhage from the ruptured tumor, highly resembling the presentation of HCC (Figure 3). Inside the white and raw fish-like liver mass existed massive necrosis. Enlarged lymph nodes were spotted in the left subdiaphragmatic region and hepatoduodenal ligament, among which three lymph nodes were positive. Unexpectedly, the postoperative pathology demonstrated poorly differentiated liver neuroendocrine carcinoma (NEC), further verified by immunohistochemistry assays (Figure 4) showing CK19 (+), CD56 (+), CgA (+), Syn (+), CK7 (-), Hep Par 1 (-), and Ki67 (+80%). The serum AFP and NSE level decreased to 276 ng/mL and 21 ng/mL on postoperative day (POD) #3 and then 20 ng/mL and 3.5 ng/mL on POD #30, respectively. To rule out extrahepatic NEC, both gastroduodenoscopy and colonoscopy were performed, but returned with negative results. Preoperative chest and abdominal CT scans were carefully reviewed by the multidisciplinary board who agreed that no extrahepatic NEC was presented. The final diagnosis was primary liver NEC. The patient recovered well from surgery, and received irinotecan plus cisplatin (IP) regimen for adjuvant chemotherapy treatment starting from POD #45.
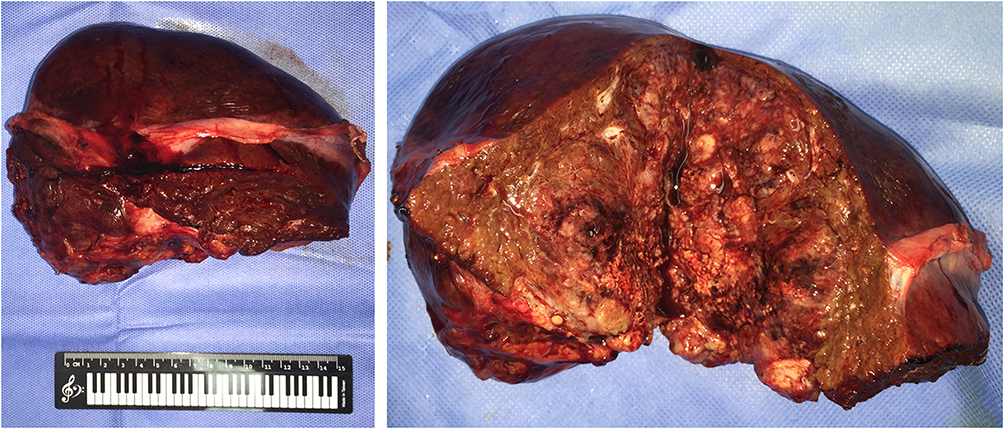 |
Figure 3 Resected tumor sample and its cross-sectional profile showing huge liver mass with diffuse necrosis and focal hemorrhage. |
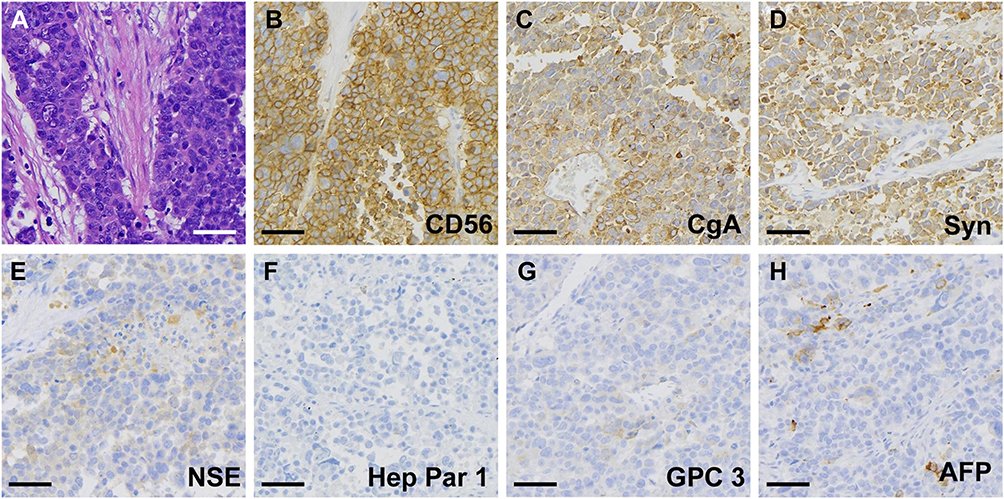 |
Figure 4 Postoperative pathological analysis revealed (A) neuroendocrine phenotype by H&E staining, (B) positive CD56, (C) positive CgA, (D) positive Syn, (E) weak positive NSE, (F) negative Hep Par 1, (G) negative GPC3, and (H) focal positive AFP. |
Discussions
Current report presents a rare, and so far the first, case of PHNET mimicking typical ruptured HCC. Although NECs of the digestive system might exhibit various behaviors,6,7 a confusing scenario to establish the correct diagnosis, like this case, has never been seen before, which raised our interests in two major aspects.
