")
Back to Journals » International Journal of Nanomedicine » Volume 13
Preparation, characterization, in vitro and in vivo anti-tumor effect of thalidomide nanoparticles on lung cancer
Authors Chen L, Ni X, Zhang H, Wu M, Liu J, Xu S , Yang L, Fu S , Wu J
Received 8 December 2017
Accepted for publication 8 March 2018
Published 23 April 2018 Volume 2018:13 Pages 2463—2476
DOI https://doi.org/10.2147/IJN.S159327
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Linlin Sun
Long Xia Chen,* Xiao Ling Ni,* Heng Zhang, Min Wu, Jing Liu, Shan Xu, Ling Lin Yang, Shao Zhi Fu, Jingbo Wu
Department of Oncology, the Affiliated Hospital of Southwest Medical University, Luzhou, People’s Republic of China
*These authors contributed equally to this work
Introduction: Thalidomide (THA) is an angiogenesis inhibitor and an efficient inhibitor of the tumor necrosis factor-α (TNF-α). However, the clinical application of THA has been limited due to hydrophobicity of the compound.
Materials and methods: To increase the water solubility of THA and in order to evaluate the anticancer abilities of this material on human lung carcinoma, methoxy poly(ethylene glycol)-poly(ε-caprolactone) nanoparticles loaded with THA (THA-NPs) were prepared. The synthesis of THA-NPs was carried out via a dialysis method with relative satisfactory encapsulation efficiency, loading capacity, size distribution, and zeta potential.
Results: A cytotoxicity assay demonstrated that THA-NPs inhibited the growth of cells in a dose-dependent manner. The evaluation of anti-tumor activity in vivo showed that THA-NPs could inhibit tumor growth and prolong the survival rate of tumor-bearing mice. Immunohistochemical analysis indicated that THA-NPs inhibited cell proliferation (Ki-67 positive rate, 32.8%±4.2%, P<0.01), and resulted in a decreased rate of the tumor tissue microvessel density (3.87%±0.77%, P<0.01), VEGF (26.67%±4.02%, P<0.01), and TNF-α (75.21±6.85 ng/mL, P<0.01).
Conclusion: In general, the drug delivery system reported herein may shed light on future targeted therapy in lung cancer treatment.
Keywords: thalidomide, nanoparticles, MPEG-PCL, lung cancer
Introduction
Lung cancer is the leading cause of cancer-related deaths in both men and women.1,2 About 85% of all lung cancer types are non-small-cell lung cancer, which mainly comprises adenocarcinoma (40%). Generally, the treatments of lung adenocarcinoma involve surgical resection, chemotherapy, radiotherapy, targeted therapy, and comprehensive tumor treatment. However, patients are often first diagnosed with an advanced disease state with primary resistance to anticancer drugs. Thus, it is of great importance to explore more efficient therapeutic strategies for the treatment of lung cancer. In general, tumor growth and systemic metastasis are greatly dependent on angiogenesis. In order to grow more than 1 mm3 in size and cause metastasis, a tumor must establish a network of blood vessels enabling an adequate supply of oxygen and nutrients. Hence, anti-angiogenesis has been regarded to be an appealing anticancer target for clinical therapy.3,4
Thalidomide (THA), an angiogenesis inhibitor, was first synthesized as a non-barbiturate sedative hypnotic drug by the German pharmaceutical company Chemie Grünenthal in the late 1950s. It was mainly administered to pregnant women for nausea relief and to alleviate morning sickness. However, the drug was withdrawn from the market in 1961 when severe teratogenic side effects were discovered. After removal of THA from the market, the compound has been identified to exhibit effects related to anti-angiogenesis, anti-inflammation, apoptosis promotion, and inhibition of cell proliferation.5–8 So far, worldwide studies on THA intervention in malignant tumors, including hematological malignancy and solid tumors, such as lung cancer, bladder cancer, pancreatic carcinoma, and hepatocellular carcinoma, have been reported.9–14 However, due to several physical properties of THA, including insolubility in water, no parenteral preparation is available and the overall applicability vastly restricted.15
In recent years, nanotechnology has been developed as an important scientific field that now attracts a significant amount of attention in drug delivery and cancer therapy. It represents an important technique to develop aqueous formulations of hydrophobic drugs.16,17 Biodegradable polymeric nanoparticles are often used to achieve a controlled release of drugs in advanced anticancer drug delivery systems.16,18,19 Therefore, in an effort to overcome the disadvantages of THA, a variety of attempts have been reported in the literature to increase the solubility in aqueous media with increased bioavailability. In this field, a temperature sensitive copolymeric nanoparticle formulation using poly (N-isopropylacrylamide-vinyl pyrrolidone-acrylic acid) has been developed.20 Moreover, nano-flakes of THA produced by a supercritical anti-solvent method and a supercritical anti-solvent with enhanced mass transfer have been reported by Jin et al, and have further been demonstrated to successfully enhance both dissolution rate and bioavailability of THA.21 Meanwhile, Suksuwan et al have synthesized nanoparticles of a molecularly imprinted polymer (MIP) enantioselective receptor for the (R)-enantiomer of THA. It was found that MIP nanoparticles of THA have the potential to attack multidrug-resistant cells.22 However, all these studies generally lacked a detailed evaluation of the anticancer effect of this nanoparticle loaded with THA (THA-NP) construct in vivo.
Furthermore, to date, no reports can be found in the literature evaluating THA/methoxy poly(ethylene glycol)-poly(ε-caprolactone) (MPEG-PCL) polymeric nanoparticle therapeutics. MPEG-PCL represents an amphiphilic diblock copolymer exhibiting distinct hydrophilic (PEG chains) and hydrophobic (PCL chains) segments. In aqueous media, the copolymer may form core-shell nanostructures with an inner hydrophobic core and outer hydrophilic shell due to high solubility.23–25 Encapsulation of hydrophobic drugs into polymeric nanoparticles may present a hydrophilic stabilizing interface that can prolong the circulation time in vivo and may further enhance the cellular uptake. More importantly, due to enhanced permeability and retention (EPR) effects, polymeric nanoparticles may improve the anti-tumor effects by passively targeting tumors.26
In this study, we decided to use THA encapsulated into the MPEG-PCL nanoparticles to form a stable water-based formulation for potential clinical applications. Furthermore, the anticancer effects of THA-NPs were evaluated both in vitro and in vivo and the THA-NPs were characterized with regards to size and surface morphology. The in vitro release profile of THA-NPs was also evaluated. Moreover, cellular uptake and MTT assays of THA-NPs were performed using A549 cells. Finally, 18F-FDG PET/CT, flow cytometry, ELISA, and immunohistochemistry were performed in A549 tumor-bearing nude mice to evaluate the anti-tumor activities in vivo.
Materials and methods
Materials
THA (≥98%) was purchased from Meilun Biotech Co. Ltd (Dalian, People’s Republic of China). ε-Caprolactone (ε-CL) was purchased from Alfa Aesar (Ward Hill, MA, USA). MPEG (Mn =2,000), stannous octoate [Sn(Oct)2], and MTT were purchased from Sigma-Aldrich Co. (St Louis, MO, USA). Dimethyl sulfoxide (DMSO), methyl alcohol, and acetonitrile (HPLC grade) were supplied by Kelong Co. Ltd. (Chengdu, People’s Republic of China). A TNF-α ELISA kit was purchased from Beijing Cheng Lin Biological Technology Co. Ltd. (Beijing, People’s Republic of China). Ki-67, CD31, and VEGF polyclonal antibody were purchased from Bioworld Technology Co. Ltd. (Nanjing, People’s Republic of China).
Cell lines and animals
Human lung carcinoma (A549) cells were obtained from the Experimental Medicine Center at the Affiliated Hospital of Southwest Medical University (Luzhou, People’s Republic of China), as approved by the Ethics Committee of Southwest Medical University. A549 cells were cultured in RPMI medium (1640, Gibco, Waltham, MA, USA) supplemented with 10% fetal bovine serum (Gibco), 100 μg/mL of streptomycin sulfate, and 100 IU/mL of penicillin G sodium at 37°C under 5% CO2 atmosphere and high humidity.
BALB/C mice (female, nu/nu, 4 weeks old, weighing 14–18 g) were purchased from Chongqing Tengxin Biotechnology Co. Ltd. (Chongqing, People’s Republic of China), housed at a controlled temperature of 20°C–22°C, with a relative humidity of 50%–60% and 12 h light/dark cycles. The mice were provided with a standard diet of laboratory chow and tap water ad libitum. Animal use was in accordance with the animal care guidelines, and the protocol was approved by the Institutional Animal Care and Treatment Committee of Southwest Medical University (Luzhou, People’s Republic of China).
