")
Back to Journals » International Journal of Nanomedicine » Volume 13
Preparation of ginsenoside compound-K mixed micelles with improved retention and antitumor efficacy
Received 7 March 2018
Accepted for publication 7 May 2018
Published 3 July 2018 Volume 2018:13 Pages 3827—3838
DOI https://doi.org/10.2147/IJN.S167529
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Linlin Sun
Xin Jin, Qing Yang, Ning Cai
Department of Hospital Pharmacy, Suqian Branch Jiangsu Province Hospital, Suqian 223800, China
Introduction: Ginsenoside compound K (CK) has effects on cell-cycle regulation, tumor growth inhibition, and apoptosis induction. However, it has limited applications in clinical settings because of its low solubility and poor absorption.
Methods: To overcome these limitations, we aimed to develop a mixed micellar system composed of phosphatidylcholine (PC) and 1,2-distearoyl-sn-glycero-3-phosphoethanolamine polyethylene glycol 2000 (DSPE PEG 2000; DP). CK encapsulated in PC/DP mixed micelles had enhanced solubility, permeability, and retention effects.
Results: Compared to free CK, the CK PC/DP micellar system exhibited improved anticancer effects in vitro, including cell-cycle arrest, apoptosis, and anti-invasion in human lung carcinoma A549 cells. The significant proapoptotic effect was reflected by increased chromosomal condensation, annexin V/propidium iodide staining, and related protein expression. In vitro cellular uptake and optical mouse imaging in vivo suggested that the improved antitumor effect was caused primarily by enhanced uptake and tumor targeting. Furthermore, an in vivo antitumor efficacy study indicated that the CK mixed micelles significantly inhibited tumor growth, thereby decreasing tumor volume at the end of the experiment as compared with that in the control mice. Histological analysis confirmed the antitumor effect with low toxicity.
Conclusion: The PC/DP micellar system was an effective drug delivery system for CK in tumor therapy.
Keywords: mixed micellar system, ginsenoside compound-K, targeted effect, antitumor, lung cancer
Introduction
Panax ginseng CA Meyer is one of the most popular traditional Chinese medicines, and ginsenosides are its main active components. Compound K (CK) was first found in the soil bacterial hydrolysates of ginsenosides in 1972.1 Although its structure had been identified at that point, CK was not considered seriously until early 1996 when the specific metabolizing pathways in the conversion of ginsenosides to CK by intestinal flora were determined.2 Ginsenosides are poorly absorbed from the gut, but their metabolite, CK, is absorbed.3 Numerous reviews have indicated that the anticancer activity of ginsenosides is inversely correlated to the number of sugar groups that they possess.4,5 CK, with just one glucopyranosyl group, is often deemed the most active component in ginseng.6 It has remarkable effects on regulation of the cell cycle,7 inhibition of tumor growth,8 induction of apoptosis,9,10 inhibition of tumor cell invasion, and metastasis.11 However, its poor water solubility and significant P-glycoprotein efflux have restricted its use in human ailments due to low oral bioavailability.12–14
Numerous drug delivery carriers – such as polymer–drug conjugates, micelles, nanoparticles, and stealth liposomes – have been investigated for efficient drug delivery.15–17 To the best of our knowledge, only polyethylene glycol–CK conjugates, hydrophilic glycol chitosan–CK conjugates, D-α-tocopheryl polyethylene glycol 1000 succinate-modified liposomes, and ascorbyl palmitate/D-α-tocopheryl polyethylene glycol 1000 succinate monoester mixed micelles were designed to enhance the efficacy of CK.18,19 In our previous studies, CK encapsulated within nanosized drug carriers had improved pharmacological efficacy compared with that of free CK.20,21 Owing to their attractive drug-loading characteristics, micellar systems can significantly increase the solubility of hydrophobic drugs.22,23 However, large particle sizes and low stability restrict the application of single polymer micelles,24 and they are gradually being replaced by binary mixed micelles. Mixed micelles with low particle sizes and improved stability could overcome the drawbacks of single polymer micelles through the synergistic effects of different excipients.25,26 Therefore, mixed micelle systems were chosen for further study.
Phosphatidylcholine (PC) and 1,2-distearoyl-sn-glycero-3-phosphoethanolamine polyethylene glycol 2000 (DSPE PEG 2000; DP) have been widely used as pharmaceutical excipients because of their proven safety.27–31 High concentrations of DP can solubilize drugs to a large extent. PC can adjust the hydrophobic segment aggregates to form a stable inner core to promote drug accommodation via hydrophobic interactions. This simple approach of combining improved solubility and stability forms the mixed micellar system. Furthermore, the hydrophilic shell – comprising PEG 2000 in DP – could endow mixed micelle systems with long circulation times and passive targeting of the tumor location.32,33 Therefore, it may be an ideal drug delivery system for the intravenous administration of CK (Figure 1).
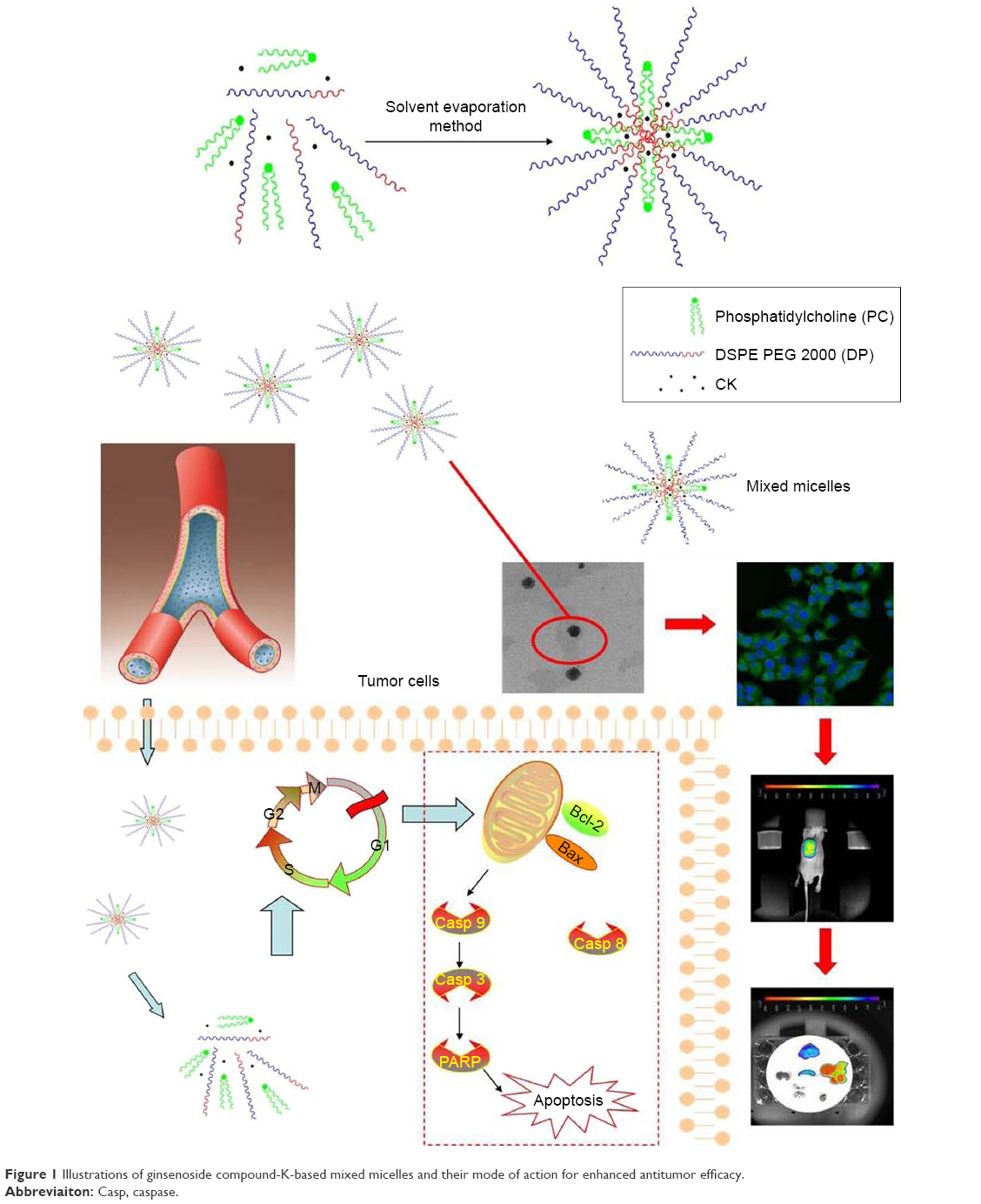 | Figure 1 Illustrations of ginsenoside compound-K-based mixed micelles and their mode of action for enhanced antitumor efficacy. |
To enhance the anticancer effect of CK and allow it to be injected, CK-loaded PC/DP mixed micelles were prepared. The human lung adenocarcinoma cell line, A549, was chosen to study the antitumor effects in vitro and in vivo. Meanwhile, the uptake and targeted tumor effects were confirmed by in vitro cellular uptake analysis and optical mouse imaging in vivo.
