Back to Journals » Infection and Drug Resistance » Volume 13
Prevalence of Potential Resistance Related Variants Among Chinese Chronic Hepatitis B Patients Not Receiving Nucleos(T)ide Analogues
Authors Qian F ![]() , Zou W
, Zou W ![]() , Jin F, Li D, Shen Y
, Jin F, Li D, Shen Y
Received 14 February 2020
Accepted for publication 24 June 2020
Published 17 July 2020 Volume 2020:13 Pages 2407—2416
DOI https://doi.org/10.2147/IDR.S249476
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Suresh Antony
Fuchu Qian,1,2 Weihua Zou,3 Fang Jin,1,2 Dongli Li,1,2 Yujuan Shen4
1Department of Precision Medicine, Huzhou Central Hospital, Affiliated Central Hospital Huzhou University, Huzhou, People’s Republic of China; 2Huzhou Key Laboratory of Molecular Medicine, Huzhou Central Hospital, Affiliated Central Hospital Huzhou University, Huzhou, People’s Republic of China; 3Department of Laboratory Medicine, Huzhou Central Hospital, Affiliated Central Hospital Huzhou University, Huzhou, People’s Republic of China; 4Department of Infectious Diseases, Huzhou Central Hospital, Affiliated Central Hospital Huzhou University, Huzhou, People’s Republic of China
Correspondence: Fuchu Qian Tel +86 572-2555801
Email [email protected]
Background and Aims: Potential drug resistance (DR) related variants in the hepatitis B virus (HBV) reverse transcriptase (RT) region may be associated with the effectiveness of antiviral drugs and disease progression. The aim of this study was to investigate the prevalence and clinical characteristics of potential DR-related variants in Chinese CHB patients not receiving nucleos(t)ide analogues (NAs).
Patients and Methods: Two hundred and six untreated CHB patients from Huzhou Central Hospital in eastern China were recruited for this study. The serum DNA was extracted and the HBV RT region was amplified using nest polymerase chain reaction (nest-PCR). The 42 potential DR-related variants were analyzed by direct sequencing.
Results: Among these CHB patients, HBV genotype B and genotype C were identified in 121 (58.7%) and 85 (41.3%) patients, respectively. Potential DR-related variants were detected in 42.7% (88/206) of patients. Primary and secondary DR variants were found in 7.3% (15/206) of patients, including rtL80I/V, rtI169T, rtV173L rtL180M, rtA181T/V, rtM204I/V, and rtN236T. The variants at rt53, rt82, rt221, rt233, rt237, and rt256 were specific for genotype B, and those at rt38, rt84, rt126, rt139, rt153, rt191, rt214, rt238, and rt242 were specific for genotype C. Moreover, the variation frequency in the A-B interdomain (3.96%) was significantly higher than that in the functional domains (1.17%) and non-A-B interdomains (1.11%). Multivariate logistic regression analysis showed that lower HBV-DNA load (< 106 IU/mL) was an independent factor associated with potential DR-related variants in untreated CHB patients (P < 0.05).
Conclusion: Potential DR-related variants were frequent and complex in untreated Chinese CHB patients. Furthermore, the variants may contribute to decreased serum HBV-DNA loads. However, the effects of potential DR-related variants on the antiviral therapy and liver disease progression require further study.
Keywords: Hepatitis B virus, potential, resistance, reverse transcriptase, variants
Introduction
Hepatitis B virus (HBV) infection is a public health issue affecting approximately 257 million people worldwide.1 It was estimated that 80 million people were infected with HBV in China.2 Nucleos(t)ide analogues (NAs) can suppress viral replication by targeting the reverse transcriptase (RT) region of HBV. However, during long-term treatment, the drug resistance (DR) occurs due to variants in the RT region of HBV, leading to the failure of anti-HBV therapy.3,4
Currently, there are four categories of RT region DR-related variants that have been reported, namely, primary variants, secondary/compensatory variants, putative resistant variants, and pretreatment variants.5 Primary and secondary RT variants have been widely investigated in chronic hepatitis B (CHB) patients treated with NAs.6–10 Several previous studies have demonstrated that the classic DR variants also exist in chronic hepatitis B (CHB) patients not receiving NAs.11–13 However, the results were quite discrepant among different areas and countries,5,14,15 and the prevalence of putative resistant variants and pretreatment variants among NAs-treated and untreated CHB patients were not well defined.
To date, the clinical factors affecting the incidence of potential DR-related variants in untreated CHB patients are still unclear. Therefore, the present study investigated the prevalence and clinical features of potential DR-related variants among Chinese CHB patients not receiving NAs.
Patients and Methods
Patients
We recruited 206 CHB patients from the Department of Infectious Diseases at Huzhou Central Hospital, China, between January 2016 and June 2018. CHB diagnosis was according to the Chinese consensus criteria suggested by the Guideline of Prevention and Treatment for Chronic Hepatitis B in 2015.2 None of the patients previously received NAs treatment at the time of blood sample collection. We confirmed that the study participants were not taking antiviral drugs by checking medical history records. The patients were excluded by other situations, including infected with hepatitis A virus, hepatitis C virus, hepatitis D virus, tuberculosis, or human immunodeficiency virus. The study was approved by the Ethics Committee of Huzhou Central Hospital in accordance with the ethical guidelines of the Declaration of Helsinki. All patients provided written informed consent. The serum samples were collected and stored at −70°C.
Detection of Serum Markers
HBsAg, anti-HBs, HBeAg, HBeAb and anti-HBc were measured using the Architect-i2000 system (Abbott Laboratories, USA). Serum alanine aminotransferase (ALT), aspartate aminotransferase (AST), alkaline phosphatase (ALP), γ-glutamyl transpeptidase (GGT) levels were measured by automated techniques (HITACHI 7600, Japan). Serum HBV-DNA was quantified by a commercial real-time polymerase chain reaction (PCR) detection kit (Liferiver, Shanghai, China), and the detection limit of the kit was 100 IU/mL. All these tests were performed in the Department of Laboratory Medicine of Huzhou Central Hospital.
Amplification of RT Region and DNA Sequencing
Serum HBV-DNA was extracted from 200μL serum by High Pure Viral Nucleic Acid Kit (ROCHE, Switzerland) according to the manufacturer’s protocol. The RT region of HBV was amplified by nest-PCR. The primers used in the first-round PCR were P1 (5ʹ-AGTCAGGAAGACAGCCTACTCC-3ʹ) and P2 (5ʹ-AGGTGAAGCGAAGTG CACAC-3ʹ)(nt2146-1596), the primers used in the second-round PCR were P3 (5ʹ-TTCCTGCTGGTGGCTCCAGTTC-3ʹ) and P4 (5ʹ-TTCCGCAGTATGGATCGGCAG-3ʹ) (nt 54–1278). The nested PCR was performed with the high-fidelity PrimeSTAR HS DNA Polymerase (Takara, Dalian, China). The PCR amplification conditions were applied as we previously described.16 The PCR products were purified using a QIAquick gel extraction kit (Qiagen, Hilden, Germany) and the bi-directional sequencing using ABI 3730xl genetic analyzer (Applied Biosystems, USA). All sequencing was performed by SunYa Applied Biotechnology Co. Ltd (Shanghai, China).