Preparation and characterization of THA-loaded NPs
MPEG-PCL (MEP, Mn =4 kDa) used in this study was synthesized by ring-opening polymerization of ε-CL on MPEG using Sn(Oct)2 as a catalyst according to a report published previously.27 The THA-NPs were prepared using a dialysis method. MPEG-PCL copolymer (MPEG2000-PCL2000) and THA at various dosages of 6 wt%, 8 wt%, or 10 wt% were used. Briefly, for 10 wt% of THA as an example, 90 mg MPEG-PCL copolymer (MPEG2000-PCL2000) and 10 mg THA were dissolved in 10 mL DMSO to form an organic phase. The organic phase was then added dropwise to 8 mL of distilled water under magnetic stirring. To allow for the formation of nanoparticles and to remove unencapsulated THA and organic solvents, the resulting mixture was then placed in a dialysis bag (MWCO: 3.5 KDa, Spectrum Laboratories, Los Angles, CA, USA) for dialysis against distilled water with stirring at 800 r/min for 48 h at room temperature. The dialysate was changed at certain time intervals (1, 2, 4, 6, 12, 24, and 36 h) and the resulting dispersions in the dialysis bag were centrifuged at 4,000 r/min for 10 min to eliminate unloaded THA and aggregated particles. The supernatant was filtered through a 450 and a 220 nm filter membrane and further lyophilized to obtain dried THA-NP powder that was stored at 4°C until further use.
The average particle size, polydispersity, and zeta potential of the THA-NPs were measured by dynamic light scattering (DLS; NanoBrook 90Plus Zeta, Brookhaven, NY, USA) at 25°C. All results were the mean of three test run repetitions.
The morphological characteristics of the THA-NPs were examined by transmission electron microscopy (Tecnai G2 F20, Hillsboro, OR, USA). THA-NPs were diluted with distilled water and placed on a copper grid covered with nitrocellulose. The THA-NPs were then negatively stained with phosphotungstic acid and dried at room temperature.
Encapsulation efficiency and drug loading
A high-performance liquid chromatography (HPLC, Agilent 1260; Agilent Technologies, Santa Clara, CA, USA) instrument equipped with a reverse phase C18 column (4.6×150 mm, 5 μm particle size) was used to measure the drug loading (DL) and encapsulation efficiency (EE). A standard curve was obtained, and the correlation coefficient of R2 was 0.9992. The THA-NPs were then dissolved in the mobile phase. The mobile phase consisted of acetonitrile/water/phosphoric acid (15/85/0.1, v/v/v), and HPLC was performed at a flow rate of 1 mL/min. The detection wavelength was 237 nm, and the column temperature was maintained at 25°C, with an injection volume of 20 μL. The DL and EE were calculated according to the following equations:
|
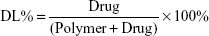
|
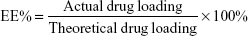
In vitro drug release studies
An in vitro drug release profile of THA from THA-NPs was carried out via a modified dialysis method. In total, 1 mL of THA-NPs (theoretical DL rate =10%) in distilled water (equal to 1 mg of THA) and 1 mL standard substance solution of THA at a concentration of 1 mg/mL were placed in a dialysis bag (MWCO: 3.5 kDa), which was immersed in 40 mL PBS (0.01 M, pH 7.4) containing Tween 80 (0.5%, wt%) at 37°C±0.5°C upon constant and gentle stirring (100 rpm). At fixed time intervals (0, 2, 4, 6, 8, 12, 24, 48, 72, 96, 120, 144, and 168 h ), 2 mL of the dialysis medium was removed and centrifuged at 12,000 r/min for 15 min to collect the supernatant. The residue was then stored at −20°C. The dialysis medium was substituted with the same volume of fresh medium and drug release was analyzed by HPLC. Each experiment was carried out in triplicates.
In vitro cytotoxicity studies
The growth inhibition of A549 cells by THA and THA-NPs was evaluated using an MTT assay. The cells in 100 μL of RPMI medium supplemented with 10% fetal bovine serum, 100 μg/mL streptomycin, sulfate and 100 IU/mL penicillin G sodium were seeded in 96-well plates at a density of 1×104 cells per well for 24 h. Subsequently, fresh medium, empty MPEG-PCL (0–1,000 μg/mL), free THA, and THA-NPs at a concentration range of 5–160 μg/mL were individually added. The cells were further cultured for 24 h. At determined time points, 20 μL of MTT (5 mg/mL) was added and the cells were then incubated for an additional 4 h. Then, the medium was removed; the formazan precipitate was dissolved in DMSO (150 μL) and the cell viability was analyzed and recorded at 490 nm using a microplate reader (iMark, MA, USA). Each treatment condition was evaluated in triplicates in three independent experiments.
Cellular uptake
In this study, a fluorescent probe Coumarin-6 (C6) was encapsulated in MPEG-PCL nanoparticles to assess the cellular uptake of MPEG-PCL NPs by A549 cells. Briefly, dichloromethane solution of C6 and DMSO solution of MPEG-PCL were mixed at a molar ratio of 4:1. Then, the mixture was added dropwise to distilled water under magnetic stirring and the resulting solution was lyophilized to obtain a dried powder that was subsequently stored at 4°C.
A549 cells were seeded in 6-well plates at a density of 105 cells per well for 24 h. Then, 10 μL of normal saline, C6-loaded MPEG-PCL NPs, the mixtures of MPEG-PCL and C6 were added and the cells were further cultured for 2 h. After washing with PBS (0.01 M, pH 7.4) for a total of three times, the cells were analyzed by fluorescence microscopy (Olympus, Tokyo, Japan).
Tumor growth inhibition study
A549 xenografts were established by subcutaneous injection of a suspension of A549 cells (106 cells in 0.1 mL PBS) into the right flank region of nude mice (weight: 18–20 g). When the tumors reached a diameter of 100–200 mm3, the mice were randomly divided into 4 groups of 12 animals each: 1) NS group, 2) MPEG-PCL group, 3) THA group, and 4) THA-NPs group. The first group was treated with normal saline. The second group was treated with empty MPEG-PCL nanoparticles. The third group was treated with free THA (20 mg/kg). The fourth group was treated with THA-NPs. The THA and THA-NPs groups were administered an equal amount of THA. The mice received an intravenous injection via tail vein injection every other day. The tumor volume was measured every other day. After treatment for a total of five times, half of the animals in each group were sacrificed and the tumor tissue was extracted. The remaining mice were used to observe tumor growth and survival time. The tumor volume was calculated using the following formula: V (mm3)=[ab2]/2, where a represents the length and b is the width of the tumor.
Micro 18F-FDG PET/CT imaging
Micro 18F-FDG PET/CT (Siemens, Munich, Germany) imaging was carried out to study the early response of tumor tissue 24 h after the last treatment. Briefly, the animals in the NS, THA, and THA-NPs groups were fasted 12 h prior to scanning, intraperitoneally anesthetized with 1% pentobarbital (5 mL/kg), and intravenously injected via tail vein injection with 200–300 μCi FDG. Micro 18F-FDG PET/CT scans and image analyses were performed 1 h after injection of a radiolabeled tracer. The parameters used were as follows: 80 kV, 500 μA, slice thickness of 1.5 mm, and 10 min of emission scan per bed position. The image plane with the largest tumor appearance on the PET/CT fusion image was selected for data collection. An irregular region of interest covering the entire tumor dimension was drawn manually to obtain the maximum standardized uptake value (SUVmax). All analyses were performed by two experienced nuclear medicine physicians.
Cell cycle and apoptosis analysis
Tumor tissue specimens were extracted from nude mice, cut into pieces, and immediately mixed with 1 mL of 0.25% trypsin/EDTA (1:1, v/v). The mixture was incubated at 37°C for 37 min and stirred once every 10 min. At a fixed time point of 37 min, the medium containing serum was added to terminate digestion. Then, the mixture was washed twice with PBS (0.01 M, pH 7.4) after centrifugation (1,000 rpm, 1 min). The precipitate was then resuspended in 4 mL of PBS and the suspension was filtered using a 70 μm nylon net to harvest tumor cells. A portion of the cells was resuspended in Annexin-V binding buffer and incubated with 5 μL Annexin V-FITC and 5 μL PI at lucifuge for 15 min, and then analyzed by flow cytometry (BD FACSVerse, BD Biosciences, San Jose, CA, USA). The other cells were analyzed by flow cytometry following the kit instructions of the BD cycle test.