Materials and methods
Materials
Ginsenoside CK was obtained from Xi’an XiaoCao Botanical Development Co., Ltd. Both PC and DP were purchased from Shanghai Advanced Vehicle Technology Pharmaceutical Co., Ltd., Shanghai, China. Coumarin-6, 1,1′-dioctadecyl-3,3,3′,3′-tetramethylindotricarbocyanine iodide (DiR), and 4′,6-diamidino-2-phenylindole (DAPI) were purchased from Shanghai Aladdin Bio-Chem Technology Co. Ltd, Shanghai, China. A549 cells, RPMI 1640 culture medium, fetal bovine serum, as well as anti-caspase 3, anti-caspase 8, anti-caspase 9, anti-poly ADP-ribose polymerase (anti-PARP), anti-Bax, and anti-Bcl-2 antibodies were obtained from Jiangsu Keygen Biotech Corp., Ltd., Nanjing, China. Nude mice were purchased from Shanghai SLAC Laboratory Animal Co., Ltd., Shanghai, China. The other solvents used were of chromatographic grade.
Animals
Male nude mice were purchased from the SLEK Lab Animal Center of Shanghai (Shanghai, China), and received care according to the Guidelines on the Use of Laboratory Animals. All animal experiments were conducted in accordance with the principles of care and use of laboratory animals, and were approved by the experimental animal administrative committee of Suqian Branch Jiangsu Province Hospital. Ethical and legal approval was obtained prior to the study.
Preparation of CK PC/DP micellar systems
The CK PC/DP micellar systems were prepared by the thin-film hydration technique. In brief, CK, PC, and DP (molar ratio = 5:18:12) were accurately weighed and completely dissolved in chloroform. The chloroform was removed by rotary evaporation at 100 rpm for 20 min at 40°C until a thin film formed in the round flask. This film was hydrated at 50 rpm by using a table-top rocker under normal pressure for 1 h at 40°C to obtain the coarse CK PC/DP micellar systems. Finally, the coarse CK PC/DP micellar systems were dispersed at 25°C in a bath sonicator at 45 W for 30 min to reduce particle size and form a homogeneous system. The coumarin-6 mixed micelles and DiR mixed micelles were prepared using the abovementioned method by substituting CK with coumarin-6 and DiR at a specified ratio of PC and DP (molar ratio = 18:12), respectively.
Characterization of CK PC/DP micellar systems
Size distribution and ζ-potential
The size distribution and ζ-potential of the CK PC/DP micellar systems were measured using a dynamic light-scattering instrument (Zetasizer Nano ZSP, Malvern Instruments, Malvern, Worcestershire, UK). After being diluted 10 times, the samples were measured at 25°C in triplicate in deionized water.
Morphological characterization
The morphological features of the CK mixed micelles were photographed with a transmission electron microscope (TEM; JEOL, Tokyo, Japan). After being diluted 10 times with distilled water, the samples were placed on a copper grid coated with carbon film, and were then air-dried. Thereafter, the samples were stained with 2% phosphotungstic acid for 3 min and air-dried before observation.
Solubility measurements
Classical approaches for measuring solubility were based on the saturation shake-flask method. Excess amounts of CK were placed into 5-mL vials containing aqueous solutions. The vials were sealed and placed into a constant temperature bath (37°C), and shaken for 24 h until equilibrium was evident. After centrifugation of the incubated suspensions at 37°C and 15,000 rpm for 15 min, the concentrations of CK in the supernatant solutions were determined by a high-performance liquid chromatography (HPLC) procedure. Furthermore, the CK concentration in the micellar system was determined after a ten- to 100-times dilution with methanol, and then analyzed for CK.
In vitro drug release
The in vitro drug release was studied by the dialysis method using a dialysis membrane with a 3.5-kDa molecular weight cutoff. The CK mixed micelle solution, or free CK at an equivalent CK concentration, was placed in the dialysis bag. Then, it was submerged in release medium (deionized water) and incubated at 37°C with continuous stirring at 100 rpm. At 1, 2, 4, 6, 8, 12, 24, 36, and 48 h, 1 mL of the dialysate was sampled and the volume of the release medium was replenished with deionized water. The sampled dialysate was filtered through a 0.45-μm membrane and analyzed by HPLC. Then, the CK release curves were calculated.
Cell proliferation assay
A549 cells were seeded in 96-well plates and treated with CK or CK mixed micelles at different concentrations. After treatment for 24 h, the drugs were replaced with blank culture medium with 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) and incubated for 4 h at 37°C. Then, the plates were shaken for 10 min at 3,000 rpm. Finally, the medium was replaced with 100 μL dimethyl sulfoxide (DMSO) to dissolve the formazan crystals that were produced. Absorbance was measured in a multi-wavelength scanning spectrophotometer (Model 680, Bio-Rad Laboratories Inc., Hercules, CA, USA) at 570 and 630 nm.
Cell-cycle analysis
Cells (1 × 106 cells/mL) were seeded into six-well plates and cultured for 24 h. Then, they were treated with CK or CK mixed micelles for 24 h at 12.15 μg/mL. The cells were washed with phosphate-buffered saline (PBS) and harvested with trypsin-ethylene diamine tetraacetic acid (EDTA). The harvested cells were counted and fixed with 70% ethanol. Samples were resuspended in 1.0-mL hypotonic propidium iodide (PI) solution and incubated in the dark for 30 min at 4°C. The PI-stained samples were analyzed with a flow cytometer (Beckman Coulter, USA).
Wound-healing assay
A549 cells were seeded in a 12-well plate and allowed to grow to confluence. A 200-μL pipette tip was used to scratch a line in the cell monolayer. The cells were washed with PBS thrice to remove floating cells, and the adherent cells were cultured for an additional 24 h with serum-free medium containing CK or CK mixed micelles at 12.15 μg/mL. The cultured cells were photographed and the width of the scratch wound was measured.
