")
Back to Journals » International Journal of Nanomedicine » Volume 9 » Issue 1
Preparation of biocompatible heat-labile enterotoxin subunit B-bovine serum albumin nanoparticles for improving tumor-targeted drug delivery via heat-labile enterotoxin subunit B mediation
Authors Zhao L, Su R, Cui W, Shi Y, Liu L, Su C
Received 15 January 2014
Accepted for publication 10 March 2014
Published 6 May 2014 Volume 2014:9(1) Pages 2149—2156
DOI https://doi.org/10.2147/IJN.S60764
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Liang Zhao,1,* Rongjian Su,2,* Wenyu Cui,3 Yijie Shi,1 Liwei Liu,1 Chang Su4
1School of Pharmacy, Liaoning Medical University, Jinzhou, People's Republic of China; 2Central Laboratory of Liaoning Medical University, Jinzhou, People’s Republic of China; 3National Vaccine and Serum Institute, Beijing, People’s Republic of China; 4School of Veterinary Medicine, Liaoning Medical University, Jinzhou, People’s Republic of China
*These authors contributed equally to this work
Abstract: Heat-labile enterotoxin subunit B (LTB) is a non-catalytic protein from a pentameric subunit of Escherichia coli. Based on its function of binding specifically to ganglioside GM1 on the surface of cells, a novel nanoparticle (NP) composed of a mixture of bovine serum albumin (BSA) and LTB was designed for targeted delivery of 5-fluorouracil to tumor cells. BSA-LTB NPs were characterized by determination of their particle size, polydispersity, morphology, drug encapsulation efficiency, and drug release behavior in vitro. The internalization of fluorescein isothiocyanate-labeled BSA-LTB NPs into cells was observed using fluorescent imaging. Results showed that BSA-LTB NPs presented a narrow size distribution with an average hydrodynamic diameter of approximately 254±19 nm and a mean zeta potential of approximately -19.95±0.94 mV. In addition, approximately 80.1% of drug was encapsulated in NPs and released in the biphasic pattern. The 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay showed that BSA-LTB NPs exhibited higher cytotoxic activity than non-targeted NPs (BSA NPs) in SMMC-7721 cells. Fluorescent imaging results proved that, compared with BSA NPs, BSA-LTB NPs could greatly enhance cellular uptake. Hence, the results indicate that BSA-LTB NPs could be a potential nanocarrier to improve targeted delivery of 5-fluorouracil to tumor cells via mediation of LTB.
Keywords: heat-labile enterotoxin subunit B, nanoparticle, bovine serum albumin, 5-fluorouracil
Introduction
Cancer is a class of disease characterized by out-of-control cell growth. There are over 100 different types of cancer, and each is classified by the type of cell that is initially affected. Although the clinical treatment for cancer has made considerable progress with improvement of surgical technique, wide application of new technology, and clinical development of new drugs, cancer remains a complicated medical problem that cannot yet be overcome and which threatens human health and lives.1 Conventional treatments of cancer generally include surgical resection, radiotherapy, and chemotherapy. Though surgical resection is helpful for removing the visible tumor tissue, it is not suitable for dealing with some symptoms, such as invisible tumor, blood tumor which invaded through blood or lymph node, and terminal cancer. Radiotherapy only works in treating a number of cancer types if they are localized to one small area of the body. In addition, it may also be used as part of adjuvant therapy to prevent tumor recurrence after removing a primary malignant tumor surgically. Moreover, radiotherapy also leads to a series of serious complications, mainly including tissue damage near the area to being irradiated, fatigue, adverse consequences of skin inflammation, hair loss, and gastrointestinal reactions.2–4 Although chemotherapeutic drugs are effective at killing tumor cells, there are some drawbacks, including nonselective distribution, drug toxicity, and unexpected side effects to normal tissues, which limit their clinical usage.5,6 Generally, it is difficult to achieve satisfactory therapeutic effects in cancer treatment with conventional treatment methods, thus it is urgent that a more active and effective way to fight against cancer is found.