Enzyme-linked immunosorbent assay for TNF-α
For this procedure, 1–2 mL of blood was collected, allowed to clot, and centrifuged at 12,000 rpm for 10 min. The serum was obtained and evaluated using a TNF-α ELISA kit (Beijing Cheng Lin Biological Technology Co. Ltd) to determine the concentration of the serum TNF-α. The procedure was carried out according to the manufacturer’s instructions, and the concentrations of unknown samples were determined by comparing the optical density of samples to the standard curve.
Immunohistochemical assessment of Ki-67, CD31, and VEGF
The harvested tumor tissues were fixed in 10% neutral buffered formalin solution, and paraffin-embedded 4 μm thick sections were prepared for immunohistochemical analysis. Ki-67, VEGF, and CD31 expressions were evaluated according to the manufacturer’s instructions (Bioworld Technology, People’s Republic of China) and photographs were taken using an optical microscope (Olympus). The Ki-67 and VEGF expressions were calculated in five randomly selected areas (magnification ×400) of each tumor sample and the number of positive cells/total were counted. The tumor tissue microvessel density (MVD) was calculated as the mean value of the CD31-positive microvessels using the same calculation method described earlier.
Statistical analysis
Statistical analysis was carried out using SPSS 22.0 software (Chicago, IL, USA). The data were performed by Student’s t-test for two groups or one-way analysis of variance for more than two groups. The means were determined to be different with P<0.05 and deemed significantly different with P<0.01.
Results
Characterization and drug loading of nanoparticles
The nanoparticles with different drug loads were prepared by varying ratios of THA and MPEG-PCL (6, 8, and 10 wt%). The particle size, DL%, and EE% were characterized by DLS and HPLC. Table 1 shows that a 10% theoretical drug load in THA-NPs actually exhibited a higher actual drug load (6.57%), a higher EE (almost 65.67%), a smaller average diameter of about 50 nm, a narrower size distribution, and a polydispersity index of nearly 0.24. Thus, THA-NPs containing 10% THA were selected for subsequent studies. Morphology and zeta potential are shown in Figure 1. The nanoparticles were spherical in shape, with a smooth surface, and were found to disperse well in aqueous solution. Furthermore, the THA-NPs exhibited a negative surface charge of −17.80 mv, ultimately increasing the circulation time of the drug.
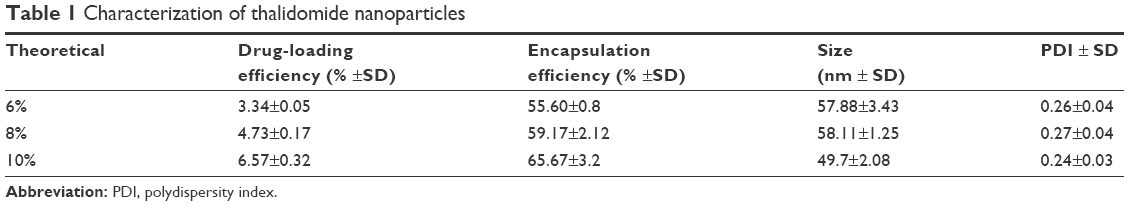 | Table 1 Characterization of thalidomide nanoparticles |
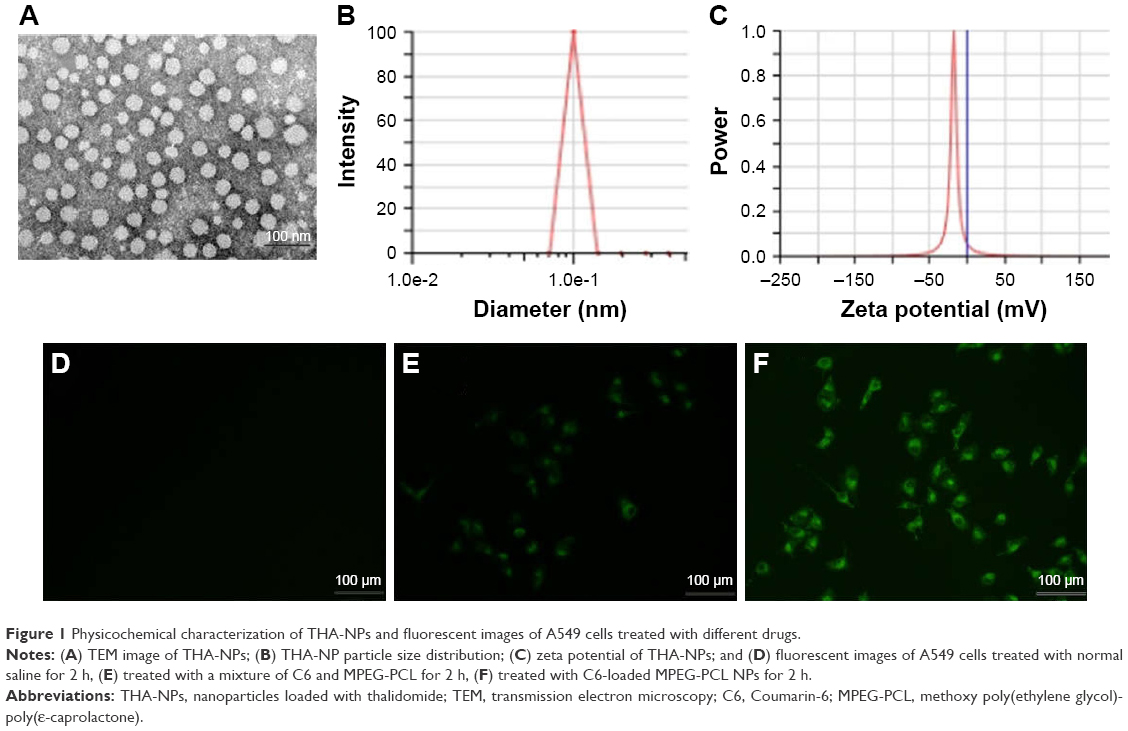 | Figure 1 Physicochemical characterization of THA-NPs and fluorescent images of A549 cells treated with different drugs. |
Cellular uptake
The cellular uptake in A549 cells after incubation for 2 h with NS, C6-loaded MPEG-PCL NPs, and the mixture of C6 and MPEG-PCL is shown in Figure 1. No fluorescence signal was observed in the control group upon treatment with normal saline (Figure 1D). Compared to the treatment with a mixture of C6 and MPEG-PCL, the fluorescence intensity of C6-loaded MPEG-PCL NPs was more remarkable, suggesting that C6-loaded MPEG-PCL NPs displayed improved cellular uptake characteristics.
In vitro cytotoxicity studies
MTT assays were conducted in order to investigate the in vitro cytotoxicity of THA-NPs. Figure 2A shows the suppression effect of free THA and THA-NPs on A549 cells and the obtained results imply a dose-dependent effect. The cell viability of THA and THA-NPs were 48.47% and 42.46%, respectively, as the concentration of THA was found to increase up to 160 μg/mL. Overall, THA-NPs exhibited a higher inhibitory effect compared to free THA.