Apoptosis analysis
Cell apoptosis was qualitatively analyzed by terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling (TUNEL). In brief, after treatment with CK or CK mixed micelles, formalin-fixed A549 cells (2 × 105 cells/mL) were permeabilized using Triton X-100 at 4°C for 5 min. The cells were washed and labeled with TUNEL reaction mixture for 60 min at 37°C in the dark. Fragmented DNA was colored brown. The cells were photographed using a light microscope.
For quantitative analysis, cell apoptosis was determined by flow cytometry. A549 cells were treated with blank culture medium, CK, or CK mixed micelles (12.15 μg/mL) for 24 h. After two washes with ice-cold PBS, the cells were harvested with trypsin-EDTA and resuspended at 1 × 106 cells/mL. The cell suspensions (100 μL) were kept in the dark and stained simultaneously with FITC-annexin V (FITC-AV) and PI for 10 min at 37°C; subsequently, apoptosis was analyzed using flow cytometry.
In vitro cellular uptake
The cell uptake was evaluated by fluorescence imaging. Coumarin-6 was chosen as the fluorescent probe because it is a substrate for P-glycoprotein, similar to CK. A549 cells were seeded in 24-well plates and incubated for 24 h. Next, medium containing coumarin-6 mixed micelles or coumarin-6 solution was added into each well and incubated at 37°C for 4 h. Subsequently, the cells were fixed with 4% formaldehyde for 10 min. The cells were then stained with DAPI for 10 min in the dark. These cells were then washed with PBS twice and observed under a fluorescence microscope (IX71; Olympus Corp, Tokyo, Japan) and by a confocal laser scanning microscope (TCS SP5; Leica, Heidelberg, Germany).
Optical in vivo imaging
DiR-loaded mixed micelles were used to investigate tumor-targeting efficacy in the A549 cell xenograft tumor model. When the tumor volume reached ~400 mm3, mice were injected with DiR or DiR mixed micelles via the tail vein at a dose of 5 mg/kg. Fluorescence intensity was visualized using the Maestro EX in vivo fluorescence imaging system (Cambridge Research & Instrumentation Inc. [CRi], Woburn, MA, USA). After in vivo imaging, the mice were euthanized at 24 h, and the major organs, such as the heart, liver, spleen, lungs, and kidneys, as well as the tumors were excised. The near-infrared fluorescence signal intensities in these excised tissues were measured.
In vivo antitumor efficacy of mixed micelles
A549 cells were implanted subcutaneously in each flank of nude mice. Once the A549 xenografts reached 50 mm3, the mice were randomized into three treatment groups (five mice per group): untreated control group, free CK group, and CK mixed micelle group. The mice were treated with the CK mixed micelle equivalent doses of 30 mg/kg free CK every 3 days for 12 consecutive days via tail-vein injection. On the fifteenth day, mice were euthanized to remove the liver, kidneys, and tumors. These tissues were stained by hematoxylin and eosin (H&E) for pathological analysis.
The tumor cell lysates (25 μg) were separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS–PAGE) and transferred onto polyvinylidene difluoride membranes. The membranes were incubated with specific primary antibodies at 4°C for 12 h and visualized by using a Western blotting detection system. Caspase-3, caspase-8, caspase-9, PARP, Bax, and Bcl-2 antibodies were obtained from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA, USA). The results of Western blotting were analyzed on the basis of the level of chemiluminescence.
High-performance liquid chromatography
The concentration of CK was determined by HPLC on an Agilent 1200 (Agilent Co. Ltd, Waldbronn, Germany) equipped with an Agilent™ RP-C18 column (150 mm × 4.6 mm, 5 μm). The mobile phase of methanol and water (60:40, v:v) was used at a flow rate of 1.0 mL/min. The UV detector was set at 230 nm to analyze the column effluent and the column temperature was set at 30°C. The standard curve of CK was A = 3.0408C+3.624. The R2 for the calibration was 0.998 and the linearity range was 2.3–57.5 μg/mL.
Statistical analysis
Data were obtained from at least three separate experiments and are presented as means ± SD. Differences between two groups were determined by Student’s t-tests. A P-value of <0.05 was considered statistically significant.
Results
Characterization of CK PC/DP micelles
The size of water-dispersed CK PC/DP micelles was determined to be 18.58 ± 2.65 nm with a low polydispersity index (PDI = 0.196; Figure 2A). The loading efficiency of CK was 11.76% ± 1.32%. Spherical morphology was observed by TEM, with well-distributed sizes (Figure 2B). The zeta potential of the CK PC/DP micelles was recorded as −35.17 ± 2.41 mV, which indicated that the micelles were stable due to the high negative surface charge (Figure 2C). Particles ranging from 10 to 200 nm can pass through the vascular bed and accumulate at the tumor location.34,35 Therefore, we can assume that CK PC/DP micelles of approximately 20 nm can passively target the tumor site. Meanwhile, the solubility of CK increased from 33.15 ± 3.82 to 2,215.67 ± 166.39 μg/mL after the constitution of the CK mixed micelle formation (P < 0.05). The solubility increased almost 66-fold. The CK PC/DP micelles had a slower in vitro release than that of free CK, as shown in Figure 2D. The results of the release suggest that CK mixed micelles will have improved absorption due to longer retention times and lower release rates.
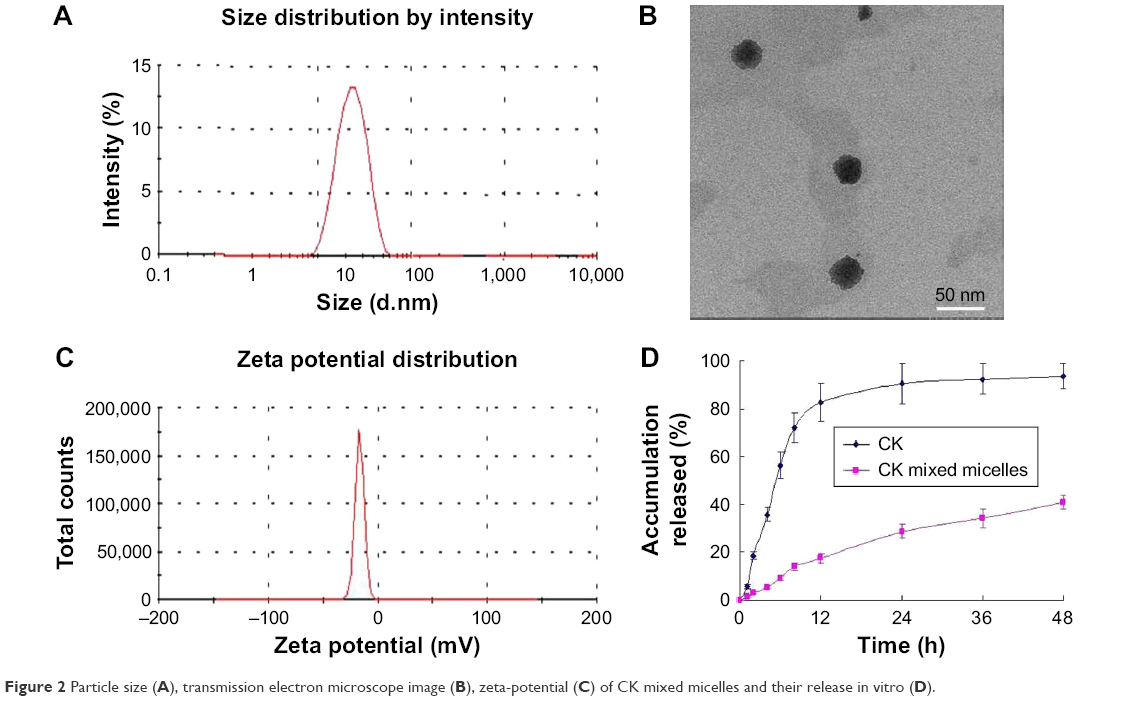 | Figure 2 Particle size (A), transmission electron microscope image (B), zeta-potential (C) of CK mixed micelles and their release in vitro (D). |
Cell proliferation assay
The cytotoxicity of CK and CK PC/DP micelles was evaluated after incubation with A549 cells for 24 h (Figure 3A). As shown in Figure 3A, cell viabilities decreased in a concentration-dependent manner. The mixed micelles resulted in greater growth inhibition of A549 cells than did free CK. The IC50 of free CK and CK mixed micelles at 24 h was 18.31 and 12.15 μg/mL (P < 0.05), respectively. The micellar system most likely promoted greater drug uptake, significantly increasing the antitumor effect.
