Back to Journals » International Journal of Nanomedicine » Volume 10 » Issue 1
Preparation, characterization, and safety evaluation of poly(lactide-co-glycolide) nanoparticles for protein delivery into macrophages
Authors Guedj A, Kell A, Barnes M, Stals S, Gonçalves D, Girard D ![]() , Lavigne C
, Lavigne C
Received 4 February 2015
Accepted for publication 26 May 2015
Published 23 September 2015 Volume 2015:10(1) Pages 5965—5979
DOI https://doi.org/10.2147/IJN.S82205
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr Thomas Webster
Anne-Sophie Guedj,1 Arnold J Kell,2 Michael Barnes,2 Sandra Stals,1 David Gonçalves,3 Denis Girard,3 Carole Lavigne1
1National Microbiology Laboratory, Public Health Agency of Canada, Winnipeg, MB, 2National Research Council of Canada, Ottawa, ON, 3Laboratoire de recherche en inflammation et physiologie des granulocytes, Université du Québec, INRS-Institut Armand-Frappier, Laval, QC, Canada
Abstract: Following infection, HIV establishes reservoirs within tissues that are inaccessible to optimal levels of antiviral drugs or within cells where HIV lies latent, thus escaping the action of anti-HIV drugs. Macrophages are a persistent reservoir for HIV and may contribute to the rebound viremia observed after antiretroviral treatment is stopped. In this study, we further investigate the potential of poly(lactic-co-glycolic) acid (PLGA)-based nanocarriers as a new strategy to enhance penetration of therapeutic molecules into macrophages. We have prepared stable PLGA nanoparticles (NPs) and evaluated their capacity to transport an active molecule into the human monocyte/macrophage cell line THP-1 using bovine serum albumin (BSA) as a proof-of-concept compound. Intracellular localization of fluorescent BSA molecules encapsulated into PLGA NPs was monitored in live cells using confocal microscopy, and cellular uptake was quantified by flow cytometry. In vitro and in vivo toxicological studies were performed to further determine the safety profile of PLGA NPs including inflammatory effects. The size of the PLGA NPs carrying BSA (PLGA-BSA) in culture medium containing 10% serum was ~126 nm in diameter, and they were negatively charged at their surface (zeta potential =-5.6 mV). Our confocal microscopy studies and flow cytometry data showed that these PLGA-BSA NPs are rapidly and efficiently taken up by THP-1 monocyte-derived macrophages (MDMs) at low doses. We found that PLGA-BSA NPs increased cellular uptake and internalization of the protein in vitro. PLGA NPs were not cytotoxic for THP-1 MDM cells, did not modulate neutrophil apoptosis in vitro, and did not show inflammatory effect in vivo in the murine air pouch model of acute inflammation. In contrast to BSA alone, BSA encapsulated into PLGA NPs increased leukocyte infiltration in vivo, suggesting the in vivo enhanced delivery and protection of the protein by the polymer nanocarrier. We demonstrated that PLGA-based nanopolymer carriers are good candidates to efficiently and safely enhance the transport of active molecules into human MDMs. In addition, we further investigated their inflammatory profile and showed that PLGA NPs have low inflammatory effects in vitro and in vivo. Thus, PLGA nanocarriers are promising as a drug delivery strategy in macrophages for prevention and eradication of intracellular pathogens such as HIV and Mycobacterium tuberculosis.
Keywords: PLGA nanoparticles, BSA delivery, inflammatory profile, neutrophil apoptosis, murine air pouch, HIV reservoir
Introduction
HIV/AIDS remains one of the world’s most significant public health challenges, particularly in low- and middle-income countries (UNAIDS, 2014). The introduction of highly active antiretroviral therapy in 1996 has, over time, been a great medical success in terms of increasing survival and improving the quality of life of the HIV-infected patient population. However, this therapy is not able to eradicate HIV from infected individuals.1–3 During the very early stages of infection, HIV establishes reservoirs within tissues that are inaccessible to optimal levels of antiviral drugs or within cells where HIV lies latent, thus escaping the action of anti-HIV drugs.4–6 Reservoir sites protect the virus from biological elimination pathways, immune response, and antiretroviral drugs; allow viral dissemination throughout the body; serve as a major source of viral rebound upon treatment failure, and contribute to the development of drug resistance.7–10 Resting CD4+ cells are known to be the main cellular reservoir for the latent HIV-1 infection due to their ability to persist for a long span, but other cellular reservoirs have been described to play an important role in HIV persistence including monocytes and macrophages, which are the most important reservoirs outside the bloodstream.7,8,11–16 Monocytes/macrophages are relatively long-lived cells since HIV has very low cytopathic effects on them, making them a persistent reservoir of HIV regardless of the presence of highly active antiretroviral therapy.8,15–17 Monocytes and macrophages express efflux transporters, which contribute to maintain subtherapeutic concentrations of antiretroviral medications in these cells and may explain why macrophages are sanctuaries for HIV.18 Therefore, eliminating or preventing viral infection in macrophages is a key element to achieve viral eradication, and new strategies to enhance penetration of antiviral therapeutics in macrophages are thus urgently needed.