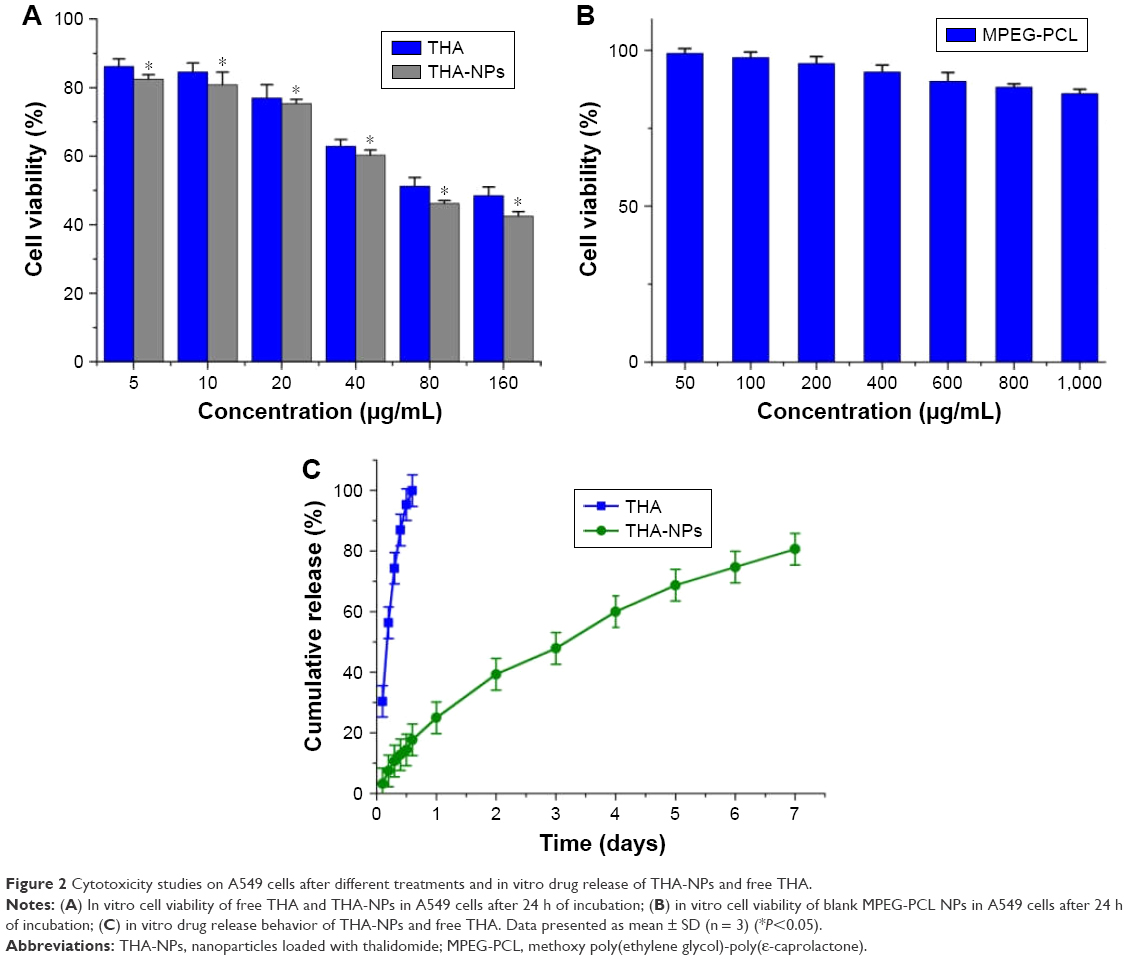 | Figure 2 Cytotoxicity studies on A549 cells after different treatments and in vitro drug release of THA-NPs and free THA. |
The cytotoxicity of blank MPEG-PCL NPs was also evaluated. Although the cell viability of A549 cells decreased after exposure to blank nanoparticles, the cell viability of A549 cells was higher than 86.21%±1.31% as the concentration of blank micelles was increased up to 1,000 μg/mL (Figure 2B). This finding indicates that the MPEG-PCL copolymer exhibited a low cytotoxicity and may therefore be considered a safe drug delivery carrier.
In vitro release studies
The in vitro release of THA-NPs (10%) was evaluated via a dialysis method. In this study, free THA showed a rapid release and more than 99% of the drug was released within 12 h (Figure 2C). On the contrary, THA-NPs exhibited a much slower cumulative release rate compared to free THA. We determined that about 160 h was required to release 80% of THA from THA-NPs, with no obvious burst effect.
In vivo anticancer evaluation
The anticancer efficacy of THA-NPs was evaluated in vivo using A549 cells xenografts in nude mice. Figure 3A shows that both THA and THA-NPs groups exhibited anti-tumor activity, whereas blank MPEG-PCL NPs did not show any significant tumor growth inhibition effect compared to the NS group (P>0.05). As shown in Figure 3A, after 30 days, the tumor volume of the NS group, the MPEG-PCL group, the THA group, and the THA-NPs group were 16.87, 16.07, 11.71, 9.29 folds, respectively, higher than the tumor volume at the beginning of the study. The latter finding indicates that THA-NPs were more efficient in inhibiting tumor growth than THA alone (P<0.05). The median survival time (cf Figure 3B) of the NS group, the MPEG-PCL group, the THA group, and the THA-NPs group were 62, 66, 86.5, and 95 days, respectively. No significant differences between the NS group and the MPEG-PCL group (P>0.05) could be observed. Compared to the THA group, the THA-NPs group showed an improved anticancer activity compared to free THA on prolonging the survival time of the tumor-bearing mice (P<0.05).
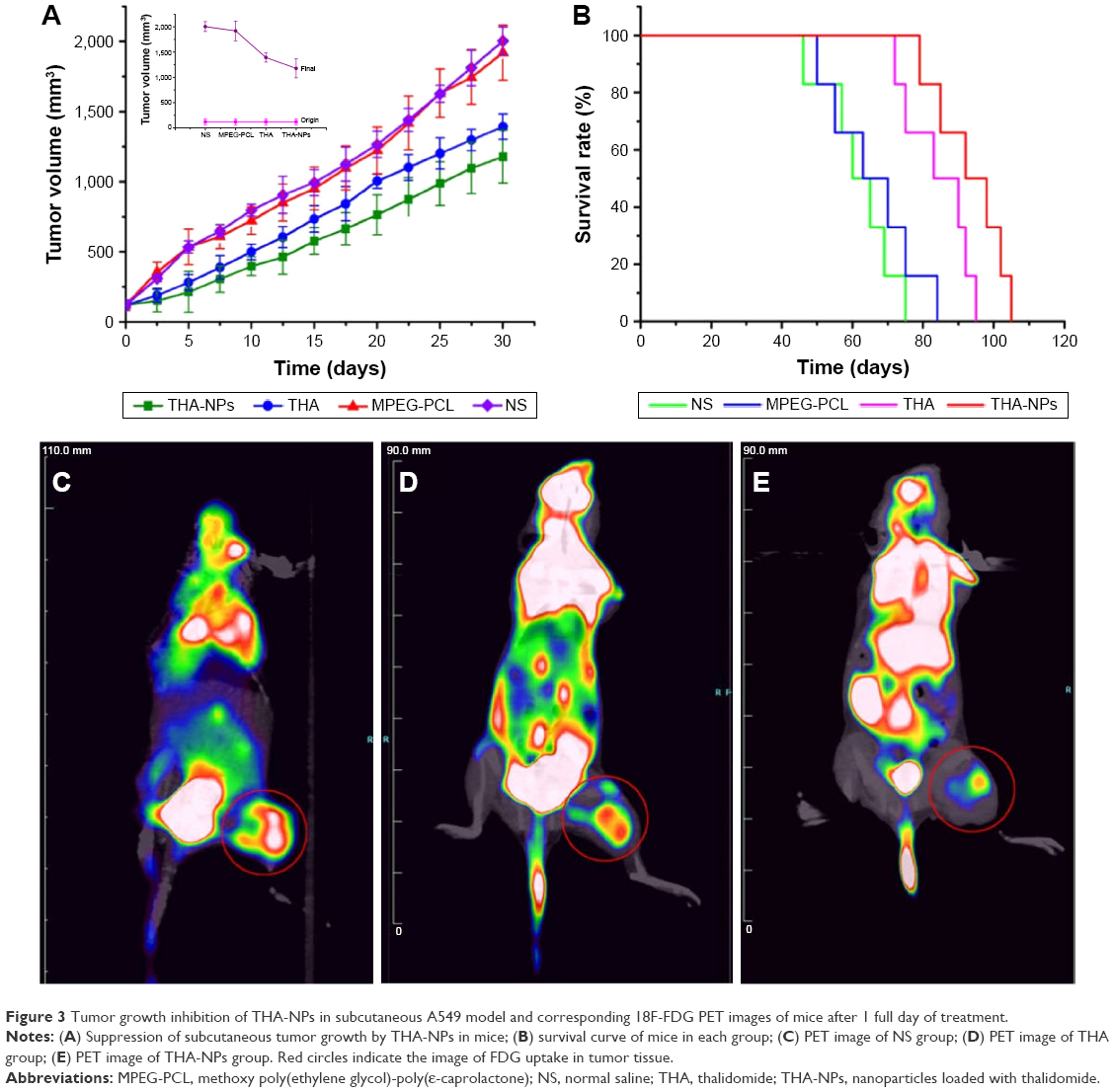 | Figure 3 Tumor growth inhibition of THA-NPs in subcutaneous A549 model and corresponding 18F-FDG PET images of mice after 1 full day of treatment. |
Micro 18F-FDG PET/CT imaging
18F-FDG PET/CT images of nude mice in the NS, THA, and THA-NPs groups were used to evaluate early anti-tumor effects. Figure 3 shows the FDG uptake of lung cancer-bearing nude mice, and a remarkable difference between the three groups could be observed. The NS group exhibited the highest signal intensity, whereas the THA-NPs group demonstrated the lowest FDG uptake, indicating a suppressed tumor metabolism. SUVmax was used as a parameter to calculate the FDG uptake in each group. The mean SUVmax of the tumor in the NS group, THA group, and THA-NPs group were 1.90±0.31, 1.01±0.08, 0.91±0.05, respectively. Compared to the NS group, both the THA (P<0.05) and THA-NPs (P<0.05) groups exhibited a decrease in SUVmax. The SUVmax in the THA-NPs group was determined to be the lowest, suggesting an improved tumor response compared to the THA group.