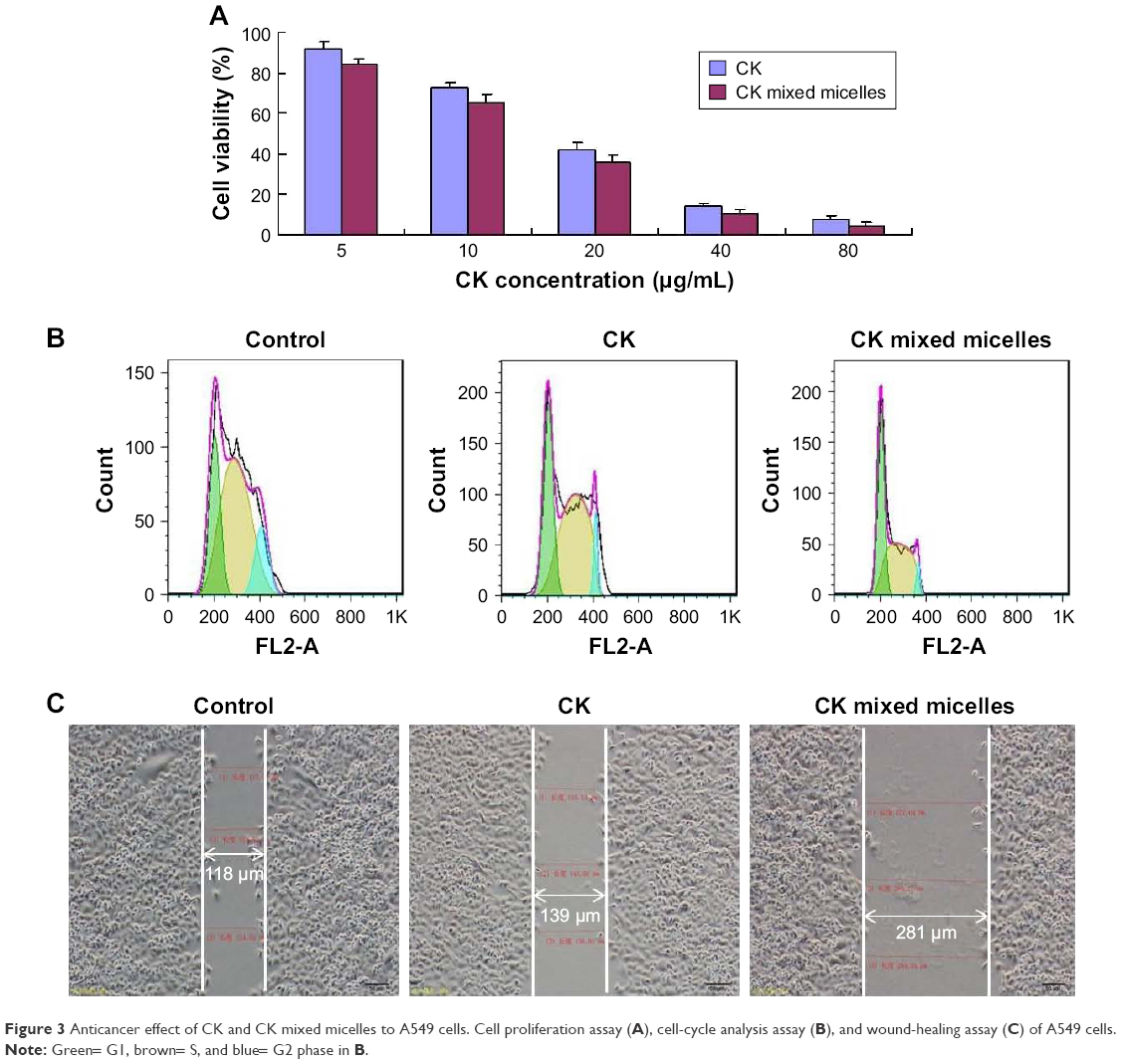 | Figure 3 Anticancer effect of CK and CK mixed micelles to A549 cells. Cell proliferation assay (A), cell-cycle analysis assay (B), and wound-healing assay (C) of A549 cells. |
Cell-cycle arrest in A549 cells
To identify the effect on the cell cycle in A549 cells, the cell-cycle distribution was assessed by flow cytometry. As shown in Figure 3B, we found that inhibition of DNA synthesis by CK arrested the cell cycle in the G1 phase, with the CK PC/DP micelles being more effective than free CK. The percentage of cells in the G1 phase increased from 31.54% ± 2.48% to 39.27% ± 4.39% after treatment with CK PC/DP micelles (P < 0.05).
Wound-healing assay
To further confirm the antitumor effect of CK PC/DP micelles, a wound-healing assay was used. As seen in Figure 3C, both CK and CK mixed micelles more strongly inhibited the invasion of A549 cells compared with that of the control. On comparing free CK and CK PC/DP micelles, the latter exhibited a significantly better inhibitory effect. Therefore, the results indicate that CK can decrease the invasiveness of tumor cells, and the effect significantly increased after loading into the PC/DP delivery system, likely because of the enhancement in uptake.
TUNEL assay and apoptosis in A549 cells
TUNEL and annexin-V/PI staining assays were used to evaluate the apoptosis-inducing effect of CK on A549 cells. The morphology of A549 cells was investigated after incubation with CK or CK mixed micelles for 24 h. Using TUNEL, positively stained cells were evident in the CK and CK mixed micelle groups, whereas stained cells were absent in the control group (Figure 4A). More apoptotic bodies were observed after treatment with CK PC/DP micelles than after treatment with CK alone. The apoptotic rates were quantified using an annexin V/PI staining assay. The apoptosis rate was highest in the CK mixed micelle group (Figure 4B). The late apoptosis rate increased from 6.7% to 19.8% after treatment with the CK PC/DP micelles (P < 0.05). In this study, qualitative and quantitative analyses were conducted to evaluate apoptosis. The results are consistent and indicate that the CK PC/DP micelle system can induce apoptosis with an improved effect over that of CK alone.
