Back to Journals » International Journal of Women's Health » Volume 18
Primary Endometrium Primitive Neuroectodermal Tumor with Diagnostic and Treatment Challenges: A Case Report
Authors Zhao H, Yang L, Shi Y, Yang L, Jiang W
Received 7 February 2026
Accepted for publication 1 May 2026
Published 12 June 2026 Volume 2026:18 602054
DOI https://doi.org/10.2147/IJWH.S602054
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Vinay Kumar
Haoyun Zhao,* Lu Yang,* Ya Shi, Lindong Yang, Weiqi Jiang
Department of Obstetrics and Gynecology, Jinling Hospital, Affiliated Hospital of Medical, Nanjing University, Nanjing, Jiangsu, 210000, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Weiqi Jiang, Email [email protected] Lindong Yang, Email [email protected]
Background: Primitive neuroectodermal tumor (PNET) is a rare and aggressive malignancy, with primary occurrence in the endometrium being uncommon. Due to nonspecific clinical and imaging features, it is frequently misdiagnosed, leading to delayed treatment and poor prognosis.
Case Presentation: We present the case of a 26-year-old woman with persistent abnormal uterine bleeding, initially misdiagnosed as benign endometrial polyp following curettage. Subsequent imaging revealed a large intrauterine mass, and definitive diagnosis of primary endometrium PNET was established through histopathological and immunohistochemical analyses, including characteristic CD99 positivity and Homer–Wright rosettes. The patient underwent modified radical hysterectomy with lymph node dissection, followed by adjuvant chemotherapy, which was discontinued due to severe toxicity.
Conclusion: This report provides further elucidation on the diagnostic challenges inherent to primary endometrium PNET, underscoring the contribution of immunohistochemical analysis in resolving differential diagnoses among small round cell tumors. Rare neoplasms should warrant consideration in young patients presenting with atypical uterine hemorrhage, particularly in scenarios where preliminary pathological findings are inconclusive. The establishment of an early and definitive diagnosis, coupled with the implementation of a multimodal treatment strategy, is fundamental to optimizing clinical outcomes for this aggressive condition.
Keywords: primitive neuroectodermal tumors, primary endometrium, prolonged menstruation, abnormal uterine bleeding, Homer–Wright rosettes
Introduction
Primitive neuroectodermal tumor (PNET) is an infrequent and highly aggressive small round cell tumor that typically originates from neuroepithelial cells in either the central nervous system or peripheral mesenchymal tissues. It is categorized into central PNET (cPNET) and peripheral PNET (pPNET).1 While PNET occurrences in the reproductive system are even more uncommon, the ovary is the most prevalent site, followed by the uterine body, cervix, and vagina.2–4 Patients commonly present with abnormal uterine bleeding and a pelvic mass. Diagnosis primarily relies on immunohistochemistry (IHC) and genetic testing.5 Treatment approaches are not well-established but typically involve surgery followed by postoperative chemotherapy or radiotherapy. There is no standardized chemotherapy protocol, and patient prognosis is generally unfavorable. Given the limited clinical data available due to its rarity, we herein report a case of advanced PNET in primary endometrium. By analyzing the clinical, pathological, and therapeutic features of this case in conjunction with a comprehensive review of the relevant literature, this study aims to deepen the understanding of this rare tumor and provide valuable insights for its clinical diagnosis and management.
Case Presentation
A 26-year-old woman presented to a local hospital in October 2021 with a chief complaint of prolonged menstrual bleeding lasting 15 days. Pelvic ultrasound at that time suggested endometrial thickening and possible submucosal fibroids. A diagnostic dilation and curettage was subsequently performed. The initial pathological diagnosis was reported as a benign endometrial polyp with necrosis, and based on this finding, the patient was prescribed estradiol valerate for two cycles.
However, her symptoms persisted. She subsequently reported intermittent, irregular, non-foul-smelling yellow vaginal discharge. A follow-up ultrasound on February 4, 2022, revealed a significant change: the uterus was enlarged and contained a large heterogeneous hypoechoic area measuring 64 × 28 mm, accompanied by uterine effusion. A repeat curettage was performed along with antibiotic therapy, but her condition showed no improvement. This marked a critical diagnostic turning point. The postoperative pathology now described endometrial polypoid hyperplasia with the presence of small, naive cells and spindle cells, raising a strong suspicion of malignancy and prompting IHC for confirmation. IHC analysis at the local hospital showed tumor cells were positive for Desmin and CD56, with a high Ki-67 index, while results for sYN, SMA, CK8/18, CD10, and CK were negative.