Cell cycle and apoptosis
Flow cytometric analysis was used to evaluate the cell cycle redistribution and cell apoptosis. Figure 4A shows the proportion of cells at different cell cycle phases (G1 phase, S phase, and G2/M phase). The G0/G1 phase was found to be effectively increased from 55.82%±2.18% to 63.77%±1.21% (P<0.05), and the S phase decreased significantly from 35.08%±1.02% to 26.95%±1.31% (P<0.05). Furthermore, THA-NPs caused a more remarkable increase of cells in the G1 phase and a decrease in the S phase.
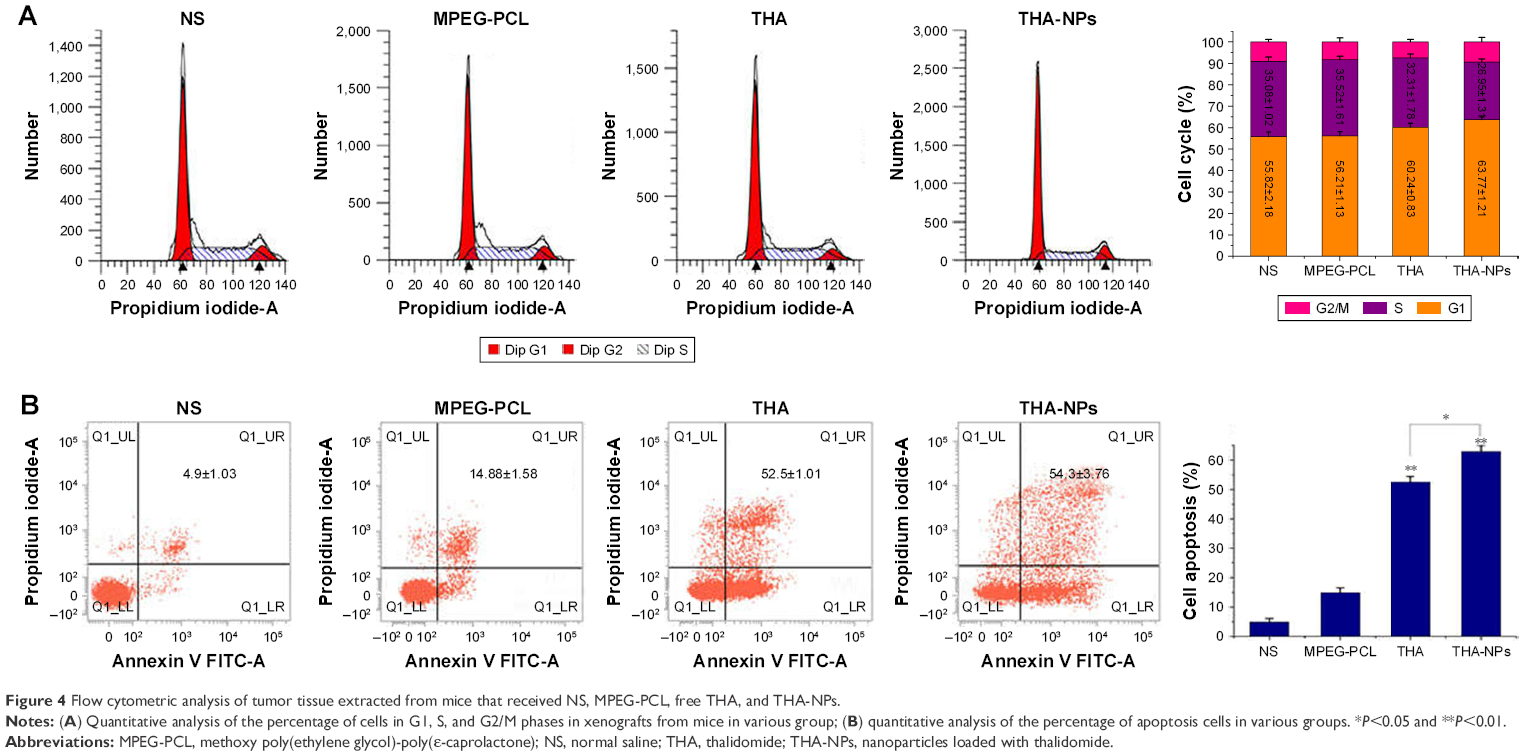 | Figure 4 Flow cytometric analysis of tumor tissue extracted from mice that received NS, MPEG-PCL, free THA, and THA-NPs. |
Figure 4B shows the proportion of apoptotic cells in each groups; NS group: 4.9%±1.03%; MPEG-PCL group: 14.88%±1.58%; THA group: 52.5%±1.01%; and THA-NPs group: 54.3%±3.76%. The percentage of apoptotic cells in the THA-NPS group and the THA group were found to be higher than those in the control group (P<0.01). Compared to the control group, the MPEG-PCL group exhibited a weak effect on apoptosis, indicating suitable biocompatibility characteristics of MPEG-PCL.
Immunohistochemistry
Immunohistochemical staining for Ki-67 was performed to assess the effect of THA-NPs on tumor proliferation. As shown in Figure 5A, the quantification of Ki-67 positive cells was lower in the THA-NPs group (32.8%±4.2%) compared to that in the THA group (42.5%±3.6%, P<0.05), the MPEG-PCL group (74.1%±3.1%, P<0.01), and the NS group (85.3%±2.5%, P<0.01).
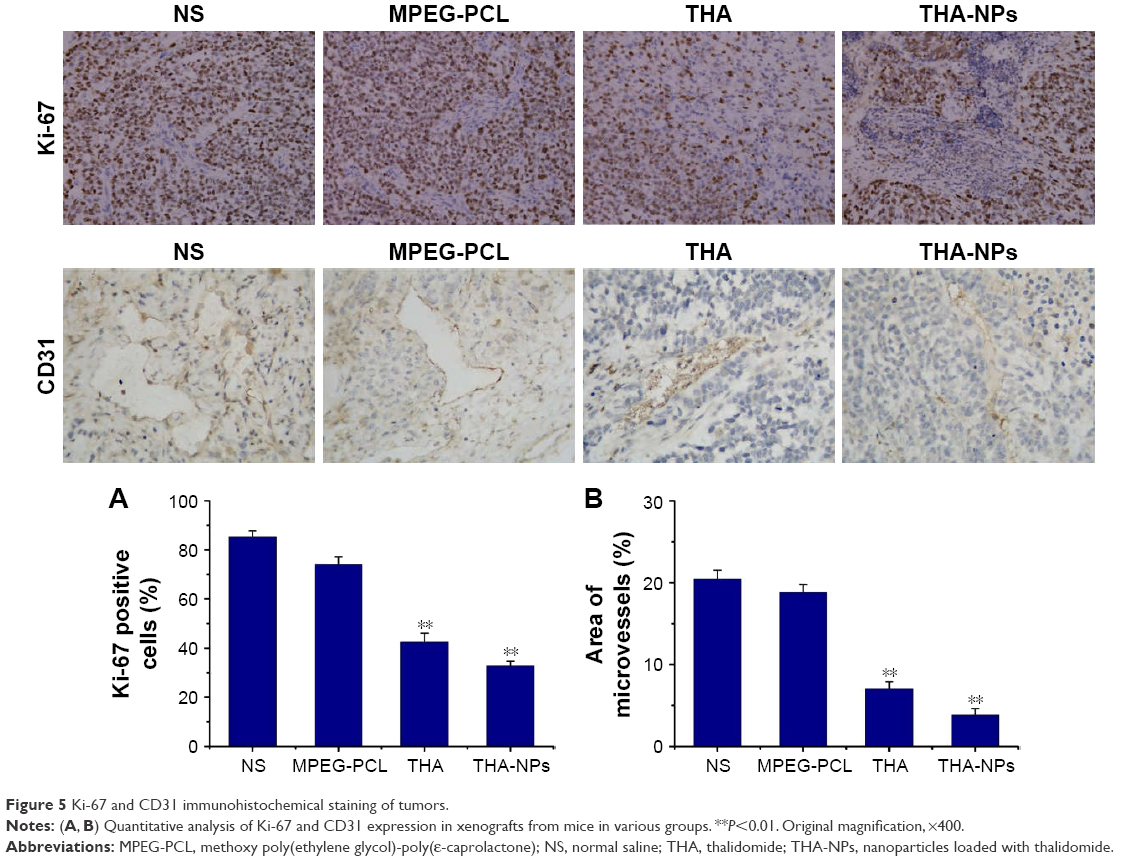 | Figure 5 Ki-67 and CD31 immunohistochemical staining of tumors. |
Immunohistochemical staining for CD31 was performed in order to assess the MVD. As shown in Figure 5B, fewer immunoreactive microvessels were observed in tumor tissues of the THA-NPs group and the overall percentage of immunoreactive microvessels area was 3.87%±0.77%, significantly lower compared to the THA group (7.06%±0.86%, P<0.05), the MPEG-PCL group (18.86%±0.93%, P<0.01), and the NS group (20.48%±1.05%, P<0.01).