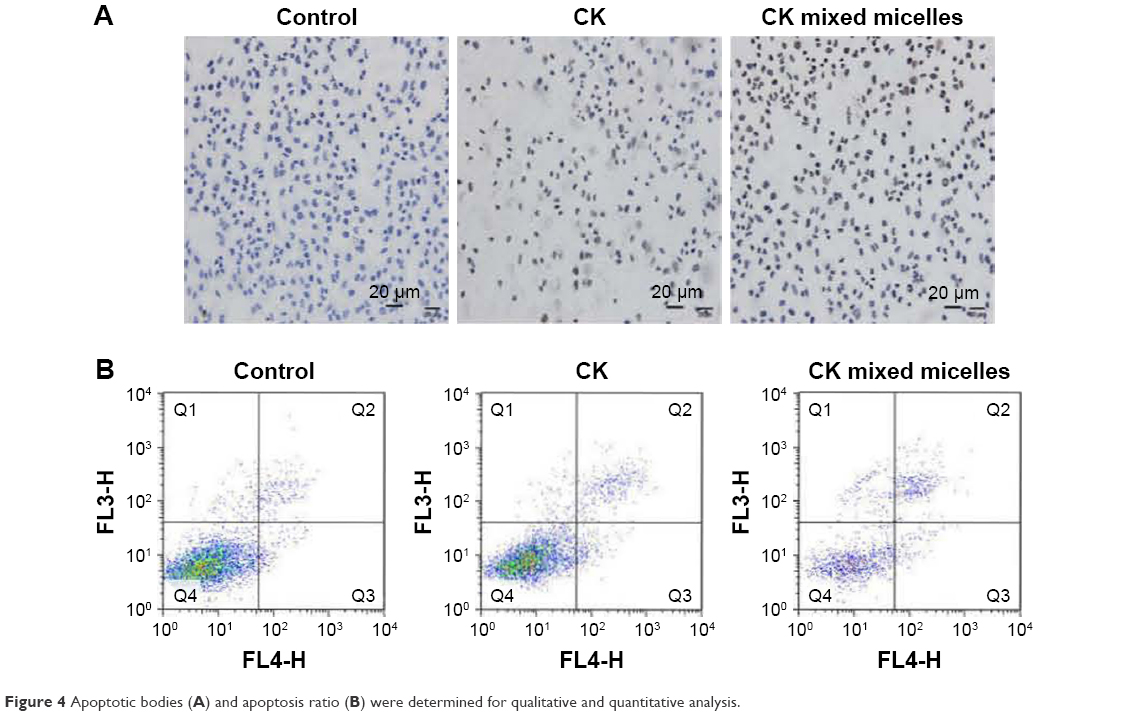 | Figure 4 Apoptotic bodies (A) and apoptosis ratio (B) were determined for qualitative and quantitative analysis. |
Cellular uptake of PC/DP micelles
Fluorescence microscopy and confocal microscopy were used to evaluate the uptake of PC/DP micelles by A549 cells. Coumarin-6 was chosen as the fluorescent dye for tracking.
The fluorescence images of A549 cells (Figure 5A and B) indicate that the fluorescence intensity of coumarin-6 is higher when delivered to the A549 cells in PC/DP micelles than when delivered as free coumarin-6. Furthermore, these images suggest that the PC/DP micelle system can enhance the uptake of drugs. Confocal microscopy (Figure 5B) indicated that the mixed micelle system delivered the coumarin-6 to the cytoplasm of the A549 cells. Therefore, the PC/DP micelle system can easily transfer drugs into tumor cells to enhance the antitumor effect.
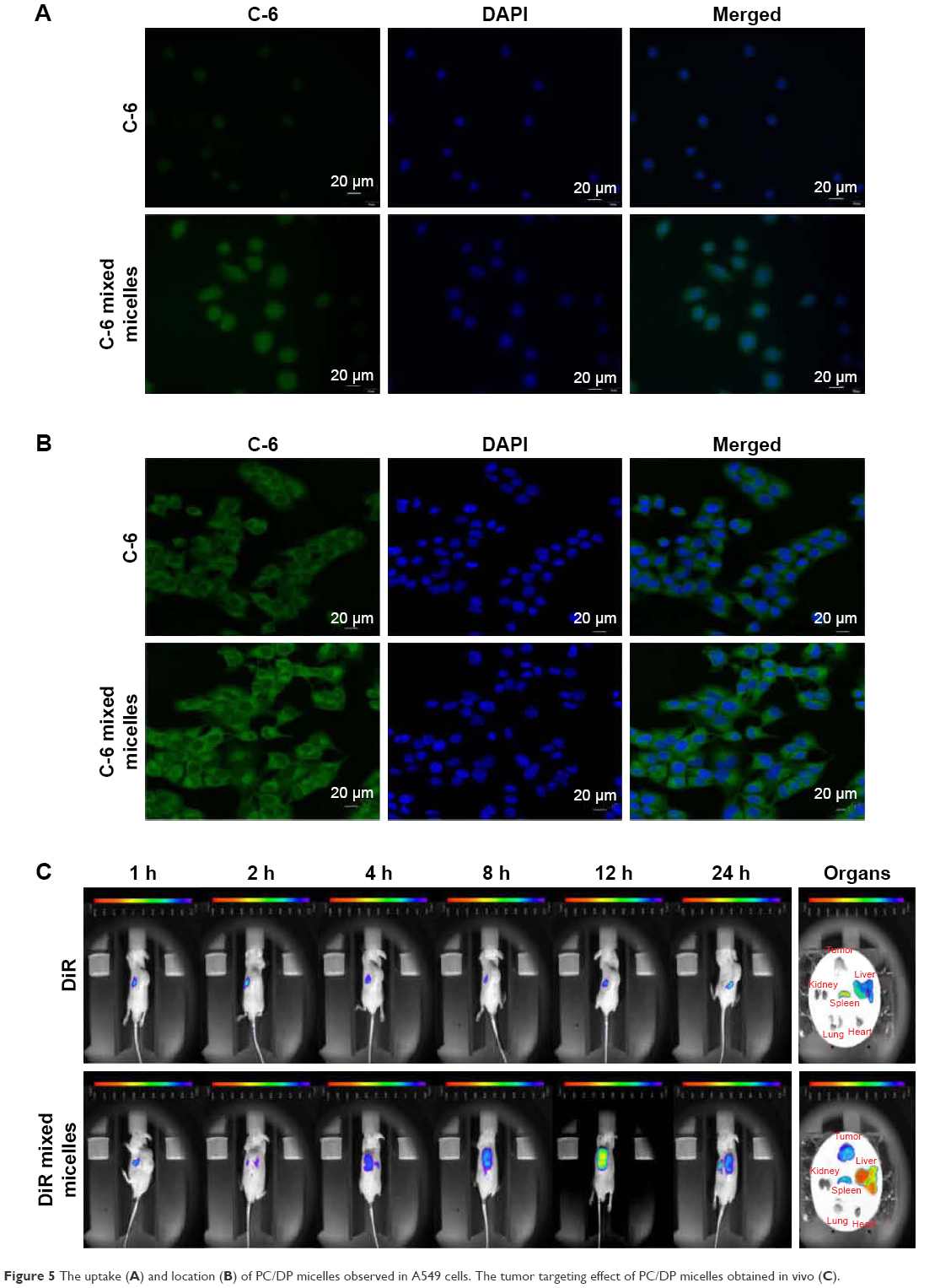 | Figure 5 The uptake (A) and location (B) of PC/DP micelles observed in A549 cells. The tumor targeting effect of PC/DP micelles obtained in vivo (C). |
Optical in vivo imaging
Figure 5C illustrates the accumulation of DiR against time in the xenograft tumors. Two hours after the administration of DiR mixed micelles, a fluorescence signal at the tumor location was observed. The maximum fluorescence intensity occurred at 12 h, and was maintained up to 24 h. However, free DiR did not exhibit a targeted tumor effect. This phenomenon illustrates that the tumor-targeting effect of the PC/DP micelle system can transfer drugs into tumor tissues, which was consistent with our in vitro results.