The patient was then referred to our tertiary hospital for further management. Upon admission, in conjunction with the Hematoxylin and Eosin (HE) staining, the present case indicates the presence of adenosarcoma with embryonic rhabdomyosarcoma differentiation (Figure 1). The HE staining results from different regions of the tumor were shown in Supplementary Figure 1. Surgical intervention was advised following admission, during which tumor marker analysis revealed a neuron-specific enolase (NSE) level of 17.02 μg/L, normal carbohydrate antigen 125 (CA125) levels, and a hemoglobin count of 71 g/L. The pathology department at our institution reviewed the case of an intrauterine (polyp) tumor, ultimately diagnosing it as a malignant adenosarcoma. IHC analysis of the original specimen indicated an undifferentiated sarcoma with a minor component displaying features of embryonal rhabdomyosarcoma, denoting a highly aggressive tumor. An exhaustive list of all IHC markers were showed in Supplementary Table 1.
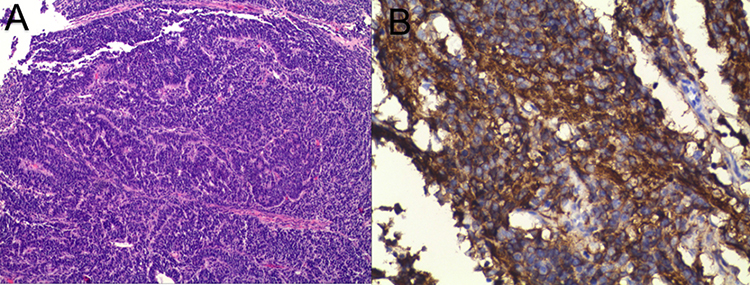 |
Figure 1 Pathological images of endometrial tissue under the microscope, including Hematoxylin and Eosin staining ×20 (A) and CD99 staining ×400 (B). |
Subsequent pelvic MRI scans (plain and contrast-enhanced) revealed a significantly enlarged uterus harboring a large irregular soft tissue mass within the uterine cavity, measuring approximately 63 × 166 mm (Figure 2). The T1-weighted signal intensity was isointense, while T2-weighted imaging exhibited slightly increased signal intensity with areas of low-signal intensity. The mass demonstrated moderate heterogeneous enhancement post-contrast administration. The tumor extended to the cervix without involvement of the bladder, rectum, or uterus, with clear organ demarcation, well-defined fat planes around the rectum, absence of pelvic lymphadenopathy, minimal pelvic fluid, normal presacral soft tissue thickness, unremarkable pelvic bone signals, and no identifiable pelvic masses.
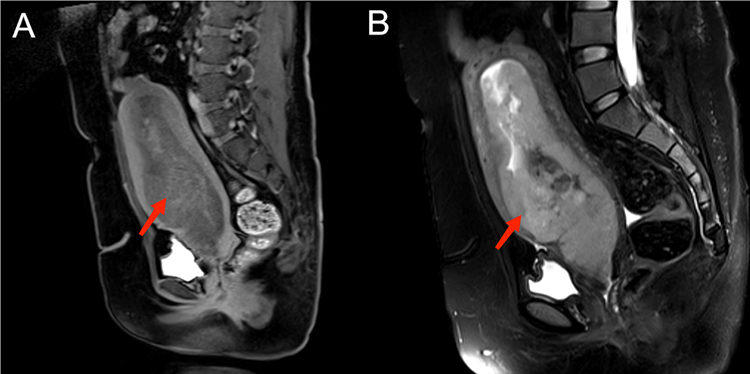 |
Figure 2 Image of pelvic MRI scans (plain and contrast-enhanced, including T1-weighted signal (A) and T2-weighted signal (B). |
On March 5, 2022, the patient underwent comprehensive surgical staging, including radical total hysterectomy, bilateral salpingo-oophorectomy, pelvic lymphadenectomy, para-aortic lymph node biopsy, and partial omentectomy. Final pathological examination confirmed a PNET of the endometrium. The tumor showed deep myometrial invasion and cervical involvement. IHC of the surgical specimen was pivotal, showing strong positivity for CD99 (+) (Figure 1). Strong positivity for CD56 (++), NKX2.2 (+), WT1 (+), P16 (+), P53 (++), β-caternin (+), desmin (+), and a high Ki-67 index (80%+) (Figure 3). Negativity for a broad panel of epithelial, myoid, and neuroendocrine markers, including PAX8 (−), SALL4 (−), ER (−), PR (−), MyoD1 (−), CD10 (−), CgA (−), CK5 (−), CK7 (−), CK20 (−), CKpan (−), S-100 (−), and Syn (−) (Supplementary Figure 2). Metastatic disease was confirmed in 15 of 43 pelvic lymph nodes, and a tumor thrombus was identified in the right iliac vessel. No tumor involvement was detected in the parametrial tissues, sacral region, or omentum. The fallopian tubes exhibited chronic inflammation, and the ovaries showed multiple follicular cysts. The final diagnosis was stage IIIC primary endometrium PNET.