Genotyping and DR-Related Variants Analysis
HBV genotyping was used online tool (http://www.ncbi.nih.gov/projects/genotyping/form.page.cgi). The sequences results were translated into amino acid (AA) sequences and aligned to different genotype reference sequences through the multiple sequence alignment by MEGA 6.0 software. The potential DR-related variants in 42 positions within the RT region were analyzed.5,6
Statistical Analysis
Statistical analyses were performed using IBM SPSS 23.0 statistics software (IBM, New York, USA). Student’s t-test was used for continuous variables, and Chi-square analysis or Fisher’s exact test was used for categorical variables. The Mann–Whitney U-test was used to compare differences between continuous variables with non-normal distribution. Univariate and multivariate logistic regression analyses were used to investigate the factors that were associated with the RT variants. P values <0.05 were considered statistically significant.
Results
Characteristics of the CHB Patients
Among the 206 NAs-naïve CHB patients, 121 were infected with HBV genotype B and 85 were infected with HBV genotype C. HBeAg-positive rate was 57.8% (119/206). A comparison of the characteristics of the HBeAg-positive group and the HBeAg-negative group did not reveal any significant differences in gender or liver function markers (P > 0.05). However, HBeAg-negative patients were older and had lower HBV-DNA loads than HBeAg-positive patients (P < 0.05). Additionally, more patients were infected with HBV genotype C in the HBeAg-positive group compared with the HBeAg-negative group (P < 0.05) (Table 1).
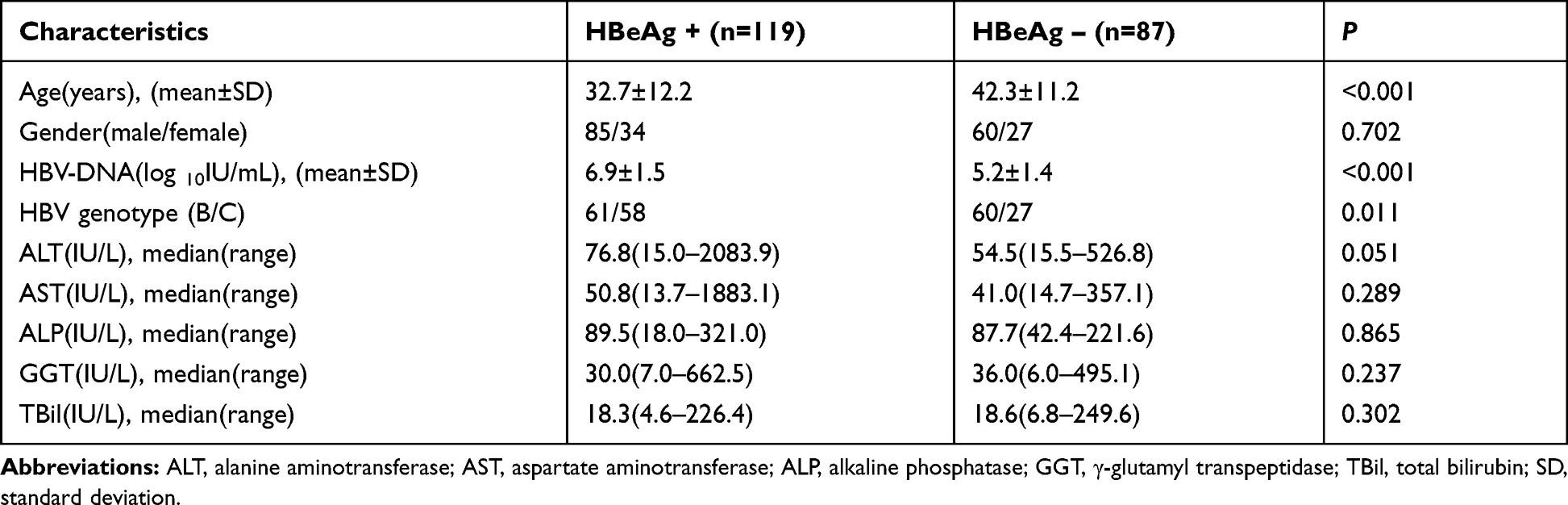 |
Table 1 Demographic and Clinical Characteristics of CHB Patients |
Prevalence of Potential DR-Related Variants in CHB Patients
In agreement with previous studies, eight sites were confirmed as genotype-dependent AA polymorphisms sites in the present study (Table 2). These data revealed that the presence of asparagine or serine at rt53, isoleucine or leucine at rt91, asparagine or tyrosine at rt124, aspartic acid or asparagine at rt134, phenylalanine or tyrosine at rt221, isoleucine or valine at rt224, histidine or asparagine at rt238, and serine or cysteine at rt256 were closely linked to genotype B or genotype C, respectively (P < 0 0.001). Therefore, the AA residue at each of the above sites was regarded as the consensus AA in B and C genotypes, respectively. The low frequencies of other AA residues at these sites were considered as variants in the present study.