In nanotechnology, a particle covers a range between 1 and 500 nm in size and usually demonstrates good transportation and special biological properties. With the rapid development of techniques for nanoparticle (NP) preparation, NPs as a key tool in targeted cancer treatment have been widely investigated. In view of their shape and size, NPs can be easily aggregated and accumulated inside tumor tissues for a long time, known as the enhanced permeability and retention effect, due to the abnormality of newly formed tumor vessels in form and architecture.7 At the same time, the bioavailability in vivo is increased by encapsulating poorly soluble drugs inside NPs. In particular, the release of drugs could be controlled depending on slow diffusion or the degradation of biomaterials, and half-life of drugs could be prolonged.8–11 However, NPs have some deficiencies that limit their application. Before reaching the target site, NPs are easily phagocytized by the reticuloendothelial system and accumulated in reticuloendothelial system-rich non-targeted organs such as liver and spleen, leading to serious side effects and lower drug concentration in the diseased organs.12–14 Therefore, it is necessary to conjugate specific targeting molecules (specific of ligands, such as monoclonal antibodies) at the surface of NPs to achieve active targeting therapy by binding with specific cell surface molecules. Commonly used ligands include specific surfactant agents, folate, transferrin proteins, and polypeptides.15–19
Lipid rafts, mainly consisting of cholesterol and GM1, which disperse in the cell membrane and show lateral drift, are conducive to the interaction between proteins and conformational changes and participate in signal transduction, material transport, and other physiological functions.20,21 It has been demonstrated that, when cholera toxin B subunit (CTB) as a marker specifically bound to ganglioside GM1 in lipid rafts, both in malignant tumor cell and normal cells, more CTB–GM1 membrane complexes were invaginated and they were further transported toward the endoplasmic reticulum and caused the death of cells by reorganization of the actin cytoskeleton.22 It has also been suggested that the amounts of GM1 in the membrane of cancer cells are greater than normal cells, therefore specific raft GM1-binding toxins like CTB could be a novel therapeutic anticancer ligand that targets the organization of lipid rafts.23–26
Heat-labile enterotoxin subunit B (LTB) is a non-catalytic protein from a pentameric subunit of Escherichia coli.27–29 Like CTB, LTB binds specifically to the branched pentasaccharide moiety of lipid-ordered domain resident entity (ganglioside GM1) on the surface of targeting cells.30–33 On the basis of the good compatibility of bovine serum albumin (BSA) with tumor tissue and tumor-targeting specificity of LTB, we designed a novel NP composed of BSA and LTB for targeted delivery of 5-fluorouracil (5-FU) to tumor cells. Systematic cellular studies and in vitro fluorescence imaging observations were performed to confirm the tumor-targeting ability of BSA-LTB NPs.
Materials and methods
Materials
BSA and fluorescein isothiocyanate (FITC) were purchased from Sigma-Aldrich (St Louis, MO, USA), LTB was a gift from National Vaccine and Serum Institute (Beijing, People’s Republic of China), 5-FU was purchased from Nantong Jinghua Pharmaceutical Co, Ltd (Nantong, People’s Republic of China). Other chemicals purchased were of analytical grade and were obtained from Sigma-Aldrich.
Preparation of BSA-LTB NPs
Ten milligrams BSA and 1 mg LTB were dissolved in 0.5 mL acetic acid (0.5%, v/v) to achieve a homogeneous protein solution. BSA-LTB NPs were prepared by quickly dropping 1.0 mL of anhydrous alcohol into 0.5 mL of protein solution at 37°C until opalescence phenomenon appeared. To prepare 5-FU loaded NPs, 1 mg of 5-FU was added into protein solution to obtain drug-loaded NPs prior to the addition of anhydrous alcohol. Ethanol was removed by rotary evaporation. Then, 8% glutaraldehyde in water (0.5 μL per mg of BSA and LTB) was added to induce particle crosslinking. The crosslinking process was performed under stirring of the suspension over a time period of 24 hours. NPs were precipitated and separated from the system by centrifugation (16,000 rpm, 20 minutes), rinsed thoroughly with deionized water, and freeze-dried for further analysis.
Characterization of BSA-LTB NPs
The hydrodynamic diameter, polydispersity, and zeta potential of 5-FU-loaded BSA-LTB NPs were determined by dynamic light scattering using a Brookhaven Zetasizer (Brookhaven Instruments Corporation, Holtsville, NY, USA). Morphology of BSA-LTB NPs was observed using a transmission electron microscope (JEM-1200EX; JEOL, Tokyo, Japan).