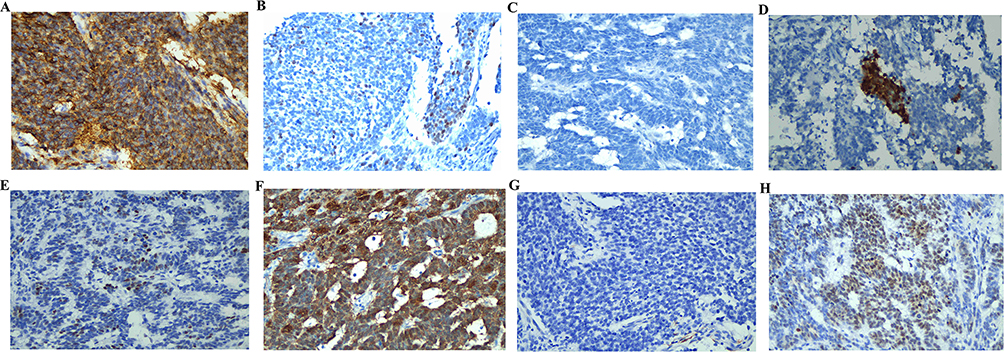 |
Figure 3 Positive immunohistochemistry results (×400), including CD56 (A), NKX2.2 (B), WT1 (C), P16 (D), P53 (E), β-caternin (F), desmin (G), and Ki-67 (H). |
Adjuvant chemotherapy was initiated on March 14, 2022, following surgical recovery. The initial regimen consisted of ifosfamide (2 g days 1–4, 1 g day 5, IV), cisplatin (90 mg day 2, intraperitoneal), and etoposide (180 mg day 1, IV). However, following the second treatment cycle, the patient developed severe bone marrow suppression, necessitating regimen modification. The adapted protocol maintained ifosfamide administration (2 g days 1–4, 1 g day 5, IV) but replaced etoposide with pirarubicin (40 mg day 1, IV) while continuing cisplatin (90 mg day 2, intraperitoneal). The patient ultimately completed four chemotherapy cycles before she and her family decided to discontinue further treatment and requested discharge for supportive care at home. Subsequently, the patient was died after one year of observation.
Discussion
In 1973, Hart and Earle initially characterized PNET as a form of small round cell neoplasm. These neoplasms consist of small round cells that derive from nerves, glia, and ependymal cells, displaying varying levels of cellular differentiation.2,3 Typically, they originate from neuroepithelial cells within the central nervous system or peripheral mesenchymal tissues, leading to their classification as cPNET or pPNET. In 2016, cPNET was reclassified as CNS embryonal tumors with NOS type and removed from the diagnostic criteria in the WHO classification of central nervous system tumors. Belonging to one of the Ewing sarcoma family of tumors, PNET shares similarities with Ewing sarcoma.6 The incidence rate of PNET is reported at 2.9 cases per million individuals. Given the rarity of pPNET and its potential occurrence at multiple anatomical sites, existing literature predominantly comprises studies with limited sample sizes, resulting in variations in clinical presentations, therapeutic approaches, and prognostic indicators across different investigations. PNET originating from the female reproductive system are exceptionally uncommon, with sporadic reports in both domestic and international literature. The ovary is the most frequently affected site, followed by the uterine body, cervix, vagina, and vulva.7–9 To date, over 50 cases of uterine PNET have been documented in the literature worldwide.