After administration of DiR or DiR mixed micelles for 24 h, the tumor and major organs were excised to undertake ex vivo optical imaging. Although the liver and spleen in the DiR and DiR mixed micelle groups exhibited fluorescence, fluorescence in the tumor tissues could only be observed in the DiR mixed micelle group. The PC/DP micelle system appears to localize at the tumor site, and this occurs for a longer duration. Therefore, the in vivo optical images and the ex vivo optical images are consistent. These results indicate that the PC/DP micelle system can carry drugs to the tumor location.
In vivo antitumor efficacy
Figure 6 illustrates the anticancer effect in xenograft tumors of different formulations. The results demonstrate that CK and CK mixed micelles significantly inhibited tumor growth (P < 0.05). The CK mixed micelles exhibited an optimum antitumor effect (52.17% ± 7.29% at Day 15) compared with that reported for the control group.
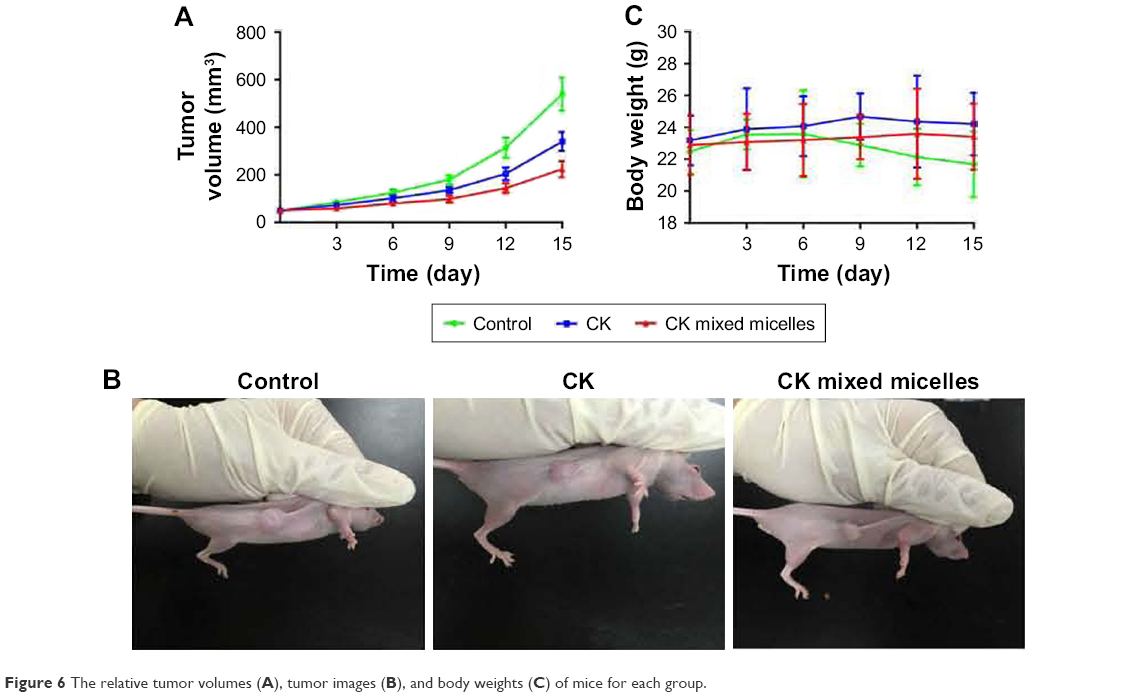 | Figure 6 The relative tumor volumes (A), tumor images (B), and body weights (C) of mice for each group. |
A loss in body weight was only observed in the tumor-bearing control group, similar to that observed in cancer patients. There was a <10% decrease in the body weights of the CK and CK mixed micelle-treated mice. This indicates the antitumor efficacy and safety of the CK and CK mixed micelles.
The safety and improved antitumor efficacy of the CK mixed micelles in the tumor-bearing nude mice was confirmed by histopathology, which was carried out to observe microscopic changes in the liver, kidney, and tumor by H&E staining (Figure 7A). The livers and kidneys were harvested on Day 15. No evidence of pathological damage was observed in the liver and kidney. Therefore, CK and the CK mixed micelle system can be used without significant toxicity to the liver and kidney. On evaluation of the tumor sections, significant apoptosis in the internal and lateral area of the tumor tissues was observed for the CK mixed micelle group, which was consistent with the in vitro study. In the control group, minor necrosis in the internal area of the tumor tissue was observed.
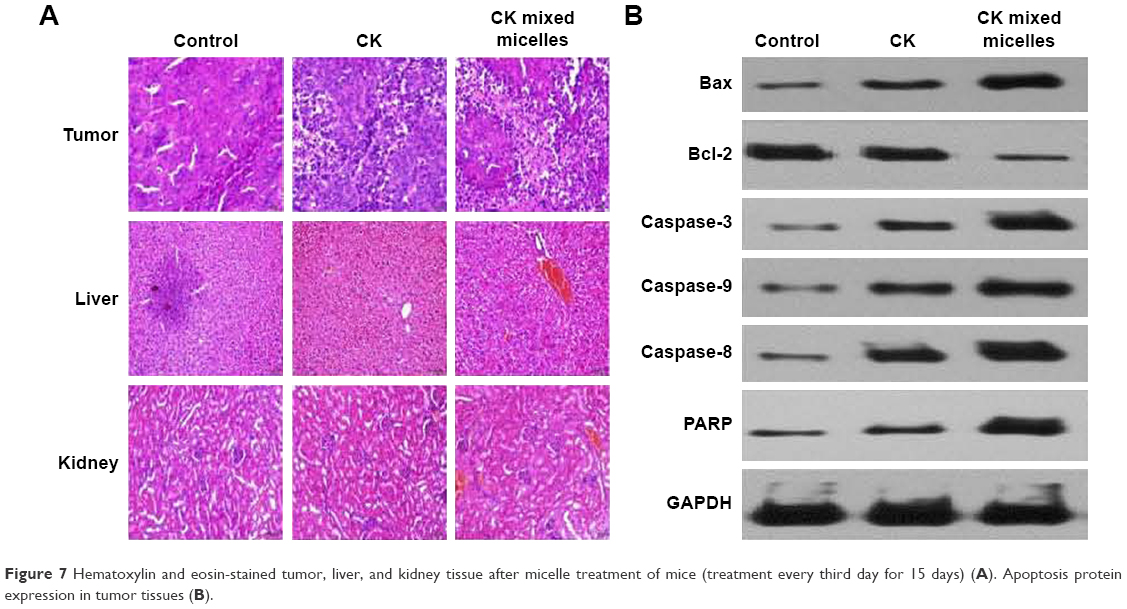 | Figure 7 Hematoxylin and eosin-stained tumor, liver, and kidney tissue after micelle treatment of mice (treatment every third day for 15 days) (A). Apoptosis protein expression in tumor tissues (B). |
We next examined Bcl-2 family proteins and caspase proteins to understand the mechanism of induced apoptosis in vivo. As shown in Figure 7B, we found that treatment with CK mixed micelles could significantly increase the BAX/Bcl-2 ratio. This demonstrated that the CK delivered to the tumor tissues by the PC/DP micelle system could promote tumor apoptosis effectively. To investigate whether the CK PC/DP micelle system could increase the apoptotic sensitivity of cancer cells, expression of caspase-3, caspase-8, caspase-9, and PARP protein in tumor tissues were investigated. After treatment with the CK mixed micelles, expression of the apoptosis-associated proteins caspase-3, caspase-8, and caspase-9 increased by 2.1-, 0.9-, and 0.7-fold, respectively. Meanwhile, PARP expression increased by 1.8-fold. A significant increase in caspase and PARP protein expression was observed after treatment with the PC/DP micelle system compared with that observed with free CK (P < 0.05). Therefore, the in vitro and in vivo studies confirmed that the CK PC/DP micelle system can induce apoptosis effectively. Furthermore, CK mixed micelles exhibit low tissue toxicity and a greater antitumor effect than that of CK alone, due particularly to apoptosis induction.