Firstly, the liver NEC had an HCC-like radiological presentation complicated by typical HCC behaviors such as PVTT and tumor rupture, with the background HBV infection and AFP elevation. The diagnosis of primary HCC could be established according to the 2017 National Guideline for Diagnosis and Treatment of Primary HCC in China,3 mainly based on epidemiological data from the large HBV-positive population in China. This exceptional misdiagnosis example indicates a rare probability of diagnosis inaccuracy and the confirmative role of pathological evidence.
Secondly, a significantly elevated serum AFP level was observed preoperatively, but the immunohistochemistry clearly showed uniform NEC rather than HCC with neuroendocrine component. Notably, the characterization of HCC-specific markers like Hep Par 1 and GPC3 were both negative, which ruled out the possibility of concurrent existence of HCC and NEC. The elevated AFP level and positive AFP immunohistochemistry outcome might be caused by secretion of AFP by other immature or hepatoblast-like hepatocytes other than HCC cells,10 since AFP is also a clinical marker of progenitor cell induction in injured or fibrotic liver.11,12
The final diagnosis was poorly differentiated PHNET (WHO grade G3). Previous publications have shown that PHNET, with an extremely rare percentage of 0.38% among all liver tumors,5 could exhibit diverse clinical manifestations, including abdominal pain, weight loss, jaundice, fatigue, and diarrhea.6,7 Immunohistochemically, most PHNET cases were reported to be strongly positive for CgA (67–100%) and Syn (92–100%).5,13–15 Several pieces of literature provided detailed serum AFP level in PHNET patients, mostly in normal range from 1.2 to 4.8 ng/mL, only one case with highly elevated AFP of 7397 IU/mL (8950 ng/mL).7,13,15 Notably, the PHNET case with significant AFP elevation showed a high tumor grade of G3 and a poor prognosis with 0.7-month survival, but no further mechanism analysis was performed.15
Another differential diagnosis in this case is HCC with neuroendocrine component or mixed tumor of HCC and NEC. Similar cases have been frequently reported.5,8,9 The immunohistochemistry analysis with negative staining of hepatocyte markers Hep Par 1 and GPC3, however, unquestionably oppose this differential diagnosis. A preoperative octreotide scan might help to establish the diagnosis of NEC. Another differential diagnosis is metastatic NEC of unknown origins, which has a relatively low incidence rate consisting 10–13% of the all neuroendocrine neoplasms.16,17 The common sites of primary NEC with liver metastases are in the intestinal tract or the pancreas, where either endoscopic studies or abdominal CT scan showed negative results. A further PET scan or somatostatin receptor imaging should be a suitable option to identify other possible NEC origins, but the patient refused the test due to concerns of excessive radiation.
There are several limitations in the treatment course. Firstly, AFP-producing neuroendocrine tumor, though rarely occurred, should also be a possible diagnosis because of the elevated NSE level. However, our multidisciplinary tumor board preferred the initial diagnosis of HCC rather than PHNET, after considering the imaging characteristics, tumor behaviors, chronic hepatitis history, elevated AFP level, and the epidemiological incidence rate. The preoperative misdiagnosis suggests that neuroendocrine malignancies be a differential diagnosis in the liver tumor patients with NSE elevation. Secondly, a somatostatin receptor-based imaging or PET scan is vital for confirmation of the PHPET diagnosis and staging. The patient refused to have the PET scan before the surgery and somatostatin receptor scan after the histological diagnosis, worrying excessive radiation. A better communication and patient education are therefore needed for better patient obedience and diagnostic accuracy.
In conclusion, a malignant PHNET case with rare tumor behaviors similar to HCC led to preoperative misdiagnosis. Even typical presentations could be deceiving especially when the possibility of NEC diagnosis has been raised, which should be carefully reviewed and identified by physicians. It is better to establish the final diagnosis of HCC with confirmative pathological evidence when conditions permit.
Ethics Approval and Consent for Publication
This study was approved by the research ethics committee of Shengzhou People’s Hospital, and written informed consent for publication of the clinical details and images was obtained from the patient.
Informed Consent Statement
Informed written consent was obtained from the patient for publication of this report and any accompanying images.
Acknowledgments
We thank the hepatobiliary and pancreatic multidisciplinary board for the assistance and helpful advice.
Author Contributions
Huang H, Zhang C, Yu J, Zhang B, and Hu Q were the patient’s surgeons, reviewed the literature; Jin P and Zhang Y reviewed the literature; Yang H and Wang J performed the pathological analyses and interpretation; Zhang X analyzed and interpreted the imaging findings; Hu Q was responsible for the revision of the manuscript for important intellectual content; all authors contributed to data analysis, drafting and revising the article, gave final approval of the version to be published, and agree to be accountable for all aspects of the work.