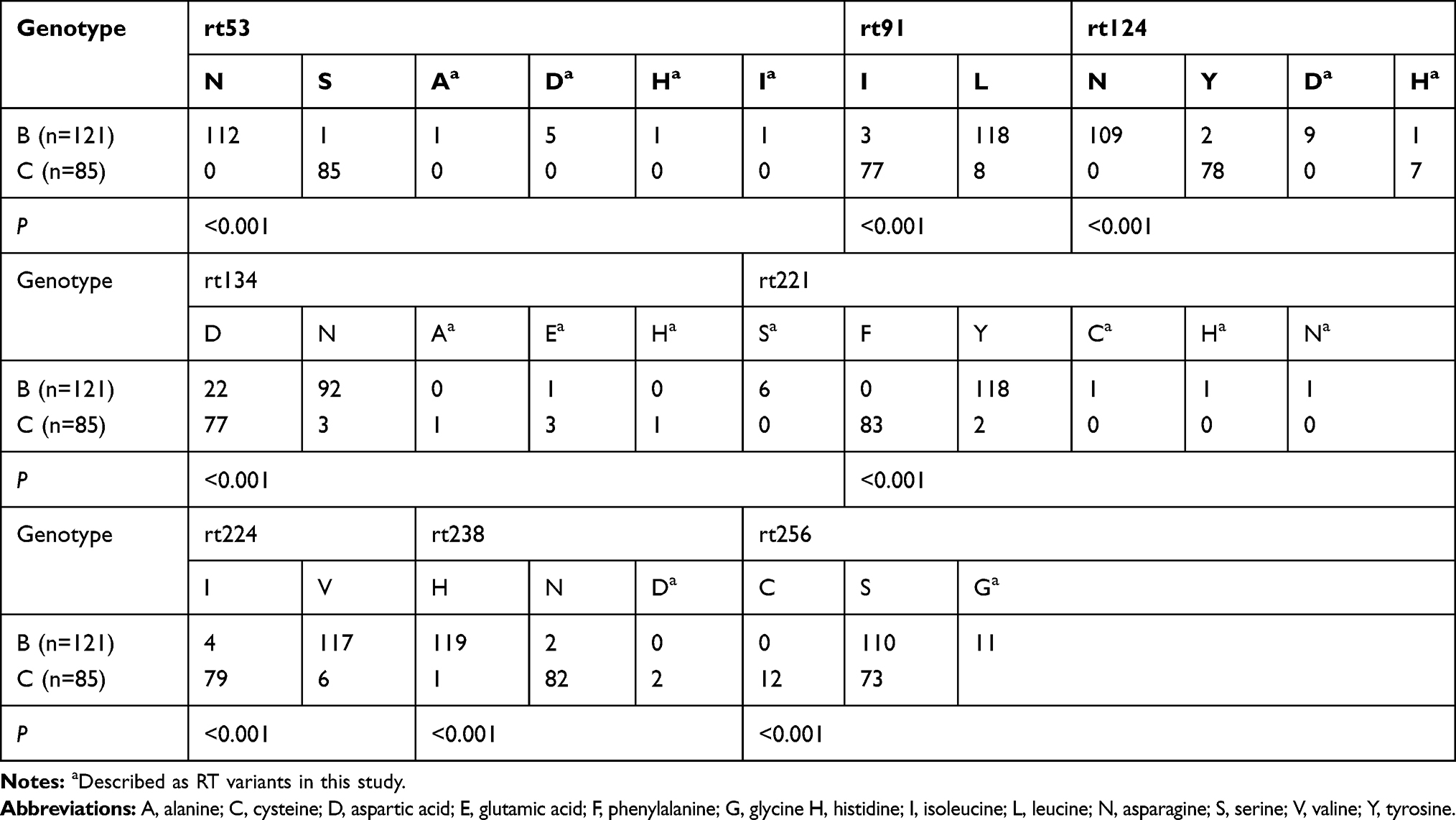 |
Table 2 Genotype-Dependent AA Polymorphic Sites in This Study |
We further analyzed 42 potential DR-related variant sites in the 206 NA-naïve CHB patients. RT variants were found in 42.7% (88/206) of CHB patients and included 29 sites. Primary and/or secondary DR variants were found in 7.3% (15/206) of patients, and included rtL80I/V, rtI169T, rtV173L, rtL180M, rtA181T/V, rtM204I/V, and rtN236T. Among these patients, seven were infected with genotype B and eight were infected with genotype C. The distributions of primary and/or secondary DR variants were not significantly different between genotype B and C (5.8% [7/121] vs 9.9% [8/81], P > 0.05). The Putative DR-related variants and pretreatment variants were found in 38.8% (80/206) of CHB patients, which included 22 AA sites and 107 variants. The variation rates of putative DR-related variants and pretreatment variants were also not significantly different between genotypes B and C (Table 3). Of note, the variants at rt53, rt82, rt221, rt233, rt237, and rt256 were specific for genotype B, and those at rt38, rt84, rt126, rt139, rt153, rt191, rt214, rt238, and rt242 were specific for genotype C (Figure 1).
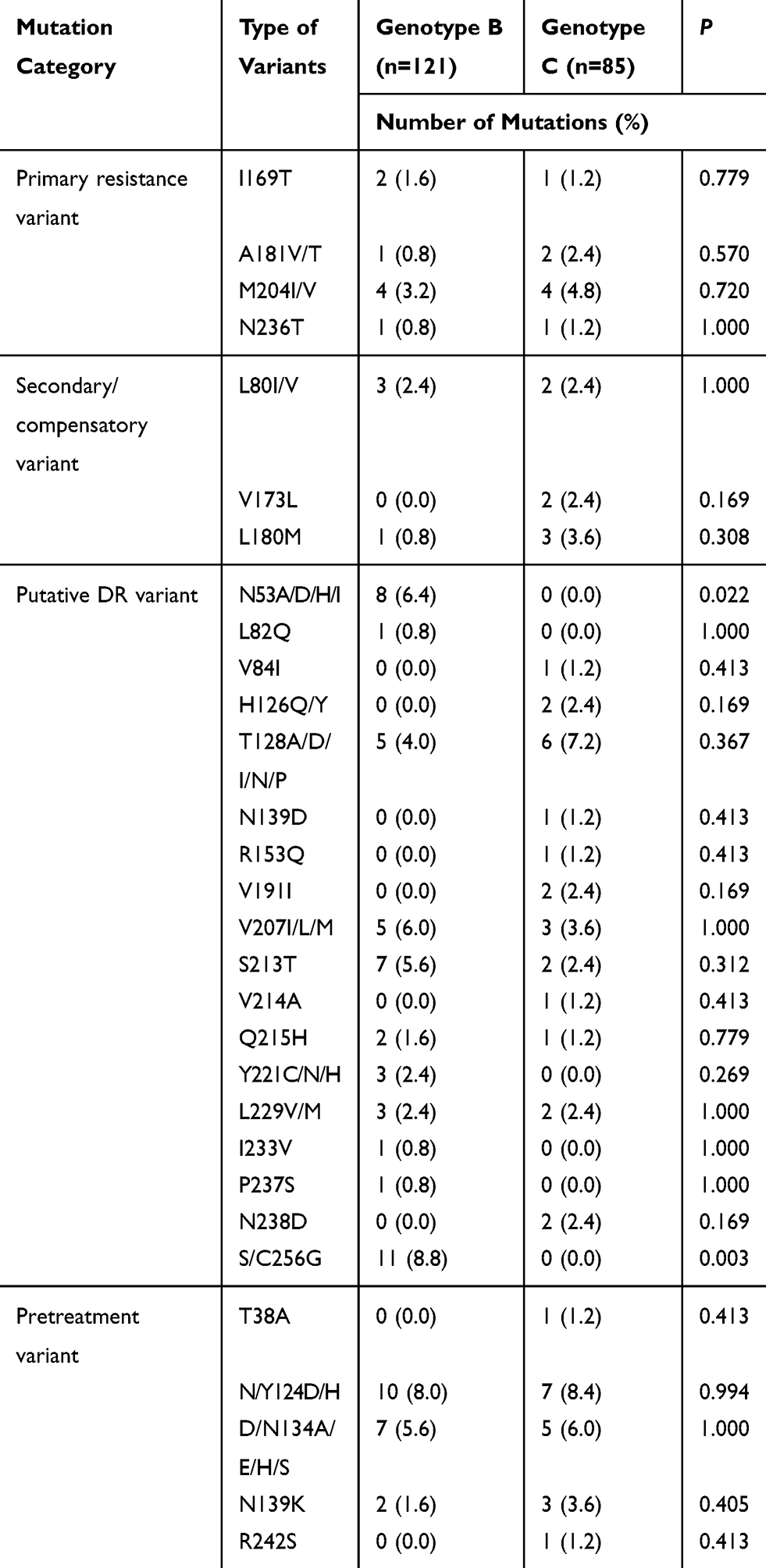 |
Table 3 Prevalence of Potential DR-Related Variants of RT Region in CHB Patients |
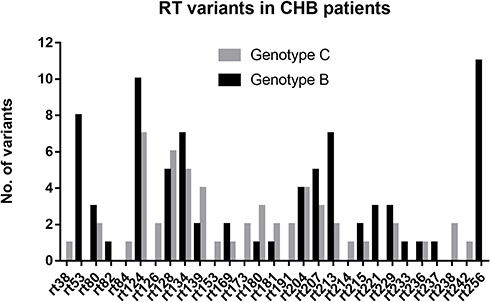 |
Figure 1 RT variants in genotype B and C HBV infected patients. Abbreviations: RT, reverse transcriptase; CHB, chronic hepatitis B; HBV, hepatitis B virus. |
Because the hepatitis B surface antigen gene overlaps with the RT gene, we analyzed this region in detail and found that the mutations at rt134, rt139, and rt153 sites overlapped with the “a” determinant of the S gene. In the present study, the rt134 and rt153 variants led to the concomitant occurrence of “a” determinant mutations, including sT126A (n=5), sT126S (n=2), sT126N (n=1), and sG145R (n=1).