Clinical Features and Imaging Examinations
In the female reproductive system, PNET can manifest variably based on the disease’s location. Symptoms may encompass an abdominal mass, abdominal discomfort, bloating, vulvar swelling, vulvar pain, and irregular uterine bleeding. Existing literature suggests that individuals with uterine PNET are frequently diagnosed at an advanced stage, in contrast to endometrial cancer, which is commonly identified early.6 Radiological assessments typically disclose findings such as a pelvic mass, uterine cavity mass, endometrial thickening, and abnormal uterine enlargement, among others. Specific imaging characteristics are not consistently observed. Research indicates that PNETs arising in the reproductive system can emerge across all age groups, including puberty, childbearing years, and menopause.10 Rare instances of uterine PNET during pregnancy have also been documented. Notably, there is no singular tumor marker specific to this tumor type. Some patients may exhibit slight elevations in CA125 or NSE levels. CA125 is commonly utilized as a prognostic indicator and for post-treatment monitoring in clinical settings.11 In the described case, the patient primarily presented with abnormal uterine bleeding and persistent menstruation. Despite pharmacological intervention, notable improvement was not observed. Ultrasound imaging revealed a mass within the uterine cavity, accompanied by elevated levels of the tumor marker NSE and normal CA125 values.
Pathological and Immunohistochemical Characteristics
The typical presentation of general pathology involves the development of tumors with varying sizes, typically ranging from 2 to 20 cm. These tumors are predominantly solid in texture and often do not have a distinct capsule. Upon examination through sectioning, they exhibit a yellowish-white appearance, frequently showing signs of hemorrhage and necrosis. Microscopically, the tumors display densely arranged small round cells that form nests and cord-like structures. A key identifying feature of PNET is the presence of H–W (Homer–Wright) rosettes in a majority of cases, although their absence is also noted in some instances.12–14 Around 50% of PNET patients demonstrate the characteristic Homer–Wright rosettes in pathological sections. Therefore, it is crucial to confirm the diagnosis by analyzing histopathological features in conjunction with IHC or ultrastructural features. The current diagnostic markers utilized for identifying Ewing’s sarcoma/PNET family tumors include MIC2 (CD99), neurofilament protein, neuron-specific enolase, vimentin, and HBA-71. These markers are known to be widely expressed on tumor cell membranes, with CD99 and FLI-1 present in over 97% of cases, while CD20 and CD15 markers are absent.15 Additionally, some tumor cells exhibit ultrastructural vimentin.16
Moreover, NSE, chromogranin, synaptophysin, and S100 exhibit high levels of expression. The majority of peripheral PNET cases, approximately 85%, have been documented to possess a balanced translocation t (11;22) (q24;q12), leading to the presence of EWS-FLI1 chimeric transcripts, a feature not observed in central PNETs. Uterine PNET are expected to display peripheral characteristics, in conjunction with EWSR1 translocation. Nevertheless, in certain instances, this chromosomal translocation may not be identified through FISH testing, necessitating the classification of the tumor as central. Elizalde et al initially proposed a classification system for uterine PNETs, distinguishing between central and peripheral types. The central type lacks chromosomal translocation, while the peripheral type is characterized by EWSR1 translocation.17 Findings from the research conducted by Chiang et al underscore the importance of utilizing a combination of morphological, immunohistochemical, and molecular genetic analyses to differentiate between central and peripheral types. Morphologically, the central type resembles central nervous system tumors like medulloblastoma, ependymoma, medullary epithelioma, glioblastoma, among others, whereas the peripheral type lacks these features.8 In gynecopathological practice, uterine PNET must be differentially diagnosed from other common small round cell tumors to avoid misdiagnosis due to morphological overlap. Although small cell carcinoma is also composed of small round cells, it typically expresses neuroendocrine markers (such as chromogranin and synaptophysin) and may also express epithelial markers like CK and TTF-1, whereas CD99 and FLI-1 are usually negative.18 Furthermore, small cell carcinoma often exhibits coarse chromatin and inconspicuous nucleoli, which differs structurally from the Homer–Wright rosettes that may be present in PNET. The tumor cells of low-grade endometrial stromal sarcoma are mostly spindle-shaped and often arranged in a woven or whorled pattern, with characteristic infiltrative growth into the myometrium forming a “tongue-like” pattern. These sarcoma cells typically express CD10, ER, and PR, while PNET-related markers such as CD99 are negative.19 Lymphoma cells show a diffuse infiltration pattern but lack the nested or cord-like arrangements commonly seen in PNET. Their immunophenotype is lineage-specific: B-cell lymphomas express CD20 and CD79a, T-cell lymphomas express CD3, among other markers, and CD45 is generally positive.20 These collective findings consolidated the diagnosis of PNET. Therefore, a comprehensive approach involving detailed morphological assessment, immunohistochemical profiling, and EWSR1 FISH analysis is essential for distinguishing between central gynecologic PNET and Ewing sarcoma/peripheral type. As previously reported, cPNET and pPNET exhibit distinct prognostic outcomes. Accurate differentiation between central and peripheral PNETs of the female reproductive system can facilitate precise diagnosis and treatment planning for patients with this rare tumor subtype.
