Back to Journals » Clinical, Cosmetic and Investigational Dermatology » Volume 16
Primary Cutaneous T-Cell/Histiocyte-Rich B-Cell Lymphoma: A Case Report and Literature Review
Authors Al Harbi SM, Al Ghamdi NJ ![]() , Elsharkawy TM, Al Hamad MA
, Elsharkawy TM, Al Hamad MA ![]() , Bajawi S
, Bajawi S
Received 1 November 2022
Accepted for publication 19 January 2023
Published 1 February 2023 Volume 2023:16 Pages 309—316
DOI https://doi.org/10.2147/CCID.S395675
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Jeffrey Weinberg
Sadan Mohammed Al Harbi,1 Nada Jomaan Al Ghamdi,1 Tarek Mohamed Elsharkawy,2 Mohammad Abdelqader Al Hamad,2 Sultan Bajawi1
1Department of Dermatology, College of Medicine, Imam Abdulrahman Bin Faisal University, Dammam, Eastern Province, Saudi Arabia; 2Department of Pathology, College of Medicine, Imam Abdulrahman Bin Faisal University, Dammam, Eastern Province, Saudi Arabia
Correspondence: Sadan Mohammed Al Harbi, Department of Dermatology, College of Medicine, Imam Abdulrahman Bin Faisal University, Post Box No. 1982, Dammam, 31441, Saudi Arabia, Email [email protected]
Abstract: T-cell/histiocyte-rich large B-cell lymphoma (THRLBCL) is a lymphoproliferative disorder in which the majority of cells are reactive T cells with only a minor population of neoplastic large B cells. THRLBCL is a very rare lymphoma, and most cases are nodal THRLBCL; an extranodal case of THRLBCL presenting primarily on the skin is an extremely rare occurrence with only a few cases reported in the literature. Here, we report a case of a primary cutaneous THRLBCL in a 41-year-old Saudi male who presented unusually with multiple skin lesions. He was successfully treated with electron beam radiotherapy and had a complete resolution with no recurrence as of his 24-month follow-up.
Keywords: T-cell-rich B-cell lymphoma, lymphoproliferative disorder, radiotherapy, neoplastic B cells
Introduction
T-cell/histiocyte-rich large B-cell lymphoma (THRLBCL) is a very rare lymphoma that can present as nodal or extra nodal.1,2 The majority (75%) of cases are nodal.3 Primary cutaneous THRLBCL, an extranodal from, is a very rare entity with only 17 cases reported in the literature.3–15 THRLBCL was first described in 1988 by Ramsay et al as a lymphoproliferative disorder with the majority of cells being reactive T-cells and neoplastic large B-cells accounting for less than 10% of the cells.16 In this case study, we describe a 41-year-old Saudi male as an additional case of primary cutaneous THRLBCL presenting unusually with multiple skin lesions.
Case Report
A 41-year-old Saudi man presented to our dermatology clinic at King Fahad Hospital of the University with multiple grouped erythematous nodular lesions on the left upper chest and another solitary erythematous nodule located on the left upper back that had been present for three years. The condition started as multiple small painless nonpruritic pink papules on his chest that had been slowly growing for over two years. He sought medical advice at a primary care center, where he was given topical steroids and topical antibiotics. Several months after his initial presentation, he discovered another slightly larger lesion on his back. Both papular lesions gradually grew into larger nodules, but the patient was completely asymptomatic. He denied any history of itching, pain, bleeding, discharge, or ulcerations within the nodules. He had no history of constitutional symptoms such as fever, night sweats, weight loss, appetite loss, or generalized body pruritus.