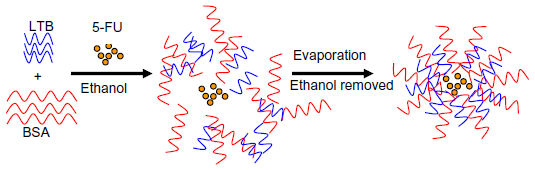 | Figure 1 Schematic formation of 5-FU-loaded BSA-LTB nanoparticles. |
Identification of LTB with immunofluorescence
BSA NPs blended or unblended with LTB were extensively washed and incubated with anti-LTB (rabbit source, Santa Cruz Biotechnology, Inc., Dallas, TX, USA) for 1 hour. These NPs were washed three times to preclude the unbound antibody and then incubated with the corresponding FITC-conjugated secondary antibody for 30 minutes. The NPs were collected by centrifuging at 12,000 rpm for 10 minutes, dropped on slides, and observed under a fluorescent microscope (DMI4000B; Leica Microsystems, Wetzlar, Germany). As shown in Figure 2, obvious green fluorescence was observed in BSA-LTB NPs, while no obvious fluorescence was observed in BSA NPs. These data indicate that LTB was successfully blended with BSA and formed NP matrix.
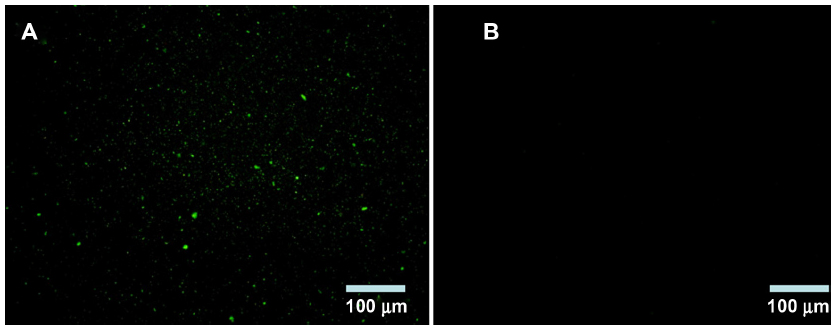 | Figure 2 Fluorescence microscope images of identification of LTB with immunofluorescence. |
5-FU release from BSA-LTB NPs
The prepared drug-loaded NPs without dialysis were centrifuged at 15,000 rpm for 15 minutes. The supernatant was collected and filtered through a 0.45 μm Millipore filter (EMD Millipore, Billerica, MA, USA) and the absorbance of the filtrate at 256 nm was determined using an ultraviolet-visible spectrophotometer (model 1601; Shimadzu, Kyoto, Japan). Encapsulation efficiency (EE%) was calculated using the equation below:

where Wtotal is the amount of initial added drug and Wfree is the amount of drug that remained in the supernatant.
The appropriate amount of freeze-dried 5-FU-loaded BSA-LTB NPs encapsulated in a dialysis bag (molecular weight [Mw] 3,000–4,000) was immersed in 50 mL of phosphate-buffered saline (PBS) and shaken constantly at 100 rpm at 37°C. At specified time points (0, 0.5, 1, 2, 4, 6, 8, 12, 24, and 48 hours), 5 mL of solution outside the dialysis bag was collected for ultraviolet-visible spectrophotometer analysis and replaced with the same volume of PBS. The cumulative amount of released 5-FU was calculated and the accumulative percentage of drug was plotted against time.
Cell culture
Human hepatocellular carcinoma cell lines SMMC-7721 were purchased from the Institute of Biochemistry and Cell Biology of Chinese Academy of Sciences (Shanghai, People’s Republic of China) and cultured in Dulbecco’s Modified Eagle’s Medium (HyClone™; Thermo Fisher Scientific, Waltham, MA, USA) containing 10% fetal bovine serum (HyClone) and penicillin/streptomycin (100 units/mL and 100 μg/mL, respectively) (Gibco®; Life Technologies/Invitrogen, Carlsbad, CA, USA). All cells were maintained at 37°C in a humidified atmosphere of 5% CO2, 95% air.
In vitro cellular uptake
The cellular uptake of FITC-labeled NPs (FITC-BSA NPs and FITC-BSA-LTB NPs) was observed by inverted fluorescent microscopy (LSM510; Carl Zeiss Meditec AG, Jena, Germany) and quantified by microplate reader (Synery-2; BioTek, Winooski, VT, USA). Briefly, confluent SMMC-7721 cells were harvested and suspended in serum-free medium at a density of 5×104/mL. The cells were dispensed into a 96-well microplate and incubated with FITC-labeled NPs. After 6 hours, cells were washed three times to remove the free NPs. The internalization of FITC-labeled NPs into cells was observed using fluorescent microscopy and the intensity of fluorescence in cells was quantified using a microplate reader. The fluorescence from FITC is excited at 485 nm and emitted at 528 nm. The cellular uptake ability for FITC-BSA NPs was analyzed at the same time as controls.