In the present case, unavailability of EWSR1 gene translocation testing precluded definitive molecular classification of PNET as peripheral or central, limiting precise treatment stratification. Nevertheless, this highlights the critical role of integrating morphological and comprehensive IHC analyses for accurate diagnosis when molecular data is lacking. Morphologically, characteristic Homer-Reit rose bunches and densely packed small round cells arranged in nests or strands were identified, thereby ruling out overlapping entities such as endometrial stromal sarcoma. By integrating morphological findings with IHC, clinicians can establish an effective diagnostic framework even in the absence of molecular information. This strategy reduces reliance on genetic testing, minimizes delays in diagnosis, decreases the risk of misdiagnosis, and helps ensure patients receive treatment within a critical window.
Treatment and Prognosis
The original neuroectodermal tumor of the female reproductive system is a relatively uncommon occurrence, and as of yet, there is no established definitive treatment protocol. Typically, surgical intervention was the primary course of treatment.6,21 Predominantly found in the ovaries and uterine body, tumors are often addressed through total hysterectomy coupled with pelvic and abdominal lymph node dissection. In cases where tumors are situated in the cervix, radical total hysterectomy with pelvic and abdominal lymph node dissection is the recommended course of action. These tumors primarily spread through the bloodstream, with a majority of patients being diagnosed with distant metastases. In addition, chemotherapy is an indispensable part, especially for advanced cases.6 Platinum drugs and etoposide are the most commonly used combination of chemotherapy drugs,4,22 which is consistent with the treatment scheme used in this case. Various combination regimens, such as VDCA, VDCA-IE, and VAC,23,24 incorporating drugs like vincristine, doxorubicin, cyclophosphamide, ifosfamide, and etoposide, are frequently utilized. In addition, stage I disease, younger age and chemotherapy based on sarcoma all show higher overall survival rate.6 Our case was diagnosed as primary endometrium PNET at an advanced stage with a poor prognosis and eventually died.
The VDC-IE regimen can lead to significant myelosuppression, necessitating careful monitoring by inexperienced gynecologists to promptly address potential adverse effects. Treatment of gynecological PNET with Ewing sarcoma chemotherapy may result in frequent discontinuation of cycles, particularly in perimenopausal women. Literature suggests that positive cytoplasmic staining of vascular endothelial growth factor antibody (VEGF) and the use of bevacizumab can be beneficial for patients.25 Radiotherapy is typically reserved for inoperable patients or those with positive margins post-surgery. The prognosis for these conditions is generally poor compared to other reproductive system malignancies, with the impact of age on prognosis remaining a topic of debate. Clinical studies have indicated that adult patients with Ewing sarcoma and pPNET have a notably worse prognosis than pediatric patients, with two-year survival rates varying by disease stage. Stage IV patients have a particularly grim prognosis, with nearly all succumbing within two years. The prognostic evaluation of this case, however, is constrained by the absence of long-term survival data, as the patient was lost to follow-up after completing only four cycles of an attenuated chemotherapy regimen. Consequently, both overall survival and progression-free survival remain undetermined, limiting insights into the natural history and therapeutic response of primary endometrium PNET in this individual. Furthermore, the chemotherapy course in this case highlights the challenges of treatment tolerance. The initial standard regimen had to be modified and ultimately discontinued due to severe toxicities, notably myelosuppression. This not only illustrates the practical obstacles in administering aggressive protocols like VDC-IE but also suggests that failure to complete a full course of standard systemic chemotherapy likely served as a key negative prognostic factor. Therefore, alongside pursuing therapeutic efficacy, effectively managing chemotherapy-related toxicity, maintaining treatment intensity, and improving patient follow-up to prevent data loss are crucial directions for improving outcomes in such patients.