Discussion
To obtain antitumor effects, CK first must be transmitted to the tumor location and traverse the cancer cell membranes to achieve effective intracellular concentrations. However, the low solubility, hepatic metabolism, and activity of P-glycoprotein have made obtaining effective concentrations of CK in the blood difficult.12,13 Moreover, because of abundant drug-efflux protein activity in cancers, CK may lack sufficient tumor accumulation, similar to that of other conventional chemotherapeutic drugs. Thus, to increase the cancer treatment efficacy of CK, it is important to enhance its serum concentration and promote its intracellular CK accumulation in tumor tissues.
Micelles are self-assembling, spherical, colloidal structures smaller than 100 nm with a narrow size distribution. They form spontaneously in aqueous conditions to produce a hydrophobic core and a hydrophilic shell36 and are used for parenteral drug administration, especially of drugs with low solubility (including anticancer drugs). Generally, they are considered a safe and biocompatible long-circulating drug delivery system when covered with a hydrophilic polymer shell.37 A number of polymeric micelles have been regarded as passive targeting agents and tested in clinical trials.38
To this end, the PC/DP micelles have emerged as potential carriers for CK delivery due to their attractive characteristics, such as their small size and high loading capacity, which allow CK accumulation in tumor tissue through leaky tumor vasculature by utilizing the enhanced permeation and retention effect. Moreover, this could minimize exposure of healthy tissues to the drug. In the present study, the solubility of CK was shown to increase from 33.15 ± 3.82 to 2,215.67 ± 166.39 μg/mL after CK mixed micelle formation. The solubility increased ~66-fold, which should allow CK to obtain a higher blood concentration than it could as a free drug. Additionally, the PC/DP mixed micelles effectively retained the CK loaded in their hydrophobic cores under a simulated physiological environment. After encapsulation in micelles, CK exhibited a sustained-release behavior in vitro. In addition, the in vivo imaging indicated that the PC/DP mixed micelles had a long retention time after injection.
Moreover, PEG coated on the surface of the mixed micelles could sterically hinder interaction with plasma proteins,39 evade macrophage engulfment in the reticuloendothelial system,40 allow extravasation across the tumor endothelium,41,42 and passively target the tumor site via the enhanced permeation and retention effect. This has been shown in the “Characterization of CK PC/DP micelles” and “Optical in vivo imaging” sections.
PEG is widely used as a hydrophilic raw material for micelles (molecular weight 2–15 kDa) due to its high water solubility, non-toxicity, and neutral charge. The hydrophilic corona formed by PEG on the micelle surface decreases nonspecific interaction with blood proteins, thereby increasing the circulation time.
Importantly, through in vivo antitumor experiments, the therapeutic effects were assessed based on the tumor volume changes at the end of the treatment. CK mixed micelles resulted in higher tumor inhibition than free CK. This was because CK mixed micelles significantly increased the CK concentration in the tumor tissues through the enhanced permeability and retention effect, which had been previously validated by optical in vivo imaging.
CK PC/DP micelles can significantly enhance the in vitro and in vivo lung cancer treatment efficacy of CK. Both PC and DP are US Food and Drug Administration-approved pharmaceutical excipients, which makes the current micellar delivery system highly translatable to the clinic. However, future studies are needed to establish the efficacy of this delivery system in the clinical setting.
Conclusion
CK mixed micelles exhibited the strongest proapoptotic effects and antitumor efficacy against A549 cells, which may be due to increased cellular uptake of, and enhanced tumor targeting by, the CK mixed micelles. These results indicate that the PC DSPE PEG 2000 mixed micelle delivery system holds great promise to be used with CK in targeted cancer therapies.
Acknowledgments
This work was supported by Project of Jiangsu Natural Science Foundation of China (grant no BK20171321), Jiangsu Youth Medical Talents Project (grant no QNRC2016482), National Natural Science Foundation of China (Grant No. 81503265), and the Science and Technology Support Project of Suqian (grant nos S201622 and S201623).
Disclosure
The authors report no conflicts of interest in this work.
References
Yosioka I, Sugawara T, Imai K, Kitagawa I. Soil bacterial hydrolysis leading to genuine aglycone. V. On ginsenosides Rbl, Rb2 and Rc of the ginseng root saponins. Chem Pharm Bull (Tokyo). 1972;20(11):2418–2421. | ||
Hasegawa H, Sung JH, Matsumiya S, Uchiyama M. Main ginseng saponin metabolites formed by intestinal bacteria. Planta Med. 1996;62(5):453–457. | ||
Akao T, Kida H, Kanaoka M, Hattori M, Kobashi K. Intestinal bacterial hydrolysis is required for the appearance of compound K in rat plasma after oral administration of ginsenoside Rb1 from Panax ginseng. J Pharm Pharmacol. 1998;50(10):1155–1160. | ||
Man S, Gao W, Zhang Y, Huang L, Liu C. Chemical study and medical application of saponins as anti-cancer agents. Fitoterapia. 2010;81(7):703–714. | ||
Qi LW, Wang CZ, Yuan CS. Ginsenosides from American ginseng: chemical and pharmacological diversity. Phytochemistry. 2011;72(8):689–699. | ||
Yu H, Zhang C, Lu M, Sun F, Fu Y, Jin F. Purification and characterization of new special ginsenosidase hydrolyzing multi-glycisides of protopanaxadiol ginsenosides, ginsenosidase type I. Chem Pharm Bull (Tokyo). 2007;55(2):231–235. | ||
Kang KA, Kim YW, Kim SU, et al. G1 phase arrest of the cell cycle by a ginseng metabolite, compound K, in U937 human monocytic leukamia cells. Arch Pharm Res. 2005;28(6):685–690. | ||
Zhang Z, Du GJ, Wang CZ, et al. Compound K, a ginsenoside metabolite, inhibits colon cancer growth via multiple pathways including p53–p21 interactions. Int J Mol Sci. 2013;14(2):2980–2995. | ||
Lee SJ, Ko WG, Kim JH, Sung JH, Moon CK, Lee BH. Induction of apoptosis by a novel intestinal metabolite of ginseng saponin via cytochrome c-mediated activation of caspase-3 protease. Biochem Pharmacol. 2000;60(5):677–685. | ||