On examination, the lesion on the left upper chest was poorly demarcated, infiltrated, and had an erythematous plaque with three small nodules on the upper medial side with a raised medial border. The plaque had a maximum diameter of 5.0 cm x 2.0 cm, with no ulceration and no tenderness (Figure 1A). The lesion on the left upper back was found to be a well demarcated erythematous nodule with maximum diameter of 2.5 cm x 2.0 cm, and it was not tender on palpation (Figure 1B). On general examination, no other skin abnormalities, hepatosplenomegaly, or lymphadenopathy were identified.
 |
Figure 1 (A) Lesion on the upper left chest showing a poorly demarcated erythematous plaque with infiltrating small nodules over the upper medial border. The plaque is 5.0 cm in maximum diameter × 2.0 cm. (B) Lesion on the upper left back showing a well demarcated erythematous nodule. The nodule is 2.5 cm in maximum diameter × 2 cm. |
The histopathological examination of skin biopsies from both sites showed epidermis and dermis. The superficial and deep dermis both showed patchy dense lymphoid infiltrate. There was no epidermotropism. The dermal infiltrate consisted of large lymphoid cells with round to oval abnormal nuclei surrounded by small mature lymphocytes in excess of 70% (Figure 2). The dermal infiltrate was also periadnexal, perivascular, and dissected the dermal collagens.
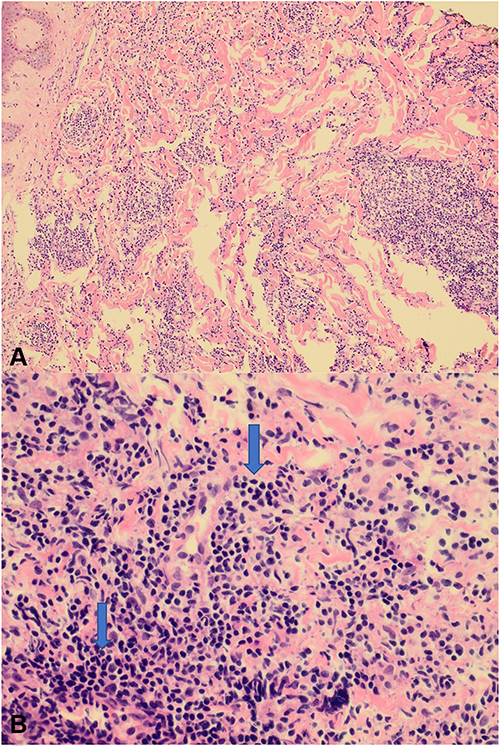 |
Figure 2 (A) (200×) The superficial and deep dermis show patchy dense lymphoid infiltrate. (B) (400×) The dermal infiltrate consists of large lymphoid cells (arrows) with round to oval abnormal nuclei surrounded by small mature lymphocytes in excess. |
Immunohistochemical stains of the large cells showed strong positivity with CD20 (Figure 3A) and CD79a, fair positivity with CD43 and paired box protein 5. The small lymphocytic populations were positive for CD3 (Figure 3B), CD5, and CD7. Immunohistochemical staining for CD30, epithelial membrane antigen (EMA), BCL2, and CD56 were all negative. A fluorescence in situ hybridization (FISH) analysis for the tumor cells revealed no evidence of rearrangement (break-apart) in IGH, BCL6, or t(14;18)(q32;q21). Checks for metastasis, including a complete blood count, test for liver and renal function, a lactate dehydrogenase level test, a peripheral blood picture, a chest x-ray, a computed tomography scan of the chest, abdomen, and pelvis, a positron emission tomography scan, and a bone marrow biopsy, were all normal and unremarkable. The patient, after being treated with electron beam radiotherapy at a dose of 24 Gy delivered in 12 daily fractions (2 Gy/fraction) and square field size of 25×25 cm2 on his left upper chest, and 20×20 cm2 on his left upper back, showed complete resolution and no recurrence within a 24-month follow-up time frame (Figure 4).