Variant Site Distribution and Frequency in Different Sections of the RT Region
The RT region consists of functional domains (G, F, A, B, C, D, and E) and interdomains (F-A, A-B, B-C, C-D and D-E).5 Our results showed that all six sites (6/6, 100%) in A-B interdomain contained variants, and the variation rate was higher than those of the functional domains (15/22, 68.2%) and non-A-B interdomains (8/14, 57.1%), but no statistically significant differences were observed (P > 0.05). Furthermore, the variation frequency was also significantly higher (3.96%) in the A-B interdomain than in the functional domains (1.17%) and non-A-B interdomains (1.11%) (P < 0.01) (Table 4). However, no significant difference was showed in the functional domains, and the A-B interdomains between genotype B and C with respect to the number and frequencies of variants (P > 0.05). The variation frequency of genotype C in the non-A-B interdomains was higher than that of genotype B (P < 0.05).
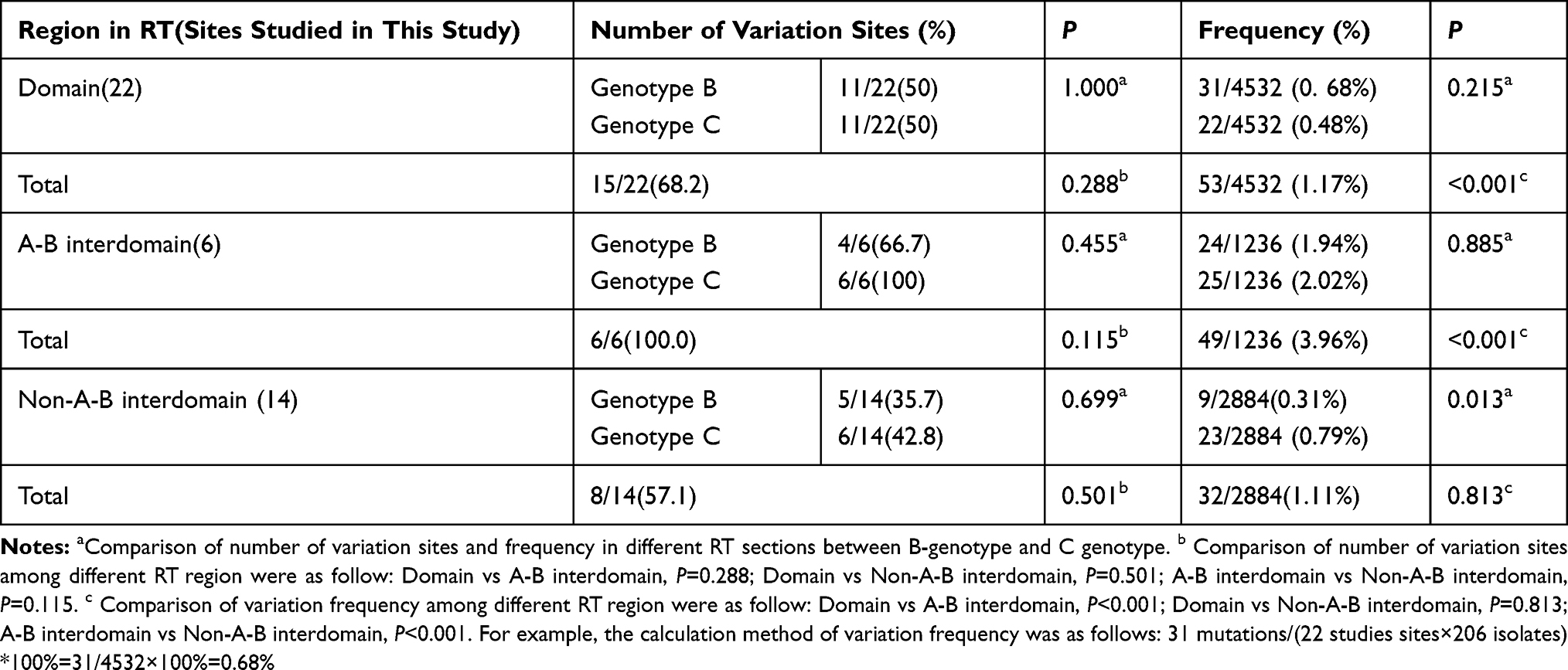 |
Table 4 Variation Distribution and Frequency in Different RT Regions |
Relationship Between Potential DR-Related Variants and Clinical Features
The characteristics were compared between CHB patients with and without RT variants. The average age of patients with RT variants was older than that of patients without RT variants (P < 0.05). The mean HBV-DNA loads were significantly lower in patients with RT variants than in patients without RT variants (P < 0.05) (Figure 2A). However, this difference was not observed between patients with variants and without variants when the patients were divided into HBeAg-positive and HBeAg-negative subgroups (P > 0.05) (Figure 2B and C). The average ALT levels of patients with variants were lower than those in patients without variants (P < 0.05). There were no significant differences in gender; AST, ALP, GGT, and TBil levels; HBeAg status; and genotype distribution between patients with and without DR-related variants (Table 5).