Immunohistochemical staining for VEGF was used to assess the expression of the VEGF protein as indicated by a brownish-yellow color in the cytoplasm. As shown in Figure 6A, the THA-NPs group exhibited 26.67%±4.02% positive cells, indicating that the corresponding VEGF protein was significantly lower than that of the THA group (32.73%±1.68%, P<0.05), the MPEG-PCL group (69.53%±3.52%, P<0.01), and the NS group (72.89%±2.56%, P<0.01).
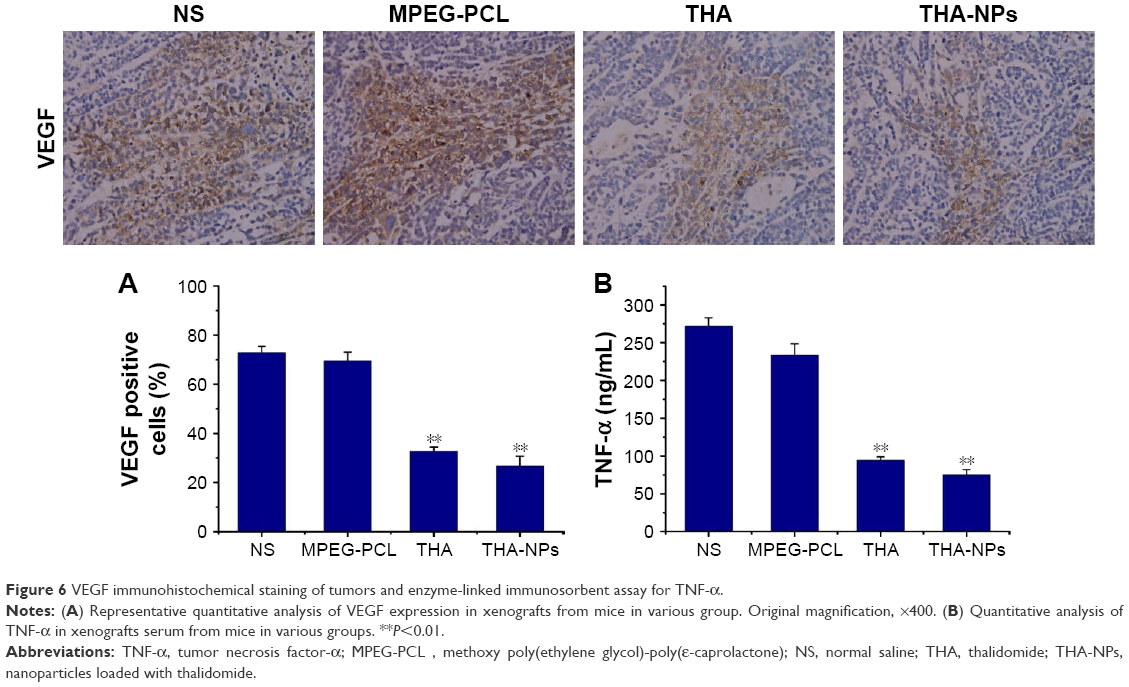 | Figure 6 VEGF immunohistochemical staining of tumors and enzyme-linked immunosorbent assay for TNF-α. |
Effects on serum TNF-α
Figure 6B shows the concentration of serum TNF-α among each group: NS group 271.71±11.30 ng/mL, MPEG-PCL group 233.84±14.62 ng/mL, THA group 94.68±4.34 ng/mL, and THA-NPs group 75.21±6.85 ng/mL. Compared to the control group, a strong suppression effect on TNF-α in the THA and THA-NPs groups could be observed (P<0.01). Furthermore, the concentration of serum TNF-α in the THA-NPs group was found to be the lowest when compared to the THA group (P<0.05).
Discussion
Lung cancer represents a major cancer type and is the leading cause of cancer-related mortality around the globe. Current chemotherapeutic approaches are often inadequate and exhibit serious undesirable side effects as well as intrinsic or acquired chemoresistance in patients.1,28,29 These side effects often restrict the frequency and dose of chemotherapeutics that can be administered. THA, a derivative of synthetic glutamic acid, contains a glutarimide moiety with a single chiral center, resulting in the formation of two possible enantiomers, S (−) and R (+). Since its introduction, the drug has been studied as a compound with anti-angiogenesis, anti-inflammatory, immunomodulatory, and cell proliferation inhibition properties for the potential use in the treatment of a variety of cancer-related diseases as well as autoimmune conditions such as experimental autoimmune encephalomyelitis, graft-versus-host disease, and Waldenstrom’s macroglobulinemia.30,31 THA has been found to exhibit preventative and therapeutic effects in various cancer species and has been confirmed to be a potent chemosensitizer.9,32 However, due to the extreme hydrophobicity and degradation characteristics exhibited by this compound in vivo, the use of THA is vastly limited and THA may not be administered via the intravenous route. Furthermore, the poor water solubility of THA limits the delivery characteristics of the drug to the tumor site at an effective concentration. To overcome the poor water solubility of this hydrophobic drug, nanotechnology may offer a feasible drug delivery method.17,33,34 The central aim of this study was to develop a drug carrier system such as THA-NPs, designed for use in intravenous injection and the improved therapeutic efficacy of THA-based therapy. Over the past few decades, biodegradable polymer nanoparticles for drug delivery have attracted a significant amount of attention in cancer-targeted therapeutics. To improve blood circulation and promote tumor accumulation of nanoparticles, the structural modification of polymers bearing hydrophilic PEG groups has been proposed.35,36 Generally, MPEG-PCL NPs exhibit a core-shell structure with a hydrophobic PCL core and a hydrophilic PEG shell. The hydrophobic core may act as a depot for a variety of hydrophobic drug species, while the hydrophilic PEG shell renders nanoparticles stable with long circulation in the blood stream. PEGylation reduces serum protein adherence and creates a stealth surface to prolong the circulation time by avoiding uptake by the reticuloendothelial systems.37 The concentrations of the encapsulated drugs selectively increase at the tumor sites by EPR effects.7,38,39 In this study, we successfully developed THA/MPEG-PCL nanoparticles used for intravenous injection. We further characterized the formulation in detail and evaluated the anticancer activity both in vitro and in vivo. We formulated three different theoretical dosages of THA-NPs using a dialysis method and demonstrated that all dosages exhibited excellent physical characteristics with the formation of monodispersed spherical particles. Stable DL characteristics for targeting drug delivery were also obtained, as shown in Table 1.
Surface charge is an important feature in determining whether the nanoparticles will cluster in the blood flow or adhere to or interact with oppositely charged cell membranes. Nanoparticles with a slightly negative surface charge may minimize nonspecific interactions with these components through electrostatic interactions. The zeta potential of THA-NPs was −17.80 mv, and the average diameter of the nanoparticles indicated that the particles may avoid renal filtration, hence facilitating effective targeting of the tumor through the EPR effect. THA indicated a pattern of steady, slow and sustained release from THA-NPs. This slow release rate of THA may in turn result in a consistent plasma concentration. An in vitro cytotoxicity assay exhibited that THA-NPs can reduce the viability of A549 cells more significantly than free THA. However, blank MPEG-PCL NPs did not show distinct cytotoxicity characteristics against A549 cells in a broad concentration range (0–1,000 μg/mL) for 24 h, indicating that blank MPEG-PCL nanoparticles exhibit low toxicity and may therefore be considered a safe drug delivery carrier. Because the use of blank MPEG-PCL nanoparticles resulted in a negligible cytotoxicity to A549 cells, the enhanced viability inhibition of THA-NPs might be attributed to an increased number of nanoparticles entering A549 cells via passive endocytosis. This finding was in accordance with fluorescent intensity analysis results obtained.