 |
Figure 3 (A) (200×) Immunohistochemical stains show strong positivity of the large cells with CD20. (B) (200×) The reactive small lymphocytic populations are positive for CD3 immunohistochemical stain. |
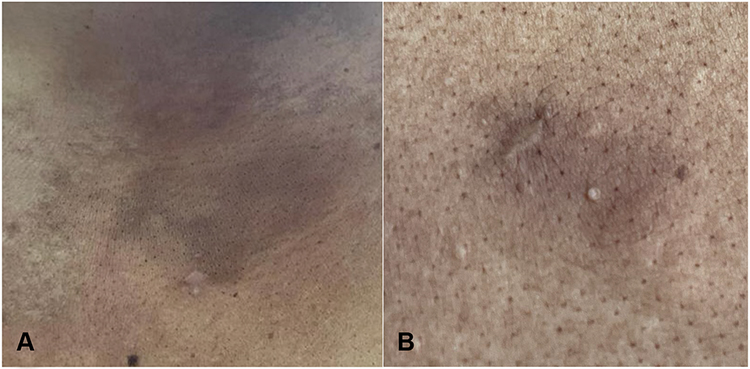 |
Figure 4 Sites of the lesions after completing 12 sessions of electron beam radiotherapy showing post-inflammatory hyperpigmented patches and scars of previous skin punch biopsies. (A) Lesion on the upper left chest. (B) Lesion on the upper left back. |
Fluorescence in situ Hybridization (FISH)
FISH for immunoglobulin heavy-chain (IGH) break-apart (Vysis LIS IGH Dual Color, Break Apart Rearrangement Probe), BCL6 break-apart (Vysis LIS BCL6 Dual Color, Break Apart Rearrangement Probe), and IGH-BCL2 chromosomal translocation (Vysis LSI IGH/BCL2 Dual Color, Dual Fusion Translocation Probe Set) was conducted on formalin-fixed paraffin-embedded tumor tissue according to the manufacturer’s protocol (Abbott). Signals were visualized under a Zeiss Axioskop microscope (Zeiss, Germany) using a Fluorescein-5-isothiocyanate/Rhodamine dual-band filter. Criteria from the American Society of Clinical Oncology were used as guidelines when analyzing the results.17 The guideline criteria stated that a result with one fusion plus one green and one orange signal is considered break-apart for IGH and BCL6, while a result with two fusion signals is considered normal. For the t(14;18)(q32;q21) probe, a result with one fusion plus one green and one orange signal is considered positive for t(14;18)(q32;q21) while a result with two green plus two orange signals is considered normal (Figure 5).
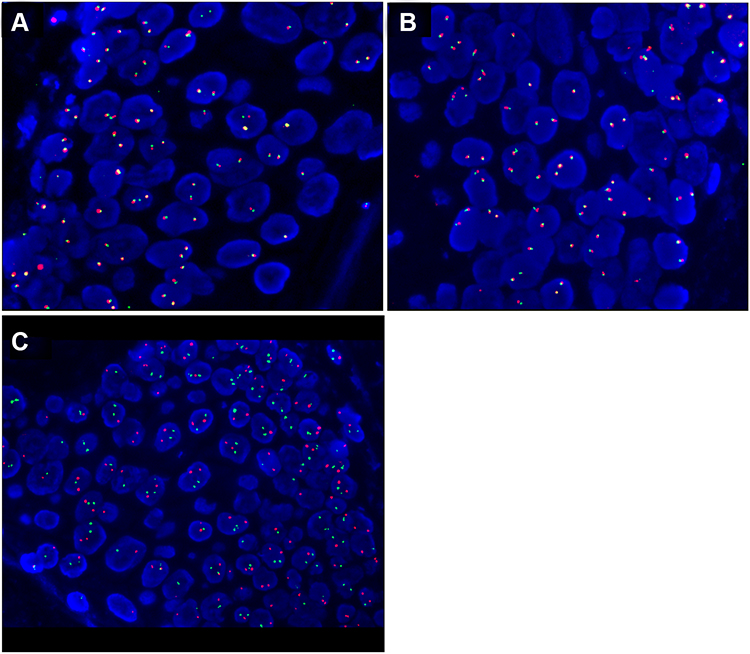 |
Figure 5 Fluorescence in situ hybridization (FISH) of (A) IGH break-apart (two fusion signals). (B) BCL6 break-apart (two fusion signals). (C) t(14;18)(q32;q21) (two green and two Orange signals). The tumor cells reveal no evidence of rearrangement (break-apart) in the IGH and BCL6 genes or in t(14;18)(q32;q21). |
Discussion