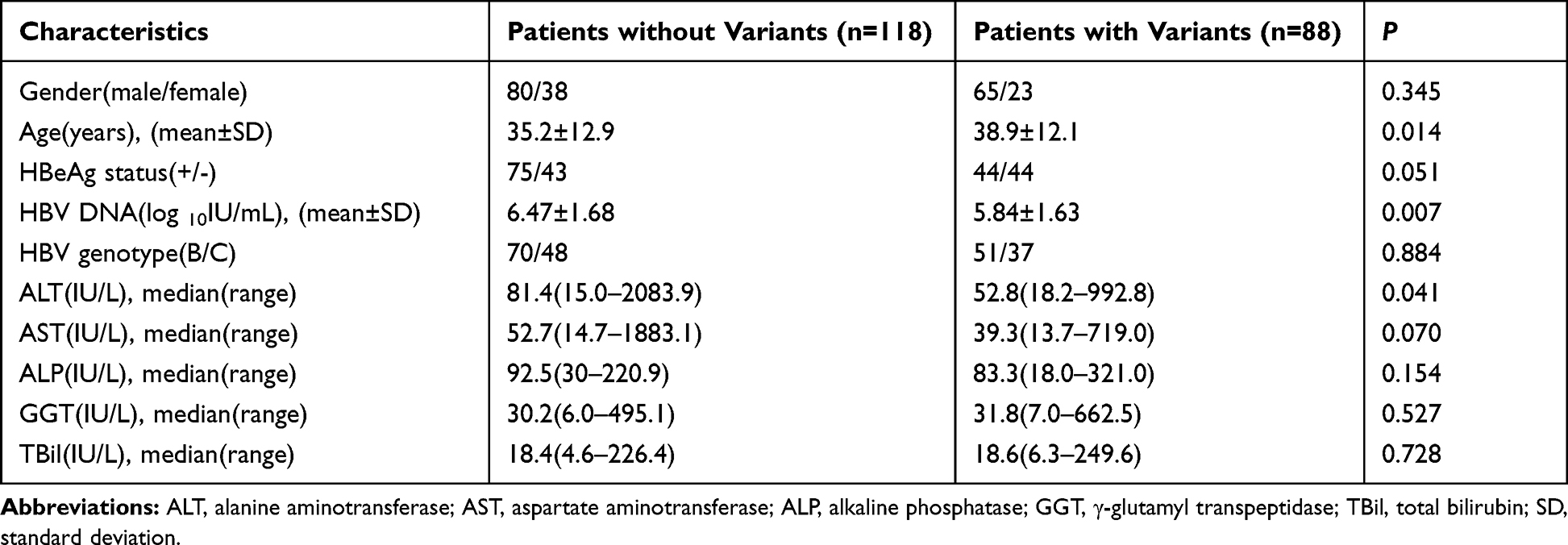 |
Table 5 Clinical Features of CHB Patients with and without Potential DR-Related Variants |
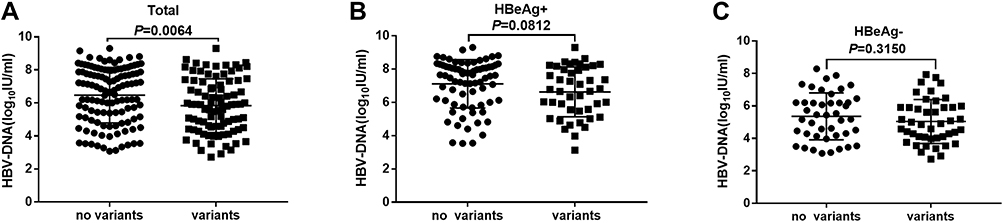 |
Figure 2 HBV-DNA loads in untreated CHB patients with or without RT variants. (A) All patients (B) Patients with HBeAg positive (C) Patients with HBeAg negative. Abbreviations: HBV, hepatitis B virus; CHB, chronic hepatitis B; RT, reverse transcriptase; HBeAg, hepatitis B virus e antigen. |
Further analysis revealed no significant differences in gender, age, HBeAg status, and liver function markers (including ALT, AST, ALP, GGT, and TBil) between patients with single and multiple variants (P > 0.05) (Table 6). Univariate logistic regression analysis results revealed that age (≥35 years old) and HBV-DNA load (<106 IU/mL) were associated with the potential DR-related variants (P < 0.05) (Table 7). No relationship was found between the other factors (gender, HBeAg status, genotype, and liver function markers) and the DR-related variants. Furthermore, multivariate logistic regression analysis showed that the HBV-DNA load (<106 IU/mL) was an independent factor associated with the potential DR-related variants in the untreated CHB patients (P < 0.05) (Table 7).
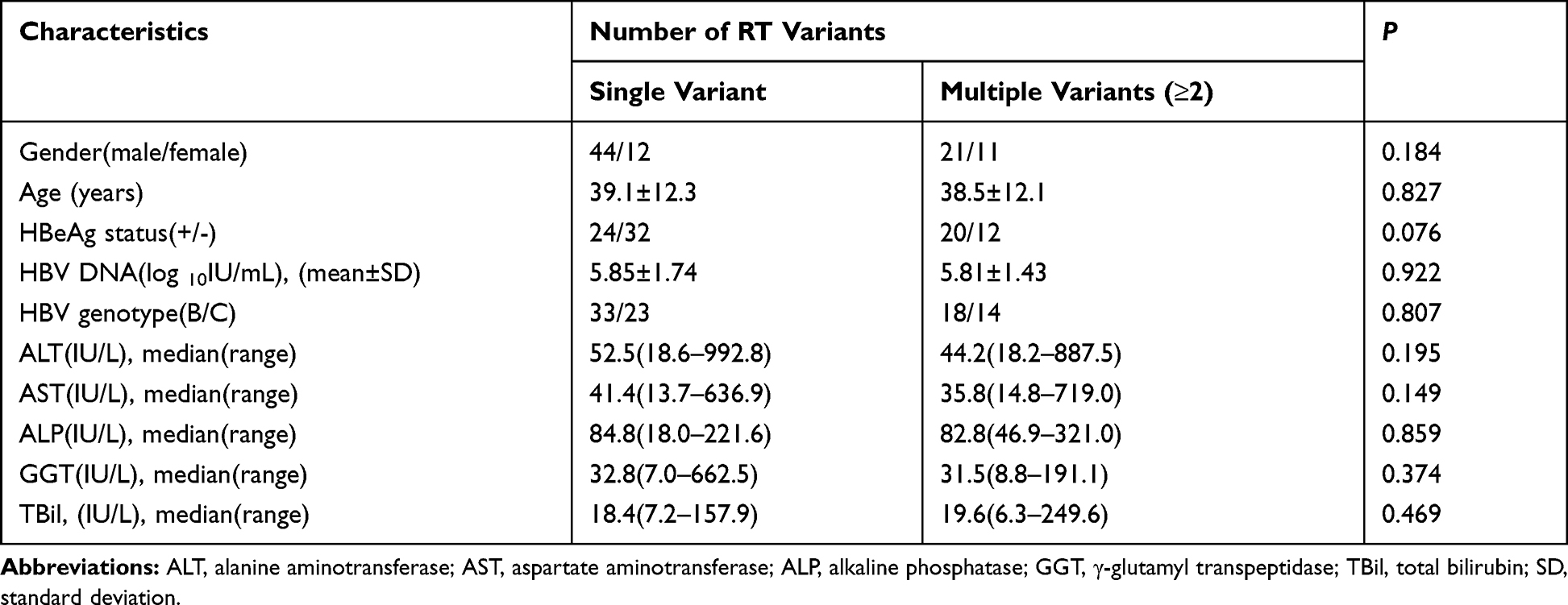 |
Table 6 Comparison of Clinical Characteristics in CHB Patients with Different Number of Potential DR-Related Variants |
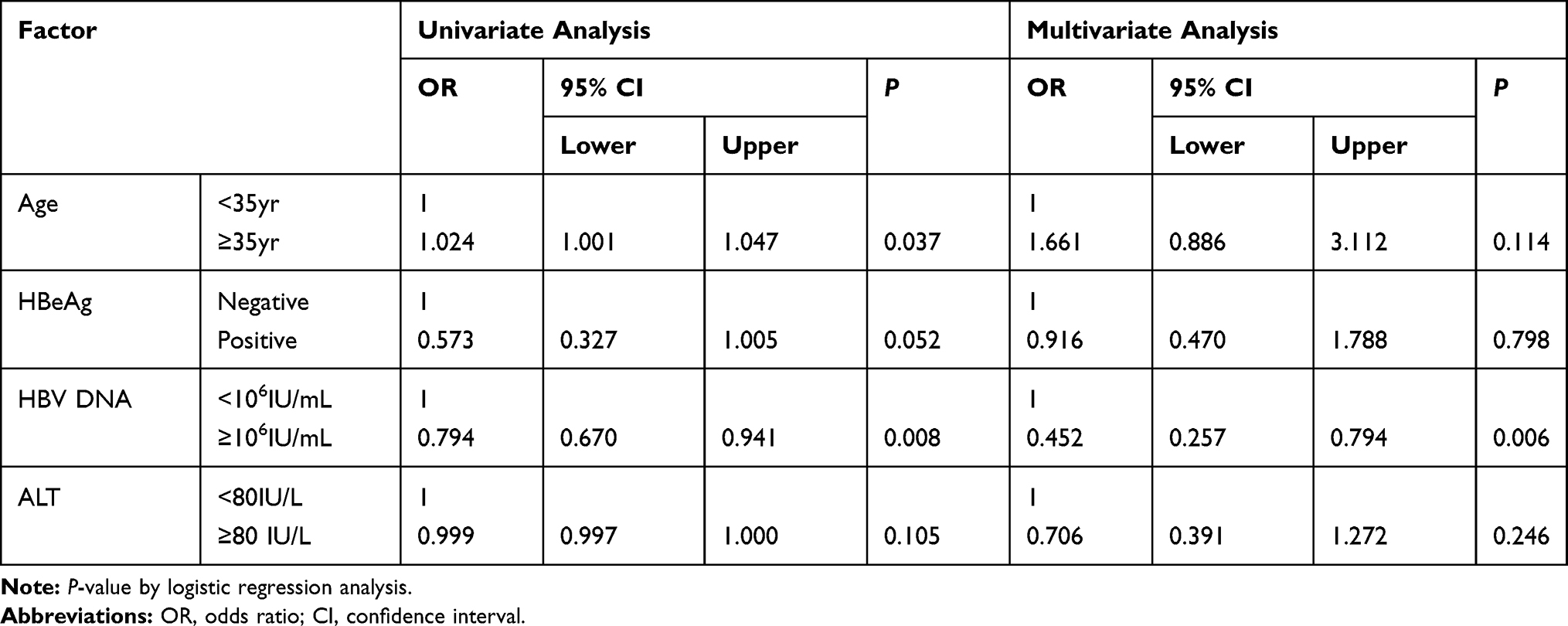 |
Table 7 Univariate and Multivariate Logistic Analyses of Factors for Potential DR-Related Variants |
Discussion
The prevalence of potential DR-related variants in the HBV RT region in NAs-untreated CHB patients has been reported in several previous studies but the results are controversial.6,7,14,17,18 In the present study, potential DR-related variants were detected in 42.7% (88/206) of untreated CHB patients, and primary and secondary DR variants were found in 7.3% (15/206) patients. Several studies have reported that the prevalence rate of primary and secondary DR variants was low or even zero in untreated patients.7,10,14,17 However, the present study revealed that primary and secondary DR variants existed in a considerable proportion of NAs-untreated CHB patients. A study in central China reported that variants associated with DR were detected in 8.9% of untreated patients.13 Another study recently reported DR variants in 6% of Brazilian treatment-naïve CHB patients,18 and a meta-analysis showed that the pooled incidence of naturally resistant variants in China was higher than that in other countries (8.00% vs 1.88%).19 Overall, the incidence of DR variants was closely related to the geographical distribution and epidemic situation of the HBV infection. For instance, the prevalence rate of DR variants in China was higher (8.00%) because HBV infection is highly endemic to China. However, in Europe, the prevalence rate of DR variants was lower (2.53%) due to low levels of endemic HBV infection.19 Furthermore, some variants were related to DR to entecavir and tenofovir disoproxil fumarate, such as rtI169T, rtl180M, rtM204I/V, rtH126Y and rtD134E, which have been previously reported,2,21 and were also found in the present study. Considering the impact of potential DR variants on treatment efficacy, it is crucial to detect the DR-related variants prior to administering anti-HBV treatment.