In vitro cell viability assay
A 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay was performed to investigate cell viability status. SMMC-7721 cells at a density of 5×104/mL were seeded into the 96-well plate (100 μL each well) and incubated for 24 hours at 37°C under 5% CO2 and 95% O2. The medium was replaced by serum-free medium in the presence of either free 5-FU, 5-FU-loaded BSA NPs, or 5-FU-loaded BSA-LTB NPs, and incubated for 72 hours. The medium was subsequently discarded and replaced with 200 μL of serum-free medium containing MTT (0.2 mg/mL; Sigma-Aldrich) and incubated for 6 hours at 37°C at 5% CO2 and 95% O2. Then, the supernatant was aspirated, 150 μL of dimethyl sulfoxide was added to each well, and the absorbance was measured at 490 nm.
Cell apoptosis study
SMMC-7721 cells were incubated with either free 5-FU, 5-FU-loaded BSA NPs, or 5-FU-loaded BSA-LTB NPs for 48 hours. After treatment, cells were collected and suspended in Nicoletti buffer (Beijing 4A Biotech Co., Ltd. Beijing, People’s Republic of China) containing propidium iodide (PI) and FITC-labeled Annexin V (AV-FITC). DNA content was determined on a fluorescence-activated cell sorter (FACSCalibur™; BD, Franklin Lakes, NJ, USA). The cells undergoing apoptosis were represented by the ratio of AV-positive cells and PI-positive cells.
Results and discussion
Physicochemical properties of NPs
The particle size and polydispersity of 5-FU-loaded BSA-LTB NPs in deionized water were determined by dynamic light scattering. The mean hydrodynamic diameter was 254±19 nm and polydispersity was 0.15±0.06 for BSA-LTB NPs. The mean zeta potential of NPs was –19.95±0.94 mV, revealing a negative surface of the NPs. Figure 3 shows that the 5-FU-loaded BSA-LTB NPs were also monodispersed spheres and the 5-FU encapsulation efficiency was 80.1%.
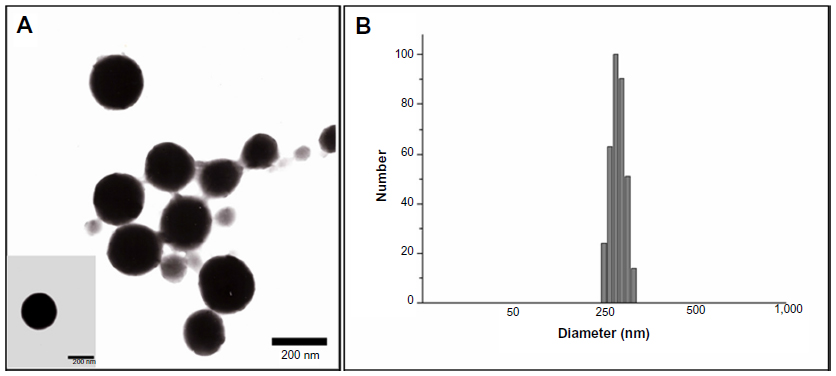 | Figure 3 Morphological shapes (A) and particle sizes (B) of 5-FU-loaded BSA-LTB nanoparticles. |
In vitro drug release study
The in vitro drug release profiles of BSA-LTB NPs in media with different pH were compared and results are shown in Figure 4. The in vitro release of 5-FU from BSA-LTB NPs was biphasic, ie, an initial burst release was followed by a slower constant release. The total accumulative amount of drug released from NPs within 48 hours in media of different pH was less than 40%. This is likely because 5-FU is a hydrophobic drug that is prone to separate and precipitate inside the NPs, thereby reducing the rate of drug release. When 5-FU-loaded BSA-LTB NPs were incubated with medium at pH 7.4, rapid release was observed between 0 and 4 hours, which was followed by a cumulative release of 17.9%, due to the diffusion of surface 5-FU into the solution. A smooth, slow-release process occurred between 4 and 48 hours. The release ratio during the first 48 hours accounted for over 38.7% of the total drug, and the surplus was released over a longer incubation time.34,35 The release rate of 5-FU from the core of NPs mainly depended on the solubility of the drug in the release medium and diffusion rate. Moreover, the increase of release rate from 33.2% in pH 5.6 to 38.7% in pH 7.4 over 48 hours is attributed to higher solubility of 5-FU in alkaline solution than in acidic solution.