According to the 2016 World Health Organization (WHO) classification of hematopoietic and lymphoid neoplasms, THRLBCL is considered a distinct category of diffuse large B cell lymphoma.18,19 THRLBCL is characterized by a paucity of neoplastic B cells scattered among abundant reactive T-cells and histiocytes.1 THRLBCL is a very rare entity.20 It makes up only 1–2% of all non-Hodgkin’s lymphoma cases, and can present as nodal or extranodal.2,3 Nodal THRLBCL is the most common presentation at 75% of all THRLBCL cases and is usually present in an advanced stage (III or IV).3,21 In 2018, the primary cutaneous B-cell lymphoma was classified by the WHO/European Organization for Research and Treatment of Cancer into six major groups: 1) primary cutaneous marginal zone B-cell lymphoma; 2) primary cutaneous follicle center lymphoma; 3) primary cutaneous diffuse large B-cell lymphoma, leg type; 4) Epstein-Barr virus (EBV+) mucocutaneous ulcer; 5) intravascular large B-cell lymphoma; 6) primary cutaneous diffuse large B-cell lymphoma, not otherwise specified (NOS).20 Primary cutaneous THRLBCL falls within the category of primary cutaneous diffuse large B-cell lymphoma, NOS.20,22 Primary cutaneous THRLBCL usually has an indolent course with a good prognosis with no tendency to metastasize compared to its nodal counterpart, and it is even rarer with only 17 cases reported in the English literature (Table 1).3–15 Histologically, it presents as diffuse dermal lymphocytic infiltration, and it may involve the subcutaneous fat. It is composed of a mixture of small reactive CD3+ T-cell lymphocytes and neoplastic large CD20+ B-cell lymphocytes. A wide range of B-cell population percentages have been described in THRLBCL. Ramsay et al, the first to use the term THRLBCL in 1988, stated that the percentage of neoplastic B cells should not exceed 10% of a total cell population, while other experts (Ng et al, U. Wollina and Watabe et al) reported a neoplastic B cell component up to 50% of the total cell.5,8,16,23 The large B cells are atypical with clear cytoplasm and occasional multilobate nuclei. Epidermotropism and Reed-Sternberg cells are almost never found.
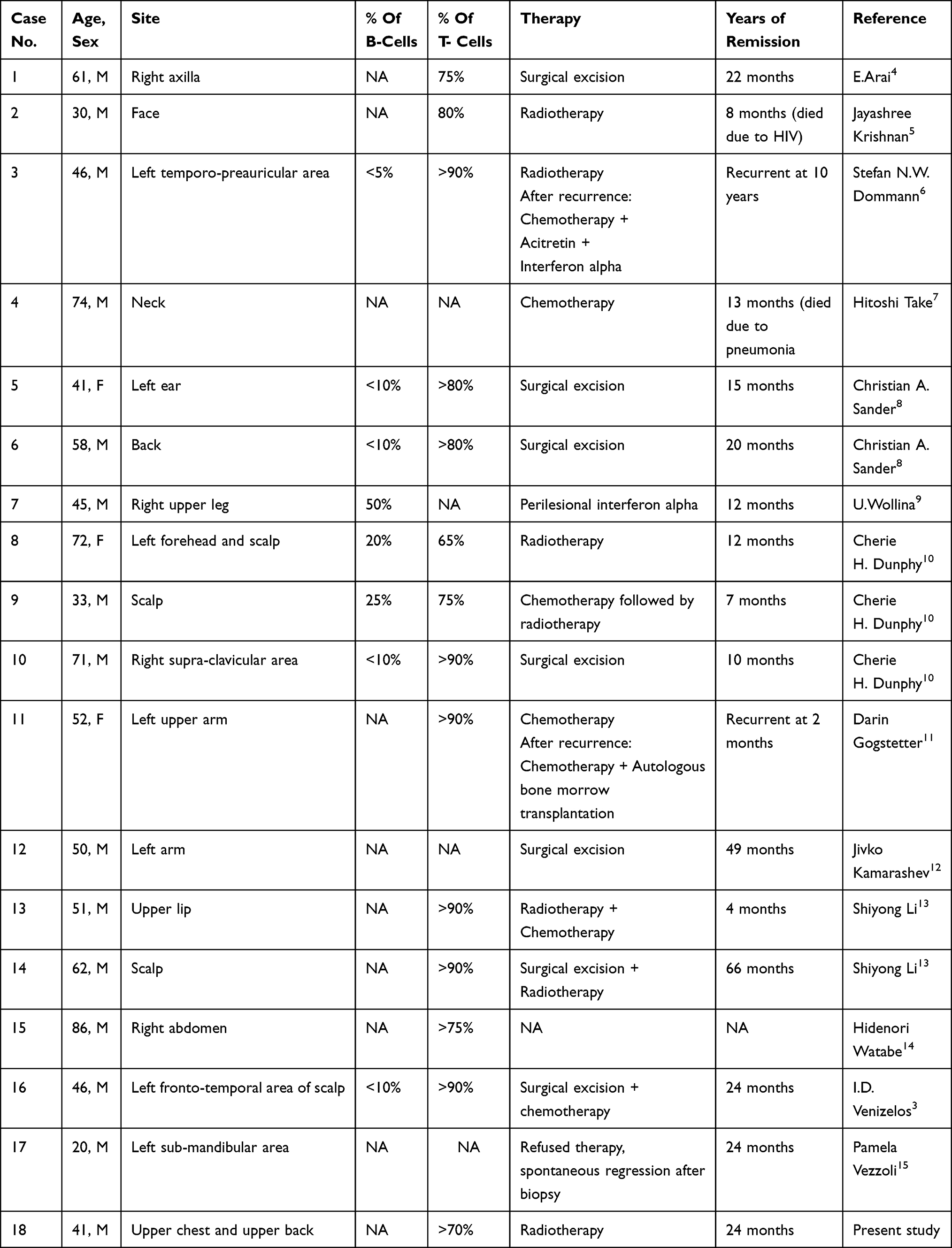 |
Table 1 Summary of the Clinical Data on Reported Cases of Primary Cutaneous THRLBCL |