Agree with previous studies,5 eight AA polymorphic positions (rt53, rt91, rt124, rt134, rt221, rt224, rt238 and rt256) were identified for genotype B and C. We further analyzed the AA polymorphic positions in the RT region for genotype A to I using an online tool (https://hivdb.stanford.edu/HBV/DB/cgi-bin/MutPrevByGenotypeRxHBV.cgi). The results showed that these eight sites were genotype-dependent AA polymorphic positions. Additionally, another study reported that AA polymorphic positions showed discrepancies in different areas in China.15 These data indicated that the definition of genotype variants needs to take into account the reference sequences of different genotypes in different areas and the consensus sequence derived from local HBV isolates. Moreover, researchers have also suggested that some AA polymorphic positions could contribute to decreased viral replication and affect drug treatment outcomes.5,8 Thus, the genotype-dependent polymorphism sites could influence the effect of antiviral treatment by regulating virus replication and/or fitness.
The RT region is separated into functional domains (G, F A, B, C, D and E) and interdomains (F-A, A-B, B-C, C-D and D-E).5,22 Previous studies have shown that the variation frequency in the A-B interdomain was higher than that in the other regions,5,23–26 and this was also observed in the present study. Moreover, when genotype B or C was analyzed separately, this difference still existed. However, there was no difference in the variation frequency between genotypes B and C in the functional domains or A-B interdomain. Overall, the A-B interdomain showed the highest frequency of variation in the RT region in NAs-naïve CHB patients. Notably, there was a significant difference in the variation frequency between genotype B and C in non-A-B interdomains. An Indonesian study also showed that potential DR-related variants were more frequent in genotype C than in genotype B.24 Hence, the driving factors and the mechanism of RT variations among different genotypes are worth further study.
An interesting finding was that some variants appeared specific to particular genotypes. Some of these positions had been reported previously, but others (rt82, rt214, and rt242) were not found in the corresponding genotype from previous studies.5,15 The rt242 variant was only found in genotype B in a previous study5 but was found only in genotype C in our study. Therefore, we speculated that different genotypes possessed different RT variants due to differences in evolutionary characteristics. Thus, the relationship between the genetic diversity of genotypes and the distribution of sites of potential DR-related variants requires further elucidation.