Conclusions
PNET originating from the internal genitalia is an uncommon neoplasm associated with a grim prognosis. Its diagnosis is complicated by the absence of distinct clinical and imaging features, rendering early detection a formidable task. Pathological examination serves as the cornerstone for diagnosis.21 Discriminating primary endometrium PNET and identifying an optimal therapeutic approach represent valuable endeavors. These efforts have the potential to enhance diagnostic precision and treatment efficacy for individuals afflicted by this infrequent malignancy.
Abbreviations
CA125, carbohydrate antigen 125; cPNET, central PNET; HE, Hematoxylin and Eosin; IHC, immunohistochemistry; NSE, neuron-specific enolase; pPNET, peripheral PNET; PNET, Primitive neuroectodermal tumor; VEGF, vascular endothelial growth factor antibody.
Data Sharing Statement
The data in this study is unavailable due to privacy of the patient.
Ethical Statement
The study was conducted in accordance with the Declaration of Helsinki and approved by the Institutional Review Board of Jinling Hospital, Affiliated Hospital of Medical, Nanjing University. The approval number was 2025-DZKY-106-01.
Informed Consent Statement
Informed written consent was obtained from the patient for the publication of this report and any accompanying images.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This work was funded by the Intramural Research Grant of Jinling Hospital Affiliated to the School of Medicine, Nanjing University (Grant No. 2023LCYYQH016 and X20241540).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Chhabda S, Carney O, D’Arco F, Jacques TS, Mankad K. The 2016 World Health Organization Classification of tumours of the Central Nervous System: what the paediatric neuroradiologist needs to know. Quant Imaging Med Surg. 2016;6(5):486–7. doi:10.21037/qims.2016.10.01
2. Khosla D, Rai B, Patel FD, Sreedharanunni S, Dey P, Sharma SC. Primitive neuroectodermal tumor of the uterine cervix diagnosed during pregnancy: a rare case with review of literature. J Obstet Gynaecol Res. 2014;40(3):878–882. doi:10.1111/jog.12238
3. Elmuti L, Amundson J, Oberman E, et al. Diffuse pediatric-type high-grade glioma arising in an ovarian mature cystic teratoma. Int J Gynecol Pathol. 2024;43(1):90–96. doi:10.1097/pgp.0000000000000954
4. Khorasanchi A, Kertowidjojo E, Kim S, Burke W, Kudelka A. Primitive neuroectodermal tumor of the uterus: excellent clinical response following a multimodal treatment approach. Gynecol Oncol Rep. 2020;32:100550. doi:10.1016/j.gore.2020.100550
5. Xiao C, Zhao J, Guo P, et al. Clinical analysis of primary primitive neuroectodermal tumors in the female genital tract. Int J Gynecological Cancer. 2014;24(3):404–409. doi:10.1097/igc.0000000000000082
6. Weinstein S, Walker AR, Barr K, et al. Case report and review of the literature regarding management of primary uterine primitive neuroectodermal tumors (PNET). Gynecol Oncol Rep. 2025;59:101775. doi:10.1016/j.gore.2025.101775
7. Feng X, Zhang L, Tan Y, et al. Primitive neuroectodermal tumor of the cervix diagnosed during pregnancy: a rare case report with discussion. BMC Pregnancy Childbirth. 2021;21(1):382. doi:10.1186/s12884-021-03859-6
8. Chiang S, Snuderl M, Kojiro-Sanada S, et al. Primitive neuroectodermal tumors of the female genital tract: a morphologic, immunohistochemical, and molecular study of 19 cases. Am J Surg Pathol. 2017;41(6):761–772. doi:10.1097/pas.0000000000000831
9. Mekheal E, Kania B, Vishwakarma U, Joseph D, Kumar V, Maroules M. A rare case of a peripheral Ewing sarcoma primitive neuroectodermal tumor of pelvic origin. Radiol Case Rep. 2023;18(4):1437–1441. doi:10.1016/j.radcr.2023.01.002