All of the 17 primary cutaneous THRLBCL cases described in the literature were stage 1E, and this is also true for our case.3–15 The clinical data of these 18 cases are summarized in Table 1. Most of the patients were male (15 males, 3 females), and age at the time of diagnosis ranged from 20 to 87 years with a median of 50.5 years (average 49.6 years).
The usual presentation of primary cutaneous THRLBCL is a solitary erythematous plaque or nodule. Our case was unusual in that it presented with multiple lesions; the first lesion appeared as erythematous to brown infiltrating papules that coalesced to form a plaque resembling a dermatofibrosarcoma protuberans morphology on the left upper chest; a few months later an erythematous nodule appeared on the upper back.
On histopathological examination, our case showed patchy dense lymphoid infiltrates involving the upper and deep dermis with no epidermotropism. The dermal infiltrate consisted of large lymphoid cells with round to oval nuclei surrounded by small mature lymphocytes in excess of 70%. The dermal infiltrate was also periadnexal, perivascular, and dissected the dermal collagens. Immunohistochemical analysis confirmed the diagnosis. The Large atypical cells were positive for CD20, CD79a, and CD43, while negative for CD30, BCL2, EMA, and Epstein-Barr encoded RNA. Kappa and lambda restriction were absent, which can be attributed to the small number of neoplastic B cells in THRLBCL. The small reactive T cells were positive for CD3, CD5, and CD7. Lastly, CD56 was negative.
Multiple bacterial and viral infections have been linked to the pathogenesis of B-cell lymphoma. In particular, the Epstein-Barr virus was reported in two primary cutaneous THRLBCL cases.7,14 However, our patient had no evidence of EBV infection.
Using FISH, we learned that the tumor cells revealed no evidence of rearrangement (break-apart) in IGH and BCL6 or t(14;18)(q32;q21). This finding is consistent with Wang et al, who described a lack of BCL2 rearrangement in 21 THRLBCL cases. However, he detected IGH rearrangement in 18 out of 21 cases.24 These findings support the heterogeneity of the proliferation of B cells in THRLBCL.