Until now, the correlation between potential DR-related variants and clinical characteristics among NAs–naïve patients remained unclear. Previous studies did not find any significant association between the presence of potential DR-related variants and the gender, age, HBeAg status, HBV-DNA loads, or ALT and AST levels.5,19 Recently, a study in China showed that the natural RT variants were associated with low HBV-DNA loads in HBeAg-negative patients.14 Our study compared the clinical characteristics between CHB patients with and without potential DR variants, and found differences in age, HBV-DNA loads, and ALT levels (Table 5). Several studies have showed that potential DR variants were associated with HBeAg-negative status.10,13 In the present study, the negative rate of HBeAg in patients with DR-related variants was higher than that in patients without DR-related variants (50% vs 36.4%), but a significant difference was not observed. The HBeAg-negative status may lead to greater host immune pressure against HBV, which could result in the generation of more variants due to the selection pressures. Another previous study showed that some naturally occurring AA substitutions in the RT region might influence the serum HBV-DNA load in HBeAg-positive CHB patients with sub-genotype B2.27 Therefore, the impact of HBeAg status on the RT variants deserves further study. A recent review also suggested that low HBV-DNA loads were associated with potential DR variants.28 In the present study, further logistic regression analysis showed that the lower HBV-DNA load was an independent factor that was related to patients with potential DR-related variants (Figure 1, Table 7). This data was consistent with several other previous studies, indicating that naturally occurring RT variants were associated with decreased HBV-DNA loads.17,26,29 The reason may be that HBV replication is often impaired by the RT variants, which decreases the activity of polymerase. Although another study indicated that patients with multiple RT variants showed decreased HBV-DNA loads compared with patients with a single RT variant,29 there was no difference in HBV-DNA loads and other clinical factors (gender, age, genotype, HBeAg, ALT, and AST levels) between the single variant and the multiple variants subgroups, implying that certain single RT variants played a crucial role in viral replication. It is also worth noting that no classical primary and secondary variants were found in several previous studies,14,29 which might have influenced the results of the analysis. Taken together, the key roles of some RT variants in viral replication and fitness require further elucidation.
Recently, the application of next-generation sequencing (NGS) was gradually introduced for the investigation of DR variants in CHB patients.30–32 This method has higher sensitivity than direct sequencing and can detect minor pre-existing DR-related variants that are undetectable using Sanger sequencing.33–36 A recent study demonstrated that NGS was more suitable for detecting low rate DR variants in untreated patients than Sanger sequencing.15 However, because the NGS technology is more costly and requires a highly technical platform in comparison with Sanger sequencing, we used Sanger sequencing to analyze the potential DR-related variants in this study. Thus, the actual prevalence rate of potential DR related variants in untreated patients might be higher than our results. Nonetheless, the current study provides a rationale for further surveillance of the prevalence of potential DR-related variants using NGS.
Conclusions
In summary, the frequency of potential DR-related variants was relatively high and their patterns were complex and diverse among NAs-naive CHB patients, which might contribute to lower HBV-DNA loads. The genotype B and C showed preferred RT variation sites. Further large-scale investigations are needed to clarify the clinical significance and evolution characteristics of potential DR-related variants in the RT region of HBV in untreated CHB patients.
Abbreviations
AA, amino acid; ALT, alanine aminotransferase; ALP, alkaline phosphatase; AST, aspartate aminotransferase; CHB, chronic hepatitis B; GGT, γ-glutamyl transpeptidase; HBV, hepatitis B virus; NAs, nucleos(t)ide analogues; PCR, polymerase chain reaction; RT, reverse-transcriptase; NGS, Next-Generation Sequencing.
Ethical Approval and Consent to Participate
This study was approved by the Ethics Committee of Huzhou Central Hospital. Written informed consent was obtained from all participants.
Acknowledgments
We thank the staffs of Biobank of Huzhou Central Hospital for their kind assistance in collecting medical record data.
Author Contributions
All authors made substantial contributions to conception and design, acquisition of data, or analysis and interpretation of data; took part in drafting the article or revising it critically for important intellectual content; gave final approval of the version to be published; and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
1. World Health Organization. Global Hepatitis Report; 2017. Available from: https://www.who.int/hepatitis/publications/global-hepatitis-report2017/en/.
2. Chinese Society of Hepatology and Chinese Society of Infectious Diseases. Chinese Medical Association. The Guideline of prevention and treatment for chronic hepatitis B (2010 version). Zhonghua Gan ZangBing Za Zhi. 2011;19:13–24.
3. Liu Y, Wang C, Zhong Y, et al. Genotypic resistance profile of hepatitis B virus (HBV) in a large cohort of nucleos(t)ide analogue-experienced Chinese patients with chronic HBV infection. J Viral Hepat. 2011;18(4):e29–39. doi:10.1111/j.1365-2893.2010.01360.x
4. Zoulim F, Locarnini S. Hepatitis B virus resistance to nucleos(t)ide analogues. Gastroenterology. 2009;137(5):1593–1608. doi:10.1053/j.gastro.2009.08.063
5. Liu BM, Li T, Xu J, et al. Characterization of potential antiviral resistance variants in hepatitis B virus reverse transcriptase sequences in treatment-naïve Chinese patients. Antiviral Res. 2010;85(3):512–519. doi:10.1016/j.antiviral.2009.12.006
6. Han Y, Huang LH, Liu CM, et al. Characterization of hepatitis B virus reverse transcriptase sequences in Chinese treatment naive patients. J Gastroenterol Hepatol. 2009;24(8):1417–1423. doi:10.1111/j.1440-1746.2009.05864.x
7. Sayan M, Akhan SC, Meric M. Naturally occurring amino-acid substitutions to nucleos(t)ide analogues in treatment naive Turkish patients with chronic hepatitis B. J Viral Hepat. 2010;17(1):23–27. doi:10.1111/j.1365-2893.2009.01149.x
8. Mirandola S, Campagnolo D, Bortoletto G, et al. Large-scale survey of naturally occurring HBV polymerase variants associated with anti-HBV drug resistance in untreated patients with chronic hepatitis B. J Viral Hepat. 2011;18(7):e212–216. doi:10.1111/j.1365-2893.2011.01435.x