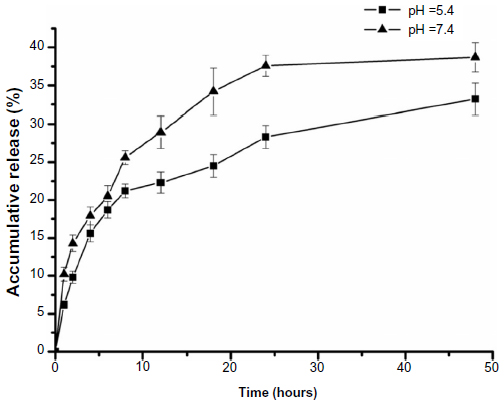 | Figure 4 In vitro release profile of BSA-LTB nanoparticles in phosphate-buffered saline (pH 5.4 and pH 7.4 at 37°C) for 48 hours. |
In vitro cellular uptake
The internalization of FITC-labeled NPs into SMMC-7721 cells was observed using fluorescent microscopy and quantified by the microplate reader. We found that FITC-labeled BSA-LTB NPs significantly enhanced the internalization of NPs into cells compared with FITC-labeled BSA NPs. As shown in Figure 5A, both FITC-labeled BSA and BSA-LTB NPs accumulated in the cytoplasm when incubated with SMMC-7721 for 6 hours. However, cells treated with FITC-labeled BSA-LTB NPs showed an increased fluorescent intensity compared with cells treated with BSA NPs, suggesting that FITC-labeled BSA-LTB NPs may be internalized more efficiently than BSA NPs. To further study the endocytosis mechanism, the internalization of NPs into cells was evaluated by determining the fluorescent intensity difference between the initially added NPs and the NPs internalized in cells. Relative fluorescent ratio (RFR%) was calculated using the equation below:
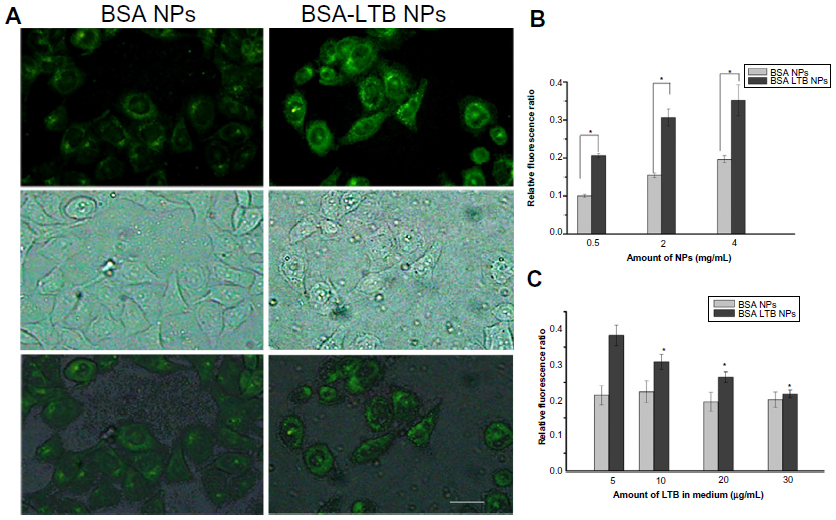 | Figure 5 In vitro uptake ability of NPs. |
where FItotal is the fluorescent intensity of the initially added FITC–NPs and FIinternalized is the fluorescent intensity of FITC–NPs internalized in cells.
Fluorescence spectrum analysis (Figure 5B) revealed that both NPs showed concentration-dependent properties on the intracellular uptake. FITC-labeled BSA-LTB NPs showed higher intracellular FITC concentration than FITC-labeled BSA NPs, indicating that the addition of LTB could significantly improve the tumor cell uptakes of NPs. To sum up, during initial contact with cells, LTB in NPs might bind to GM1 in lipid raft to form GM1–lipid membrane complexes localized at the outer leaflet of the cell membrane, meaning that NPs are internalized into cells rapidly. To investigate the competing effects of LTB on the cellular internalization of BSA-LTB NPs, cells were incubated with FITC-labeled BSA NPs and BSA-LTB NPs, respectively, in media containing different concentrations of free LTB for 6 hours at 37°C. An LTB competing study on the intracellular uptake also further proved that active transportation of BSA-LTB NPs into cells was inhibited with increasing addition of free LTB in medium (Figure 5C). In contrast, the addition of free LTB in medium led to no obvious improving effects on the internalization of BSA NPs.