Funding
This work was financially supported by the National Natural Science Foundation of China (Nos. 81972207, 81830089, 81537009, 81502026, and 51573161), the Zhejiang Natural Science Foundation (Nos. LQ16H18000245 and LY18H160026), and the Shengzhou Technology Program (No. 201909).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2019. CA Cancer J Clin. 2019;69:7–34. doi:10.3322/caac.21551
2. Zheng R, Qu C, Zhang S, et al. Liver cancer incidence and mortality in China: temporal trends and projections to 2030. Chin J Cancer Res. 2018;30:571–579. doi:10.21147/j.issn.1000-9604.2018.06.01
3. Zhou J, Sun HC, Wang Z, et al. Guidelines for diagnosis and treatment of primary liver cancer in China (2017 Edition). Liver Cancer. 2018;7:235–260. doi:10.1159/000488035
4. Villanueva A. Hepatocellular carcinoma. N Engl J Med. 2019;380:1450–1462. doi:10.1056/NEJMra1713263
5. Nomura Y, Nakashima O, Akiba J, et al. Clinicopathological features of neoplasms with neuroendocrine differentiation occurring in the liver. J Clin Pathol. 2017;70:563–570. doi:10.1136/jclinpath-2016-203941
6. Gravante G, De Liguori Carino N, Overton J, Manzia TM, Orlando G. Primary carcinoids of the liver: a review of symptoms, diagnosis and treatments. Dig Surg. 2008;25:364–368. doi:10.1159/000167021
7. Li MX, Li QY, Xiao M, et al. Survival comparison between primary hepatic neuroendocrine neoplasms and primary pancreatic neuroendocrine neoplasms and the analysis on prognosis-related factors. Hepatobiliary Pancreat Dis Int. 2019;18:538–545. doi:10.1016/j.hbpd.2019.03.009
8. Yamaguchi R, Nakashima O, Ogata T, Hanada K, Kumabe T, Kojiro M. Hepatocellular carcinoma with an unusual neuroendocrine component. Pathol Int. 2004;54:861–865. doi:10.1111/j.1440-1827.2004.01770.x
9. Ruck P, Harms D, Kaiserling E. Neuroendocrine differentiation in hepatoblastoma. An immunohistochemical investigation. Am J Surg Pathol. 1990;14:847–855. doi:10.1097/00000478-199009000-00006
10. Tomizawa M, Shinozaki F, Motoyoshi Y, Sugiyama T, Yamamoto S, Ishige N. An optimal medium supplementation regimen for initiation of hepatocyte differentiation in human induced pluripotent stem cells. J Cell Biochem. 2015;116:1479–1489. doi:10.1002/jcb.25139
11. Alison MR, Islam S, Lim S. Stem cells in liver regeneration, fibrosis and cancer: the good, the bad and the ugly. J Pathol. 2009;217:282–298. doi:10.1002/path.2453
12. Kuhlmann WD, Peschke P. Hepatic progenitor cells, stem cells, and AFP expression in models of liver injury. Int J Exp Pathol. 2006;87:343–359. doi:10.1111/j.1365-2613.2006.00485.x
13. Pilichowska M, Kimura N, Ouchi A, Lin H, Mizuno Y, Nagura H. Primary hepatic carcinoid and neuroendocrine carcinoma: clinicopathological and immunohistochemical study of five cases. Pathol Int. 1999;49:318–324. doi:10.1046/j.1440-1827.1999.00866.x
14. Burad DK, Kodiatte TA, Rajeeb SM, Goel A, Eapen CE, Ramakrishna B. Neuroendocrine neoplasms of liver - A 5-year retrospective clinico-pathological study applying World Health Organization 2010 classification. World J Gastroenterol. 2016;22:8956–8966. doi:10.3748/wjg.v22.i40.8956
15. Park CH, Chung JW, Jang SJ, et al. Clinical features and outcomes of primary hepatic neuroendocrine carcinomas. J Gastroenterol Hepatol. 2012;27:1306–1311. doi:10.1111/j.1440-1746.2012.07117.x
16. Yao JC, Hassan M, Phan A, et al. One hundred years after “carcinoid”: epidemiology of and prognostic factors for neuroendocrine tumors in 35,825 cases in the United States. J Clin Oncol. 2008;26:3063–3072. doi:10.1200/JCO.2007.15.4377
17. Catena L, Bichisao E, Milione M, et al. Neuroendocrine tumors of unknown primary site: gold dust or misdiagnosed neoplasms? Tumori. 2011;97:564–567. doi:10.1177/030089161109700504
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.