Our review of the literature has revealed no well documented first-line therapy for primary cutaneous THRLBCL. Surgical excisions, radiotherapy, and chemotherapy have all been used. In all cases where surgical excision was done, there was no evidence of recurrence. Pamela Vezzoli et al reported a case where a patient refused therapy and spontaneous remission occurred.4 Intralesional interferon alpha was used to treat one patient and resulted in a complete resolution with no recurrence at a 1-year follow-up.9 As mentioned earlier, the treatment in this case was electron beam radiotherapy at a dose of 24 Gy delivered in 12 daily fractions of 2 Gy each. The patient had a complete resolution with no evidence of recurrence throughout a follow-up lasting 24 months.
Conclusion
T-cell/histiocyte-rich large B-cell lymphoma is a very rare lymphoma in which nodal THRLBCL forms the majority of reported cases. Primary cutaneous THRLBCL is a very rare entity with only 17 cases reported in the English literature. We report this case of a 41-year-old Saudi man with primary cutaneous THRLBCL for its rarity, presentation of unusual multiple skin lesions, and positive treatment results.
Abbreviations
THRLBCL, T-cell/histiocyte-rich B-cell lymphoma; EMA, epithelial membrane antigen; FISH, fluorescence in situ hybridization; IGH, immunoglobulin heavy-chain; WHO, World Health Organization; EBV, Epstein-Barr virus; NOS, not otherwise specified.
Data Sharing Statement
All data for this scientific paper are provided within the article.
Ethics Approval and Consent for Publication
This case report has been performed in accordance with the principles stated in the Declaration of Helsinki. A written informed consent, provided by the Taylor & Francis group® for publication of this case report and including photography and medical information, was signed by the patient. Institutional ethical approval was not required to publish this case details.
Acknowledgments
The authors would like to thank all department colleagues who contributed to this case study.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis, and interpretation, or in all these areas; took part in drafting, revising, or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interests in this reported work.
References
1. Ott G. Aggressive B-cell lymphomas in the update of the 4th edition of the World Health Organization classification of haematopoietic and lymphatic tissues: refinements of the classification, new entities and genetic findings. Br J Haematol. 2017;178(6):871–887. doi:10.1111/bjh.14744
2. Lima M. Cutaneous primary B-cell lymphomas: from diagnosis to treatment. An Bras Dermatol. 2015;90(5):687–706. doi:10.1590/abd1806-4841.20153638
3. Venizelos ID, Tatsiou ZA, Mandala E. Primary cutaneous T-cell-rich B-cell lymphoma: a case report and literature review. Acta Dermatovenerol Alp Pannonica Adriat. 2008;17(4):177–181.
4. Arai E, Sakurai M, Nakayama H, Morinaga S, Katayama I. Primary cutaneous T-cell-rich B-cell lymphoma. Br J Dermatol. 1993;129(2):196–200. doi:10.1111/j.1365-2133.1993.tb03529.x
5. Krishnan J, Wallberg K, Frizzera G. T-cell-rich large B-cell lymphoma. A study of 30 cases, supporting its histologic heterogeneity and lack of clinical distinctiveness. Am J Surg Pathol. 1994;18(5):455–465. doi:10.1097/00000478-199405000-00004
6. Dommann SN, Dommann-Scherrer CC, Zimmerman D, Dours-Zimmermann MT, Hassam S, Burg G. Primary cutaneous T-cell-rich B-cell lymphoma. A case report with a 13-year follow-up. Am J Dermatopathol. 1995;17(6):618–624. doi:10.1097/00000372-199512000-00017
7. Take H, Kubota K, Fukuda T, Shinonome S, Ishikawa O, Shirakura T. An indolent type of Epstein-Barr virus-associated T-cell-rich B-cell lymphoma of the skin: report of a case. Am J Hematol. 1996;52(3):221–223. doi:10.1002/(SICI)1096-8652(199607)52:3<221::AID-AJH16>3.0.CO;2-I