Cellular viability study
The cytotoxic effect of free 5-FU, 5-FU-BSA NPs, and 5-FU-loaded BSA-LTB NPs against SMMC-7721 cells was estimated in vitro by MTT assay. Cells were treated with various concentrations of 5-FU ranging from 3.75–30 μg/mL. As shown in Figure 6, treatment of SMMC-7721 cells with 5-FU-loaded BSA-LTB NPs caused a significant decrease in the cell viability at 24, 48, and 72 hours compared with that of 5-FU-loaded BSA NPs. The 50% of maximal inhibitory concentration (IC50) values in cells treated with 5-FU-loaded BSA-LTB NPs were 23 μg/mL at 48 hours and 15.3 μg/mL at 72 hours compared with 30 μg/mL at 48 hours and 21.7 μg/mL at 72 hours in cells treated with 5-FU-loaded BSA NPs. Neither 5-FU-loaded BSA-LTB nor BSA NPs had any obvious cytotoxic effects on SMMC-7721 cells at 24 hours. By contrast, the IC50 values of free 5-FU were 18.5 μg/mL at 48 hours and 14.7 μg/mL at 72 hours. 5-FU also had no obvious cytotoxic effects on SMMC-7221 cells at 24 hours.
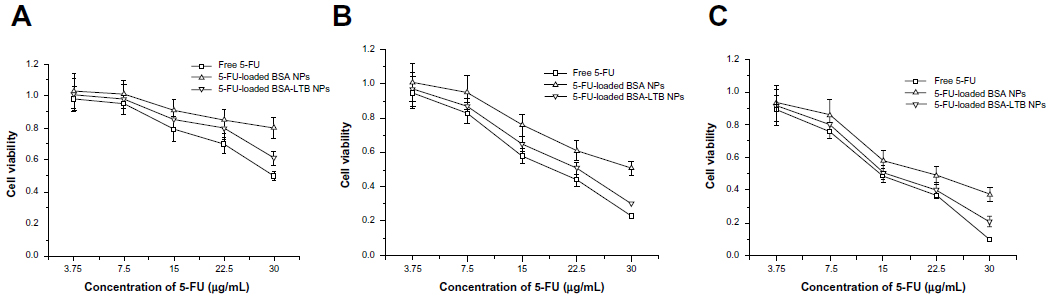 | Figure 6 Viability of cells after incubation with either free 5-FU, 5-FU-loaded BSA NPs, or 5-FU-loaded BSA-LTB NPs, at various 5-FU concentrations (3.75–30 μg/mL). |
Cell apoptosis and necrosis
To further analyze the apoptosis of SMMC-7721 cells induced by free 5-FU, 5-FU-loaded BSA NPs, and 5-FU-loaded BSA-LTB NPs, Annexin V-FITC/PI staining assay was performed and the apoptotic and necrotic cells were quantified by flow cytometry. The percentages of early apoptotic, late apoptotic, necrotic, and live cells are shown in Figure 7. Flow cytometry analysis revealed that the ratio of AV-positive and PI-positive cells treated with 5-FU-BSA-LTB NPs was 36.1%, compared with that of free 5-FU-treated cells (46.9%). It is worth noting that the ratio of AV-positive and PI-positive fractions in cells treated with 5-FU-loaded BSA-LTB NPs was markedly higher than that in cells treated with 5-FU-BSA NPs (18.1%). The results of flow cytometry were consistent with the MTT results.
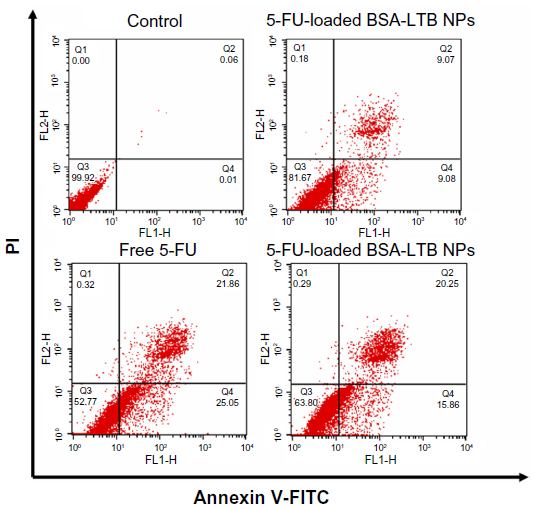 | Figure 7 Cell apoptosis determined by Annexin V-FITC staining. |
Conclusion
LTB as a targeting group was successfully blended with BSA to prepare the biocompatible NPs for cancer therapy. BSA-LTB NPs showed good monodispersity, negative charge, and homogeneous particle size. The encapsulating efficiency of 5-FU and the release pattern were investigated in vitro. MTT assay showed that BSA-LTB NPs exhibited higher cytotoxic activity than non-targeted ones (BSA NPs) in SMMC-7721 cells. Fluorescent imaging results proved that, compared with BSA NPs, BSA-LTB NPs could greatly enhance cellular uptake. These results suggest that BSA-LTB NPs may be a promising active tumor-targeting carrier candidate via LTB mediation.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (no 81172048), the Natural Science Foundation of Liaoning Province (no 2013022035), and a grant from Liaoning Medical University (no XZJJ20130104-05). We thank Professor Yunhong Jia and Dr Bo Zhang for critical reading of the manuscript and kind corrections.