9. Salpini R, Svicher V, Cento V, et al. Characterization of drug-resistance variants in HBV D-genotype chronically infected patients, naïve to antiviral drugs. Antiviral Res. 2011;92(2):382–385. doi:10.1016/j.antiviral.2011.08.013
10. Vutien P, Trinh HN, Garcia RT, et al. Variants in HBV DNA polymerase associated with nucleos(t)ide resistance are rare in treatment-naive patients. Clin Gastroenterol Hepatol. 2014;12(8):1363–1370. doi:10.1016/j.cgh.2013.11.036
11. Li X, Liu Y, Zhao P, et al. Investigation into drug-resistant variants of HBV from 845 nucleoside/nucleotide analogue-naive Chinese patients with chronic HBV infection. Antivir Ther. 2015;20(2):141–147. doi:10.3851/IMP2813
12. Gomes-Gouvêa MS, Ferreira AC, Teixeira R. HBV carrying drug-resistance variants in chronically infected treatment-naive patients. Antivir Ther. 2015;20(4):387–395. doi:10.3851/IMP2938
13. Zhao Y, Wu J, Sun L, et al. Prevalence of variants in HBV DNA polymerase gene associated with nucleos(t)ide resistance in treatment-naive patients with Chronic Hepatitis B in Central China. Braz J Infect Dis. 2016;20(2):173–178. doi:10.1016/j.bjid.2015.12.006
14. Xu J, Wu B, Wang JH, et al. Pre-existing variants in reverse transcriptase of hepatitis B virus in treatment-naive Chinese patients with chronic hepatitis B. PLoS One. 2015;10(3):e0117429. doi:10.1371/journal.pone.0117429
15. Fu Y, Zeng Y, Chen T, et al. Characterization and clinical significance of natural variability in Hepatitis B virus reverse transcriptase in treatment-naive chinese patients by Sanger sequencing and next-generation sequencing. J Clin Microbiol. 2019;57(8):e00119–e00219. doi:10.1128/JCM.00119-19
16. Qian F, Qin J, Li D, et al. Monitoring of genotypic resistance profile in chronic hepatitis B patients receiving nucleos(t)ide analogues in Huzhou, China. J Infect Dev Ctries. 2016;10(9):996–1002. doi:10.3855/jidc.8020
17. Fan J, Zhang Y, Xiong H, et al. Nucleotide analogue-resistant variants in hepatitis B viral genomes found in hepatitis B patients. J Gen Virol. 2015;96(Pt 3):663–670. doi:10.1099/jgv.0.000010
18. Pacheco SR, Dos Santos MIMA, Stocker A, et al. Genotyping of HBV and tracking of resistance variants in treatment-naïve patients with chronic hepatitis B. Infect Drug Resist. 2017;5(10):201–207. doi:10.2147/IDR.S135420
19. Zhang Q, Liao Y, Cai B, et al. Incidence of natural resistance variants in naïve chronic hepatitis B patients: a systematic review and meta-analysis. J Gastroenterol Hepatol. 2015;30(2):252–261. doi:10.1111/jgh.12831
20. Liu, Zhou Y, Li X, et al. Hepatitis B virus mutation pattern rtL180M+A181C+M204V may contribute to entecavir resistance in clinical practice. Emerg Microbes Infect. 2019;8(1):354–365. doi:10.1080/22221751.2019.1584018
21. Park ES, Lee AR, Kim DH, et al. Identification of a quadruple mutation that confers tenofovir resistance in chronic hepatitis B patients. J Hepatol. 2019;70(6):1093–1102. doi:10.1016/j.jhep.2019.02.006
22. Stuyver LJ, Locarnini SA, Lok A, et al. Nomenclature for antiviral-resistant human hepatitis B virus variants in the polymerase region. Hepatology. 2001;33(3):751–757. doi:10.1053/jhep.2001.22166
23. Li H, Song XF, Hu TT, et al. A strong conservative tendency in HBV transcriptase (RT): a majority of natural RT variants derived from the S gene. Liver Int. 2016;36(7):963–970. doi:10.1111/liv.13051
24. Yamani LN, Yano Y, Utsumi T, et al. Profile of variants in the reverse transcriptase and overlapping surface genes of Hepatitis B Virus (HBV) in treatment-Naïve Indonesian HBV carriers. Jpn J Infect Dis. 2017;70(6):647–655. doi:10.7883/yoken.JJID.2017.078
25. Zheng J, Zeng Z, Zhang D, et al. Prevalence and significance of Hepatitis B reverse transcriptase mutants in different disease stages of untreated patients. Liver Int. 2012;32(10):1535–1542. doi:10.1111/j.1478-3231.2012.02859.x
26. Kim JE, Lee SY, Kim H, et al. Naturally occurring variants in the reverse transcriptase region of hepatitis B virus polymerase from treatment-naïve Korean patients infected with genotype C2. World J Gastroenterol. 2017;23(23):4222–4232. doi:10.3748/wjg.v23.i23.4222
27. Su M, Xiang K, Li Y, et al. Higher detection rates of amino acid substitutions in HBV reverse transcriptase/surface protein overlapping sequence is correlated with lower serum HBV DNA and HBsAg levels in HBeAg-positive chronic hepatitis B patients with subgenotype B2. Infect Genet Evol. 2016;40:275–281. doi:10.1016/j.meegid.2016.03.019
28. Choi YM, Lee SY, Kim BJ. Naturally occurring hepatitis B virus reverse transcriptase variants related to potential antiviral drug resistance and liver disease progression. World J Gastroenterol. 2018;24(16):28. doi:10.3748/wjg.v24.i16.1708
29. Zhu B, Wang T, Wei X, et al. Accumulation of variants in reverse transcriptase of hepatitis B virus is associated with liver disease severity in treatment-naïve Chinese patients with chronic hepatitis B. Adv Clin Exp Med. 2017;26(7):1123–1129. doi:10.17219/acem/63998
30. Nishijima N, Marusawa H, Ueda Y, et al. Dynamics of hepatitis B virus quasispecies in association with nucleos(t)ide analogue treatment determined by ultra-deep sequencing. PLoS One. 2012;7(4):e35052. doi:10.1371/journal.pone.0035052
31. Ciftci S, Keskin F, Cakiris A, et al. Analysis of potential antiviral resistance mutation profiles within the HBV reverse transcriptase in untreated chronic hepatitis B patients using an ultra-deep pyrosequencing method. Diagn Microbiol Infect Dis. 2014;79(1):25–30. doi:10.1016/j.diagmicrobio.2014.01.005
32. Zhang X, Li M, Xi H, et al. Pre-existing variants related to tenofovir in chronic hepatitis B patients with long-term nucleos(t)ide analogue drugs treatment by ultra-deep pyrosequencing. Oncotarget. 2016;7(43):70264–70275. doi:10.18632/oncotarget.11840
33. Ko SY, Oh HB, Park CW, et al. Analysis of hepatitis B virus drug-resistant mutant haplotypes by ultra-deep pyrosequencing. Clin Microbiol Infect. 2012;18(10):E404–411. doi:10.1111/j.1469-0691.2012.03951.x
34. Widasari DI, Yano Y, Heriyanto DS, et al. A deep-sequencing method detects drug-resistant variants in the hepatitis B virus in Indonesians. Intervirology. 2014;57(6):384–392. doi:10.1159/000366420
35. Jones LR, Sede M, Manrique JM, et al. Hepatitis B virus resistance substitutions: long-term analysis by next-generation sequencing. Arch Virol. 2016;161(10):2885–2891. doi:10.1007/s00705-016-2959-8
36. Margeridon-Thermet S, Shulman NS, Ahmed A, et al. Ultra-deep pyrosequencing of hepatitis B virus quasispecies from nucleoside and nucleotide reverse-transcriptase inhibitor (NRTI)-treated patients and NRTI-naive patients. J Infect Dis. 2009;199(9):1275–1285. doi:10.1086/597808
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.