We also evaluated the anticancer effect of THA-NPs on mouse xenograft models of lung cancer in vivo. In animal models, transplantation procedures, including subcutaneous xenograft (SX) and orthotopic xenograft (OX) methods, are widely used to investigate pathology, diagnosis, and therapy for different cancer types. As different research studies have shown, OX models are similar to the primary site of tumors in the human body, providing a microenvironment equivalent or similar to that in the human body. This microenvironment promotes tumor invasion or metastasis. However, OX models are technically challenging and labor intensive. Preclinical testing most commonly relies on subcutaneously implanted human xenografts, which lack the ability of metastatic development due to the fact that the technique of subcutaneous injection is fairly straightforward. Unfortunately, SX models do not represent the most advantageous models and frequently fail to represent in situ (orthotopic) lung cancer. In our study, we selected an SX model to evaluate the anticancer effects of THA-NPs.40,41 Compared to equivalent doses of THA, the use of THA-NPs resulted in a longer median survival time in mice as is shown in Figure 4B (P<0.05). This further illustrates that THA encapsulated in THA-NPs was more effective than free THA in the treatment of lung cancer-bearing mice. In our study, 18F-FDG PET/CT was used for monitoring early therapy response. 18F-FDG is taken up by cells through glucose transporters and is subsequently phosphorylated by hexokinase. The compound then remains trapped in the cells and cannot be further metabolized. Therefore, a higher FDG uptake in tumors may indicate a poor therapeutic response and overall treatment prognosis. On the contrary, tumors with a lower 18F-FDG uptake may suggest a better treatment response. Thus, compared with the control group, a lower 18F-FDG uptake observed in the treatment groups might suggest that THA was effective on suppressing the tumor metabolism. The lowest FDG uptake was observed in THA-NPs group, suggesting that THA-NPs were more effective than THA on suppressing the tumor metabolism.
THA has been identified to exhibit anti-angiogenesis, anti-inflammation, anti-proliferation, and apoptosis promoting properties.8,42 Despite the fact that the THA anticancer effects may not be explained by a single mechanism, our study clearly demonstrated a variety of anti-tumor effects resulting in cell cycle arrest, apoptosis, proliferation suppression, and microvessel inhibition. Apoptosis represents a type of programmed cell death and plays a vital role in the enhancement of drug treatment. Previous studies have shown that THA can inhibit the growth of tumor cells through the inhibition of cell cycle progression. The susceptibility of cancer cells to apoptosis may be increased by downregulating the anti-apoptosis protein and cytokines.43,44 Our results also demonstrated that THA may increase the accumulation of cells in the G0/G1 phase cell cycle, and may further accelerate cell apoptosis. Tumor growth and systemic metastasis highly depend on angiogenesis.31 Of the secreted angiogenic factors, VEGF is perhaps the most specific factor for endothelial cells. VEGF triggers signaling pathways that result in endothelial cell migration, proliferation, differentiation, promoted vascular permeability, and the release of endothelial cell precursors from the bone marrow when bound to its receptor.45 In addition, angiogenesis plays an essential role in tumor vascular targeting therapy. TNF-α, an important inflammatory factor, has been shown to be overproduced in numerous malignancies. High levels of pharmacological TNF-α induce vasculotoxic tumor-regressing effects. However, pathophysiological doses of endogenous TNF-α accelerate angiogenesis, tumor genesis, and growth. Previous studies have shown that the anti-inflammatory, anti-angiogenic, and anti-tumor effects of THA may be due to its ability to block the production of TNF-α.46 THA is thought to inhibit angiogenesis by reducing levels of VEGF as well as TNF-α. Ki 67, a cell nuclear antigen, is associated with cancer cell proliferation. CD31 is an endothelial cell surface molecule that can be used to visualize the microvascular density. Our results exhibited a remarkable suppression of both CD31 and Ki 67 as well as TNF-α and VEGF in the THA-treated groups. Compared to the THA group, the THA-NPs group exhibited an improved suppression of these factors, suggesting that THA-NPs is a potent inhibitor of angiogenesis and proliferation.
In summary, we have successfully prepared injectable THA-NPs. Our findings indicate that the use of THA-NPs may exhibit superior anticancer characteristics with potential applications in anticancer treatment. In particular, the material may be useful as a chemoradiation sensitizer for the combination with chemotherapy or radiotherapy in the future. However, we also recognize limitations in this study. The EE of THA-NPs was not as good as we anticipated it to be. Furthermore, more detailed studies on drug distribution and side effects in vivo have to be performed to draw definitive conclusions on the applicability of THA-NPs. Further studies to obtain more conclusive data are currently underway in our laboratory and representative results on these topics will be published shortly.
Conclusion
In this study, a drug delivery system based on NPs with loaded THA was successfully developed and its anti-tumor effect was evaluated both in vivo and in vitro. The obtained nanoparticles exhibited a spherical morphology, monodispersity, and negative zeta potential. In vitro drug release experiments demonstrated that THA was released from the nanoparticles both long-term and with a sustained-release pattern. THA-NPs exhibited a concentration-dependent cytotoxicity against A549 cells and also increased the cellular uptake via passive endocytosis in vitro. Based on tumor volume measurements, the medium survival time and PET/CT imaging on the A549 xenograft mice indicated that THA-NPs featured a superior in vivo anti-tumor activity. Furthermore, a potentially promising therapeutic applicability was demonstrated due to the material’s abilities to induce cell apoptosis, cell cycle arrest at G1 phase, and inhibition of cell proliferation and microvessel generation. In summary, THA-NPs may find potential applications in pulmonary carcinoma therapy as well as therapeutic resistance and radiotherapy sensitization.
Acknowledgment
This work was supported by the projects from the General Fund Project of Sichuan Provincial Department of Education (No 17ZB0468); Health and Family Planning Commission of Sichuan Province (No 17PJ557); the research project from the Office of Science and Technology and Intellectual Property of Luzhou (No 2017), the Union Project of Luzhou and Southwest Medical University under Grant (No14JC0144 and 2013LZLY-J40).
Disclosure
The authors report no conflicts of interest in this work.