Disclosure
The authors report no conflicts of interest in this work.
References
Liotta LA, Steeg PS, Stetler-Stevenson WG. Cancer metastasis and angiogenesis: an imbalance of positive and negative regulation. Cell. 1991;64(2):327–336. | |
Jensen AB, Hansen O, Jørgensen K, Bastholt L. Influence of late side-effects upon daily life after radiotherapy for laryngeal and pharyngeal cancer. Acta Oncol. 1994;33(5):487–491. | |
Marijnen CA, Kapiteijn E, van de Velde CJ, et al; Cooperative Investigators of the Dutch Colorectal Cancer Group. Acute side effects and complications after short-term preoperative radiotherapy combined with total mesorectal excision in primary rectal cancer: report of a multicenter randomized trial. J Clin Oncol. 2002;20(3):817–825. | |
Zachariah B, Balducci L, Venkattaramanabalaji GV, Casey L, Greenberg HM, DelRegato JA. Radiotherapy for cancer patients aged 80 and older: a study of effectiveness and side effects. Int J Radiat Oncol Biol Phys. 1997;39(5):1125–1129. | |
Chen H, Yang W, Chen H, et al. Surface modification of mitoxantrone-loaded PLGA nanospheres with chitosan. Colloids Surf B Biointerfaces. 2009;73(2):212–218. | |
Das M, Mohanty C, Sahoo SK. Ligand-based targeted therapy for cancer tissue. Expert Opin Drug Deliv. 2009;6(3):285–304. | |
Acharya S, Sahoo SK. PLGA nanoparticles containing various anticancer agents and tumour delivery by EPR effect. Adv Drug Deliv Rev. 2011;63(3):170–183. | |
Ferrari M. Cancer nanotechnology: opportunities and challenges. Nat Rev Cancer. 2005;5(3):161–171. | |
Farokhzad OC, Langer R. Nanomedicine: developing smarter therapeutic and diagnostic modalities. Adv Drug Deliv Rev. 2006;58(14):1456–1459. | |
Allen TM, Cullis PR. Drug delivery systems: entering the mainstream. Science. 2004;303(5665):1818–1822. | |
Peer D, Karp JM, Hong S, Farokhzad OC, Margalit R, Langer R. Nanocarriers as an emerging platform for cancer therapy. Nat Nanotechnol. 2007;2(12):751–760. | |
Mohapatra S, Mallick SK, Maiti TK, Ghosh SK, Pramanik P. Synthesis of highly stable folic acid conjugated magnetite nanoparticles for targeting cancer cells. Nanotechnology. 2007;18(38):385102. | |
Kim J, Lee JE, Lee SH, et al. Designed fabrication of a multifunctional polymer nanomedical platform for simultaneous cancer-targeted imaging and magnetically guided drug delivery. Adv Mater. 2008;20(3):478–483. | |
Hong G, Yuan R, Liang B, Shen J, Yang X, Shuai X. Folate-functionalized polymeric micelle as hepatic carcinoma-targeted, MRI-ultrasensitive delivery system of antitumor drugs. Biomed Microdevices. 2008;10(5):693–700. | |
Jain RK, Stylianopoulos T. Delivering nanomedicine to solid tumors. Nat Rev Clin Oncol. 2010;7(11):653–664. | |
Bae YH. Drug targeting and tumor heterogeneity. J Control Release. 2009;133(1):2–3. | |
Chen H, Pazicni S, Krett NL, et al. Coencapsulation of arsenic- and platinum-based drugs for targeted cancer treatment. Angew Chem Int Ed Engl. 2009;48(49):9295–9299. | |
Neff CP, Zhou J, Remling L, et al. An aptamer-siRNA chimera suppresses HIV-1 viral loads and protects from helper CD4+ T cell decline in humanized mice. Sci Transl Med. 2011;3(66):66ra6. | |
Kim Y, Pourgholami MH, Morris DL, Stenzel MH. An optimized RGD-decorated micellar drug delivery system for albendazole for the treatment of ovarian cancer: from RAFT polymer synthesis to cellular uptake. Macromol Biosci. 2011;11(2):219–233. | |