10. Snijders-Keilholz A, Ewing P, Seynaeve C, Burger CW. Primitive neuroectodermal tumor of the cervix uteri: a case report -- changing concepts in therapy. Gynecologic Oncol. 2005;98(3):516–519. doi:10.1016/j.ygyno.2005.05.020
11. Smoligová V, Kosťun J, Stráník P, Presl J. Včasná detekce rekurentního karcinomu vaječníků, současné využití onkomarkerů, zobrazovací metody a budoucí perspektivy [Early detection of recurrent ovarian cancer, current use of oncomarkers, imaging methods, and future perspectives]. Ceska Gynekol. 2025;90(4):333–338. doi:10.48095/cccg2025333
12. Jia P, Cui R, Zhang Y. Primary Ewing sarcoma in the uterine cervix with multiple bone metastases. Arch Gynecol Obstetrics. 2022;306(3):911–912. doi:10.1007/s00404-022-06613-1
13. Han LM, Weiel JJ, Longacre TA, Folkins AK. DICER1-associated tumors in the female genital tract: molecular basis, clinicopathologic features, and differential diagnosis. Adv Anat Pathol. 2022;29(5):297–308. doi:10.1097/pap.0000000000000351
14. Benitez Delgado T, Laseca-Modrego M, Gonzalez Garcia-Cano D, Rave Ramirez A, Arencibia-Sánchez O. Uterine primitive neuroectodermal tumor. Cureus. 2021;13(7):e16437. doi:10.7759/cureus.16437
15. Tintila A, Doroftei B, Grab D, et al. Importance of studying primitive neuroectodermal tumors and extraosseous Ewings sarcoma of the vagina and vulva. Oncol Lett. 2021;21(2):171. doi:10.3892/ol.2021.12432
16. Rekhi B, Qureshi S, Basak R, et al. Primary vaginal Ewing’s sarcoma or primitive neuroectodermal tumor in a 17-year-old woman: a case report. J Med Case Rep. 2010;4:88. doi:10.1186/1752-1947-4-88
17. Jay V, Pienkowska M, Becker L, Zielenska M. Primitive neuroectodermal tumors of the cerebrum and cerebellum: absence of t(11;22) translocation by RT-PCR analysis. Mod Pathol. 1995;8(5):488–491.
18. Stachs A, Makovitzky J, Briese V. Small cell carcinoma of the endometrium: light microscopic and immunohistochemical study of a case. Anticancer Res. 2005;25(3a):1823–1825.
19. Zhao X, Chen C, Xie T, Wang L. Low-grade endometrial stromal sarcoma: a case report and literature review. Front Oncol. 2025;15:1652010. doi:10.3389/fonc.2025.1652010
20. Cho J. Basic immunohistochemistry for lymphoma diagnosis. Blood Res. 2022;57(S1):55–61. doi:10.5045/br.2022.2022037
21. Prasad I, Meena A, Kumari M, Singh P, Ismail AA. Primary uterine primitive neuroectodermal tumour mistaken for leiomyosarcoma in an adolescent girl: a very rare case with many diagnostic and therapeutic challenges. Cureus. 2026;18(3):e105020. doi:10.7759/cureus.105020
22. Tek Z, Laibangyang A, Odujoko O, Khandpur B, Doo D. Ewing sarcoma of the uterus: a case report. Case Rep Womens Health. 2024;43:e00640. doi:10.1016/j.crwh.2024.e00640
23. Grier HE, Krailo MD, Tarbell NJ, et al. Addition of ifosfamide and etoposide to standard chemotherapy for Ewing’s sarcoma and primitive neuroectodermal tumor of bone. New Engl J Med. 2003;348(8):694–701. doi:10.1056/NEJMoa020890
24. Ehrlich Y, Beck SDW, Ulbright TM, et al. Outcome analysis of patients with transformed teratoma to primitive neuroectodermal tumor. Ann Oncol. 2010;21(9):1846–1850. doi:10.1093/annonc/mdq045
25. Novo J, Bitterman P, Guirguis A. Central-type primitive neuroectodermal tumor of the uterus: case report of remission of stage IV disease using adjuvant cisplatin/etoposide/bevacizumab chemotherapy and review of the literature. Gynecol Oncol Rep. 2015;14:26–30. doi:10.1016/j.gore.2015.09.002
 © 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.