8. Sander CA, Kaudewitz P, Kutzner H, et al. T-cell-rich B-cell lymphoma presenting in skin. A clinicopathologic analysis of six cases. J Cutan Pathol. 1996;23(2):101–108. doi:10.1111/j.1600-0560.1996.tb01282.x
9. Wollina U. Complete response of a primary cutaneous T-cell-rich B cell lymphoma treated with interferon alpha2a. J Cancer Res Clin Oncol. 1998;124(2):127–129. doi:10.1007/s004320050144
10. Dunphy CH, Nahass GT. Primary cutaneous T-cell-rich B-cell lymphomas with flow cytometric immunophenotypic findings. Report of 3 cases and review of the literature. Arch Pathol Lab Med. 1999;123(12):1236–1240. doi:10.5858/1999-123-1236-PCTCRB
11. Gogstetter D, Brown M, Seab J, Scott G. Angiocentric primary cutaneous T-cell-rich B-cell lymphoma: a case report and review of the literature. J Cutan Pathol. 2000;27(10):516–525. doi:10.1034/j.1600-0560.2000.027010516.x
12. Kamarashev J, Dummer R, Schmidt MH, Kempf W, Kurrer MO, Burg G. Primary cutaneous T-cell-rich B-cell lymphoma and Hodgkin’s disease in a patient with Gardner’s syndrome. Dermatology. 2000;201(4):362–365. doi:10.1159/000051557
13. Li S, Griffin CA, Mann RB, Borowitz MJ. Primary cutaneous T-cell-rich B-cell lymphoma: clinically distinct from its nodal counterpart? Mod Pathol. 2001;14(1):10–13. doi:10.1038/modpathol.3880250
14. Watabe H, Kawakami T, Soma Y, Baba T, Mizoguchi M. Primary cutaneous T-cell-rich B-cell lymphoma in a zosteriform distribution associated with Epstein-Barr virus infection. J Dermatol. 2002;29(11):748–753. doi:10.1111/j.1346-8138.2002.tb00215.x
15. Vezzoli P, Fiorani R, Girgenti V, et al. Cutaneous T-cell/histiocyte-rich B-cell lymphoma: a case report and review of the literature. Dermatology. 2011;222(3):225–230. doi:10.1159/000327376
16. Ramsay AD, Smith WJ, Isaacson PG. T-cell-rich B-cell lymphoma. Am J Surg Pathol. 1988;12(6):433–443. doi:10.1097/00000478-198806000-00003
17. Wolff DJ, Bagg A, Cooley LD. Guidance for fluorescence in situ hybridization testing in hematologic disorders. J Mol Diagn. 2007;9(2):134–143. doi:10.2353/jmoldx.2007.060128
18. Campo E, Swerdlow SH, Harris NL, Pileri S, Stein H, Jaffe ES. The 2008 WHO classification of lymphoid neoplasms and beyond: evolving concepts and practical applications. Blood. 2011;117(19):5019–5032. doi:10.1182/blood-2011-01-293050
19. Swerdlow SH, Campo E, Pileri SA, et al. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood. 2016;127(20):2375–2390. doi:10.1182/blood-2016-01-643569
20. Willemze R, Cerroni L, Kempf W, et al. The 2018 update of the WHO-EORTC classification for primary cutaneous lymphomas [published correction appears in Blood. 2019 Sep 26;134(13):1112]. Blood. 2019;133(16):1703–1714. doi:10.1182/blood-2018-11-881268
21. Grimm KE, O’Malley DP. Aggressive B cell lymphomas in the 2017 revised WHO classification of tumors of hematopoietic and lymphoid tissues. Ann Diagn Pathol. 2019;38:6–10. doi:10.1016/j.anndiagpath.2018.09.014
22. Selva R, Violetti SA, Delfino C, et al. A literature revision in primary cutaneous B-cell lymphoma. Indian J Dermatol. 2017;62(2):146–157. doi:10.4103/ijd.IJD_74_17
23. Ng CS, Chan JK, Hui PK, Lau WH. Large B-cell lymphomas with a high content of reactive T cells. Hum Pathol. 1989;20(12):1145–1154. doi:10.1016/s0046-8177(89)80004-8
24. Wang J, Sun NCJ, Chen YY, Weiss LM. T-cell /histiocyte-rich B cell lymphoma displays a heterogeneity similar to diffuse large B cell lymphoma: a clinicopathologic, Immunohistochemical, Molecular study of 30 cases. Appl Immunohistochem Mol Morphol. 2005;13(2):109–115. doi:10.1097/01.pai.0000132199.47017.35
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.