Cho SH, Chung KS, Choi JH, Kim DH, Lee KT. Compound K, a metabolite of ginseng saponin, induces apoptosis via caspase-8-dependent pathway in HL-60 human leukemia cells. BMC Cancer. 2009;9:449. | ||
Ming Y, Chen Z, Chen L, et al. Ginsenoside compound K attenuates metastatic growth of hepatocellular carcinoma, which is associated with the translocation of nuclear factor-kB p65 and reduction of matrix metalloproteinase-2/9. Planta Med. 2011;77(5):428–433. | ||
Yang Z, Wang JR, Niu T, et al. Inhibition of P-glycoprotein leads to improved oral bioavailability of compound K, an anticancer metabolite of red ginseng extract produced by gut microflora. Drug Metab Dispos. 2012;40(8):1538-1544. | ||
Paek IB, Moon Y, Kim J, et al. Pharmacokinetics of a ginseng saponin metabolite compound K in rats. Biopharm Drug Dispos. 2006;27(1):39-45. | ||
Lee PS, Song TW, Sung JH, Moon DC, Song S, Chung YB. Pharmacokinetic characteristics and hepatic distribution of IH-901, a novel intestinal metabolite of ginseng saponin, in rats. Planta Med. 2006;72(3):204-210. | ||
Jiang T, Zhang Z, Zhang Y, et al. Dual-functional liposomes based on pH-responsive cell-penetrating peptide and hyaluronic acid for tumor-targeted anticancer drug delivery. Biomaterials. 2012;33(36):9246–9258. | ||
Zhang Z, Chen Y, Deng J, Jia X, Zhou J, Lv H. Solid dispersion of berberine-phospholipid complex/TPGS 1000/SiO2: preparation, characterization and in vivo studies. Int J Pharm. 2014;465(1–2):306–316. | ||
Wang W, Li M, Zhang Z, et al. Design, synthesis and evaluation of multi-functional tLyP-1-hyaluronic acid-paclitaxel conjugate endowed with broad anticancer scope. Carbohydr Polym. 2017;156:97–107. | ||
Mathiyalagan R, Subramaniyam S, Kim YJ, et al. Synthesis and pharmacokinetic characterization of a pH-sensitive polyethylene glycol ginsenoside CK (PEG-CK) conjugate. Biosci Biotechnol Biochem. 2014;78(3):466–468. | ||
Mathiyalagan R, Subramaniyam S, Kim YJ, Kim YC, Yang DC. Ginsenoside compound K-bearing glycol chitosan conjugates: synthesis, physicochemical characterization, and in vitro biological studies. Carbohydr Polym. 2014;112:359–366. | ||
Yang L, Xin J, Zhang Z, et al. TPGS-modified liposomes for the delivery of ginsenoside compound K against non-small cell lung cancer: formulation design and its evaluation in vitro and in vivo. J Pharm Pharmacol. 2016;68(9):1109–1118. | ||
Zhang Y, Tong D, Che D, et al. Ascorbyl palmitate/d-α-tocopheryl polyethylene glycol 1000 succinate monoester mixed micelles for prolonged circulation and targeted delivery of compound K for antilung cancer therapy in vitro and in vivo. Int J Nanomedicine. 2017;12:605–614. | ||
Mohamed EA, Abu Hashim II, Yusif RM, et al. Polymeric micelles for potentiated antiulcer and anticancer activities of naringin. Int J Nanomedicine. 2018;13:1009–1027. | ||
Lin J, Zhao C, Liu C, et al. Redox-responsive F127-folate/F127-disulfide bond-d-α-tocopheryl polyethylene glycol 1000 succinate/P123 mixed micelles loaded with paclitaxel for the reversal of multidrug resistance in tumors. Int J Nanomedicine. 2018;13:805–830. | ||
Jin X, Zhang Y, Zhang Z, Che D, Lv H. Juglone loaded poloxamer 188/phospholipid mixed micelles evaluated in vitro and in vivo in breast cancer. Int J Pharm. 2016;515(1–2):359–366. | ||
Duan Y, Zhang B, Chu L, Tong HH, Liu W, Zhai G. Evaluation in vitro and in vivo of curcumin-loaded mPEG-PLA/TPGS mixed micelles for oral administration. Colloids Surf B Biointerfaces. 2016;141:345–354. | ||
Gao ZG, Fain HD, Rapoport N. Controlled and targeted tumor chemotherapy by micellar-encapsulated drug and ultrasound. J Control Release. 2005;102(1):203–222. | ||
Dong J, Song X, Lian X, Fu Y, Gong T. Subcutaneously injected ivermectin-loaded mixed micelles: formulation, pharmacokinetics and local irritation study. Drug Deliv. 2016;23(7):2220–2227. | ||
Gill KK, Kaddoumi A, Nazzal S. PEG-lipid micelles as drug carriers: physiochemical attributes, formulation principles and biological implication. J Drug Target. 2015;23(3):222–231. | ||
Che J, Okeke CI, Hu ZB, Xu J. DSPE-PEG: a distinctive component in drug delivery system. Curr Pharm Des. 2015;21(12):1598–1605. | ||
Yang C, Wu T, Qin Y, et al. A facile doxorubicin-dichloroacetate conjugate nanomedicine with high drug loading for safe drug delivery. Int J Nanomedicine. 2018;13:1281–1293. | ||
Haghiralsadat F, Amoabediny G, Helder MN, et al. A comprehensive mathematical model of drug release kinetics from nano-liposomes, derived from optimization studies of cationic PEGylated liposomal doxorubicin formulations for drug-gene delivery. Artif Cells Nanomed Biotechnol. 2018;46(1):169–177. | ||
Rydberg HA, Yanez Arteta M, Berg S, Lindfors L, Sigfridsson K. Probing adsorption of DSPE-PEG2000 and DSPE-PEG5000 to the surface of felodipine and griseofulvin nanocrystals. Int J Pharm. 2016;510(1):232–239. | ||
Jin Y, Zhang Z, Zhao T, Liu X, Jian L. Mixed micelles of doxorubicin overcome multidrug resistance by inhibiting the expression of P-glycoprotein. J Biomed Nanotechnol. 2015;11(8):1330–1338. | ||
Bae YH, Park K. Targeted drug delivery to tumors: myths, reality and possibility. J Control Release. 2011;153(3):198–205. | ||
Torchilin V. Tumor delivery of macromolecular drugs based on the EPR effect. Adv Drug Deliv Rev. 2011;63(3):131–135. | ||
Zhang Y, Ren T, Gou J, et al. Strategies for improving the payload of small molecular drugs in polymeric micelles. J Control Release. 2017;261:352–366. | ||
Manaia EB, Abuçafy MP, Chiari-Andréo BG, Silva BL, Oshiro Junior JA, Chiavacci LA. Physicochemical characterization of drug nanocarriers. Int J Nanomedicine. 2017;12:4991–5011. | ||
Deshmukh AS, Chauhan PN, Noolvi MN, et al. Polymeric micelles: basic research to clinical practice. Int J Pharm. 2017;532(1):249–268. | ||
Papi M, Caputo D, Palmieri V, et al. Clinically approved PEGylated nanoparticles are covered by a protein corona that boosts the uptake by cancer cells. Nanoscale. 2017;9(29):10327–10334. | ||
Maruyama K. Intracellular targeting delivery of liposomal drugs to solid tumors based on EPR effects. Adv Drug Deliv Rev. 2011;63(3):161–169. | ||
Taurin S, Nehoff H, Greish K. Anticancer nanomedicine and tumor vascular permeability; where is the missing link? J Control Release. 2012;164(3):265–275. | ||
Zou L, Wang D, Hu Y, et al. Drug resistance reversal in ovarian cancer cells of paclitaxel and borneol combination therapy mediated by PEG-PAMAM. Oncotarget. 2017;8(36):60453–60468. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.