Li YC, Park MJ, Ye SK, Kim CW, Kim YN. Elevated levels of cholesterol-rich lipid rafts in cancer cells are correlated with apoptosis sensitivity induced by cholesterol-depleting agents. Am J Pathol. 2006; 168(4):1107–1118. | |
Johannes L, Decaudin D. Protein toxins: intracellular trafficking for targeted therapy. Gene Ther. 2005;12(18):1360–1368. | |
Kabaso D, Lokar M, Kralj-Igli V, Verani P, Igli A. Temperature and cholera toxin B are factors that influence formation of membrane nanotubes in RT4 and T24 urothelial cancer cell lines. Int J Nanomedicine. 2011;6:495–509. | |
Zhuang L, Lin J, Lu ML, Solomon KR, Freeman MR. Cholesterol-rich lipid rafts mediate akt-regulated survival in prostate cancer cells. Cancer Res. 2002;62(8):2227–2231. | |
Sarnataro D, Grimaldi C, Pisanti S, et al. Plasma membrane and lysosomal localization of CB1 cannabinoid receptor are dependent on lipid rafts and regulated by anandamide in human breast cancer cells. FEBS Lett. 2005;579(28):6343–6349. | |
Merritt EA, Sarfaty S, van den Akker F, L’Hoir C, Martial JA, Hol WG. Crystal structure of cholera toxin B-pentamer bound to receptor GM1 pentasaccharide. Protein Sci. 1994;3(2):166–175. | |
Wernick NL, Chinnapen DJ, Cho JA, Lencer WI. Cholera toxin: an intracellular journey into the cytosol by way of the endoplasmic reticulum. Toxins (Basel). 2010;2(3):310–325. | |
Hirst TR, Hardy SJ, Randall LL. Assembly in vivo of enterotoxin from Escherichia coli: formation of the B subunit oligomer. J Bacteriol. 1983;153(1):21–26. | |
Hofstra H, Witholt B. Heat-labile enterotoxin in Escherichia coli. Kinetics of association of subunits into periplasmic holotoxin. J Biol Chem. 1985;260(29):16037–16044. | |
Hirst TR, Sanchez J, Kaper JB, Hardy SJ, Holmgren J. Mechanism of toxin secretion by Vibrio cholerae investigated in strains harboring plasmids that encode heat-labile enterotoxins of Escherichia coli. Proc Natl Acad Sci U S A. 1984;81(24):7752–7756. | |
Balanzino LE, Barra JL, Galván EM, Roth GA, Monferran CG. Interaction of cholera toxin and Escherichia coli heat-labile enterotoxin with glycoconjugates from rabbit intestinal brush border membranes: relationship with ABH blood group determinants. Mol Cell Biochem. 1999;194(1–2):53–62. | |
Galván EM, Roth GA, Monferran CG. Participation of ABH glycoconjugates in the secretory response to Escherichia coli heat-labile toxin in rabbit intestine. J Infect Dis. 1999;180(2):419–425. | |
Galván EM, Roth GA, Monferran CG. Functional interaction of Escherichia coli heat-labile enterotoxin with blood group A-active glycoconjugates from differentiated HT29 cells. FEBS J. 2006;273(15):3444–3453. | |
Teneberg S, Hirst TR, Angström J, Karlsson KA. Comparison of the glycolipid-binding specificities of cholera toxin and porcine Escherichia coli heat-labile enterotoxin: identification of a receptor-active non-ganglioside glycolipid for the heat-labile toxin in infant rabbit small intestine. Glycoconj J. 1994;11(6):533–540. | |
Yang L, Cui F, Cun D, Tao A, Shi K, Lin W. Preparation, characterization and biodistribution of the lactone form of 10-hydroxycamptothecin (HCPT)-loaded bovine serum albumin (BSA) nanoparticles. Int J Pharm. 2007;340(1):163–172. | |
Zu Y, Zhang Y, Zhao X, Zhang Q, Liu Y, Jiang R. Optimization of the preparation process of vinblastine sulfate (VBLS)-loaded folate-conjugated bovine serum albumin (BSA) nanoparticles for tumor-targeted drug delivery using response surface methodology (RSM). Int J Nanomedicine. 2009;4:321–323. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.