References
Chen W, Zheng R, Baade PD, et al. Cancer statistics in China, 2015. CA Cancer J Clin. 2016;66(2):115–132. | ||
Torre LA, Siegel RL, Jemal A. Lung cancer statistics. Adv Exp Med Biol. 2016;893:1–19. | ||
Scharovsky OG, Matar P, Rozados VR, et al. [Immunomodulation and antiangiogenesis in cancer therapy. From basic to clinical research]. Medicina (B Aires). 2012;72(1):47–57. Spanish [with English abstract]. | ||
Nowak-Sliwinska P, Griffioen AW. Angiogenesis inhibitors in combinatorial approaches. Angiogenesis. 2017;20(2):183–184. | ||
Zhou S, Wang FF, Hsieh TC, Wu JM, Wu E. Thalidomide–a notorious sedative to a wonder anticancer drug. Curr Med Chem. 2013;20(33):4102–4108. | ||
Vargesson N. Thalidomide-induced teratogenesis: history and mechanisms. Birth Defects Res C Embryo Today. 2015;105(2):140–156. | ||
Wang D, Fu J, Shi Y, et al. The modulation of tumor vessel permeability by thalidomide and its impacts on different types of targeted drug delivery systems in a sarcoma mouse model. J Control Release. 2016;238:186–196. | ||
Vargesson N. Thalidomide embryopathy: an enigmatic challenge. ISRN Dev Biol. 2013;2013(3):1–18. | ||
Huang YT, Cheng CC, Chiu TH, Lai PC. Therapeutic potential of thalidomide for gemcitabine-resistant bladder cancer. Int J Oncol. 2015;47(5):1711–1724. | ||
Hoang T, Dahlberg SE, Schiller JH, et al. Randomized phase III study of thoracic radiation in combination with paclitaxel and carboplatin with or without thalidomide in patients with stage III non-small-cell lung cancer: the ECOG 3598 study. J Clin Oncol. 2012;30(6):616–622. | ||
Qiao Z, Yuan J, Shen J, et al. Effect of thalidomide in combination with gemcitabine on human pancreatic carcinoma SW-1990 cell lines in vitro and in vivo. Oncol Lett. 2015;9(5):2353–2360. | ||
Cao DD, Xu HL, Liu L, et al. Thalidomide combined with transcatheter arterial chemoembolization for primary hepatocellular carcinoma: a systematic review and meta-analysis. Oncotarget. 2017;8(27):44976–44993. | ||
Hungria VT, Crusoé EQ, Maiolino A, et al. Phase 3 trial of three thalidomide-containing regimens in patients with newly diagnosed multiple myeloma not transplant-eligible. Ann Hematol. 2016;95(2):271–278. | ||
Gao M, Kong Y, Wang H, et al. Thalidomide treatment for patients with previously untreated multiple myeloma: a meta-analysis of randomized controlled trials. Tumour Biol. 2016;37(8):11081–11098. | ||
Ozanic Bulic S, Fassihi H, Mellerio JE, McGrath JA, Atherton DJ. Thalidomide in the management of epidermolysis bullosa pruriginosa. Br J Dermatol. 2005;152(6):1332–1334. | ||
Fu S, Xia J, Wu J. Functional chitosan nanoparticles in cancer treatment. J Biomed Nanotechnol. 2016;12:1585–1603. | ||
Xu H, Hou Z, Zhang H, et al. An efficient Trojan delivery of tetrandrine by poly(N-vinylpyrrolidone)-block-poly(ε-caprolactone) (PVP-b-PCL) nanoparticles shows enhanced apoptotic induction of lung cancer cells and inhibition of its migration and invasion. Int J Nanomedicine. 2014;9(1):231–242. | ||
Dinarvand R, Sepehri N, Manoochehri S, Rouhani H, Atyabi F. Polylactide-co-glycolide nanoparticles for controlled delivery of anticancer agents. Int J Nanomedicine. 2011;6:877–895. | ||
Derakhshandeh K, Soheili M, Dadashzadeh S, Saghiri R. Preparation and in vitro characterization of 9-nitrocamptothecin-loaded long circulating nanoparticles for delivery in cancer patients. Int J Nanomedicine. 2010;5(1):463–471. | ||
Chen H, Gu Y, Hu Y. Comparison of two polymeric carrier formulations for controlled release of hydrophilic and hydrophobic drugs. J Mater Sci Mater Med. 2008;19(2):651–658. | ||
Jin H, Hemingway M, Gupta RB, Xia F, Zhao Y. Preparation of thalidomide nano-flakes by supercritical antisolvent with enhanced mass transfer. Particuology. 2012;10(1):17–23. | ||
Suksuwan A, Lomlim L, Rungrotmongkol T, Nakpheng T, Dickert FL, Suedee R. The composite nanomaterials containing (R)-thalidomide-molecularly imprinted polymers as a recognition system for enantioselective-controlled release and targeted drug delivery. J Appl Polym Sci. 2015;132(18):14368–14373. | ||
Yu Y, Xu S, You H, et al. In vivo synergistic anti-tumor effect of paclitaxel nanoparticles combined with radiotherapy on human cervical carcinoma. Drug Deliv. 2017;24(1):75–82. | ||
Zhang H, Li X, Ding J, et al. Delivery of ursolic acid (UA) in polymeric nanoparticles effectively promotes the apoptosis of gastric cancer cells through enhanced inhibition of cyclooxygenase 2 (COX-2). Int J Pharm. 2013;441(1–2):261–268. | ||
Tan LW, Ma BY, Zhao Q, et al. Toxicity evaluation and anti-tumor study of docetaxel loaded mpeg-polyester micelles for breast cancer therapy. J Biomed Nanotechnol. 2017;13(4):393–408. | ||
Jung B, Shim MK, Park MJ, et al. Hydrophobically modified polysaccharide-based on polysialic acid nanoparticles as carriers for anticancer drugs. Int J Pharm. 2017;520(1–2):111–118. | ||
Diab R, Hamoudeh M, Boyron O, Elaissari A, Fessi H. Microencapsulation of cytarabine using poly(ethylene glycol)-poly(epsilon-caprolactone) diblock copolymers as surfactant agents. Drug Dev Ind Pharm. 2010;36(4):456–469. | ||
El-Aarag B, Kasai T, Masuda J, Agwa H, Zahran M, Seno M. Anticancer effects of novel thalidomide analogs in A549 cells through inhibition of vascular endothelial growth factor and matrix metalloproteinase-2. Biomed Pharmacother. 2017;85:549–555. | ||
Chang L, Gong F, Cai H, Li Z, Cui Y. Combined RNAi targeting human Stat3 and ADAM9 as gene therapy for non-small cell lung cancer. Oncol Lett. 2016;11(2):1242–1250. | ||
Cipriani P, Smith CY. Characterization of thalidomide using Raman spectroscopy. Spectrochim Acta A Mol Biomol Spectrosc. 2008;69(2):333–337. | ||
Sherbet GV. Therapeutic potential of thalidomide and its analogues in the treatment of cancer. Anticancer Res. 2015;35(11):5767–5772. | ||
Calabrese L, Resztak K. Thalidomide revisited: pharmacology and clinical applications. Expert Opin Investig Drugs. 1998;7(12):2043–2060. | ||
Xiong W, Peng L, Chen H, Li Q. Surface modification of MPEG-b-PCL-based nanoparticles via oxidative self-polymerization of dopamine for malignant melanoma therapy. Int J Nanomedicine. 2015;10:2985–2996. | ||
Liu B, Han L, Liu J, Han S, Chen Z, Jiang L. Co-delivery of paclitaxel and TOS-cisplatin via TAT-targeted solid lipid nanoparticles with synergistic antitumor activity against cervical cancer. Int J Nanomedicine. 2017;12:955–968. | ||
Wei M, Xu Y, Zou Q, et al. Hepatocellular carcinoma targeting effect of pegylated liposomes modified with lactoferrin. Eur J Pharm Sci. 2012;46(3):131–141. | ||
Rafiei P, Haddadi A. Docetaxel-loaded PLGA and PLGA-PEG nanoparticles for intravenous application: pharmacokinetics and biodistribution profile. Int J Nanomedicine. 2017;12:935–947. | ||
Voon SH, Tiew SX, Kue CS, et al. Chitosan-coated poly (lactic-co-glycolic acid)-diiodinated boron-dipyrromethene nanoparticles improve tumor selectivity and stealth properties in photodynamic cancer therapy. J Biomed Nanotechnol. 2016;12(7):1431–1452. | ||
Liu Q, Li R, Zhu Z, et al. Enhanced antitumor efficacy, biodistribution and penetration of docetaxel-loaded biodegradable nanoparticles. Int J Pharm. 2012;430(1–2):350–358. | ||
Liu L, Sun L, Wu Q, et al. Curcumin loaded polymeric micelles inhibit breast tumor growth and spontaneous pulmonary metastasis. Int J Pharm. 2013;443(1–2):175–182. | ||
Taromi S, Kayser G, von Elverfeldt D, et al. An orthotopic mouse model of small cell lung cancer reflects the clinical course in patients. Clin Exp Metastasis. 2016;33(7):651–660. | ||
Guo J, Cai J, Zhang Y, Zhu Y, Yang P, Wang Z. Establishment of two ovarian cancer orthotopic xenograft mouse models for in vivo imaging: a comparative study. Int J Oncol. 2017;51(4):1199–1208. | ||
Wang X, Shen Y, Li S, et al. Importance of the interaction between immune cells and tumor vasculature mediated by thalidomide in cancer treatment (review). Int J Mol Med. 2016;38(4):1021–1029. | ||
Marriott JB, Clarke IA, Czajka A, et al. A novel subclass of thalidomide analogue with anti-solid tumor activity in which caspase-dependent apoptosis is associated with altered expression of bcl-2 family proteins. Cancer Res. 2003;63(3):593–599. | ||
Keller M, Sollberger G, Beer HD. Thalidomide inhibits activation of caspase-1. J Immunol. 2009;183(9):5593–5599. | ||
Dredge K, Dalgleish AG, Marriott JB. Angiogenesis inhibitors in cancer therapy. Curr Opin Investig Drugs. 2003;4(6):667–674. | ||
Piura B, Medina L, Rabinovich A, Dyomin V, Huleihel M. Thalidomide distinctly affected TNF-α, IL-6 and MMP secretion by an ovarian cancer cell line (SKOV-3) and primary ovarian cancer cells. Eur Cytokine Netw. 2013;24(3):122–129. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.