One promising approach to target HIV in reservoir sites is the use of nanomedicine.19–24 The potential advantages of using nanoformulations over conventional HIV therapies include the capacity to incorporate, encapsulate, or conjugate a variety of drugs to offer tunable and site-specific targeting and drug release.25 In addition, the use of nanosystems to deliver a therapeutic molecule can overcome biological barriers; improve drug stability, solubility, and bioavailability; and reduce side effects.26–28 A wide range of nanomaterials have been used and evaluated for their efficiency to deliver HIV-1 therapeutics.25,29 Particular interest has been focused on polymeric particles prepared from polyesters such as poly(lactic-co-glycolic) acid (PLGA), poly(lactide), and polyglycolide due to their biocompatibility and biodegradability and their approval by the US Food and Drug Administration for use in drug delivery and biomaterial applications.30–32 Various polymeric nanomedicines have been developed for the delivery of antiretroviral drugs in HIV reservoir sites, giving some hope for viral eradication.20,24 However, despite major breakthroughs in the development of nanopolymeric particles for vaccine and drug delivery into macrophages, there are still many limitations to overcome before their use in clinical applications.33,34 Although PLGA co-polymer has been used in several studies as micro/nanoparticles (NPs) delivery systems with a certain degree of success20,30,32,35–37 including delivery of antimicrobial drugs into monocytes/macrophages,29,38–42 their safety for therapeutic application is still unknown. Indeed, few studies have focused on the inflammatory profile of PLGA nanopolymeric carriers and, to our knowledge, no study has yet investigated the effect of PLGA-based NPs on polymorphonuclear neutrophil (PMN) apoptosis and recruitment. PMNs are key players in innate immunity and inflammation, thus determining whether PLGA-based NPs interfere with neutrophil function is crucial for the further development of PLGA-based nanomedicines.
Therefore, the aim of this research was to further evaluate the potential of PLGA-based NPs in delivery strategies to target HIV in the macrophage cellular reservoir and to further investigate their inflammatory properties by determining their effect on PMNs. We designed and characterized a PLGA-based nanocarrier for the delivery of therapeutic agents in human monocyte-derived macrophages (MDMs). Bovine serum albumin (BSA) was used as the model protein/drug to investigate more specifically the potential of PLGA nanomaterials to serve as nanocarriers for antimicrobial or antigenic proteins for future microbicide and vaccine development, respectively. The PLGA NPs were prepared by the double emulsion method and characterized in terms of various physicochemical properties, cellular uptake and internalization, cytotoxicity, and inflammatory properties including interference with neutrophil apoptosis and recruitment in a pro-inflammation animal model.
Materials and methods
Materials
Biodegradable PLGA 50:50 polymer of 7,000–17,000 daltons average molecular weight (MW), BSA, and fluorescein isothiocyanate (FITC)-labeled BSA were all purchased from Sigma-Aldrich Co. (St Louis, MO, USA). Polyvinyl alcohol (PVA) of 6,000 daltons average MW was purchased from Polysciences, Inc. (Warrington, PA, USA). Titanium dioxide (TiO2) was obtained from Vive Nano (Toronto, ON, Canada). Bradford Protein Assay kit was purchased from Bio-Rad Laboratories Inc. (Hercules, CA, USA). Roswell Park Memorial Institute (RPMI) 1640 cell culture medium, phosphate buffered saline (PBS), and Hank’s Balanced Salt Solution (HBSS) were all purchased from Thermo Fisher Scientific (Waltham, MA, USA). CellTiter-Blue® Cell Viability Assay was obtained from Promega Corporation (Fitchburg, WI, USA). Milli-Q and deionized water were produced by a Millipore water purification system (EMD Millipore, Billerica, MA, USA).
Preparation of nanoparticles
Antigenic protein-loaded biodegradable NPs were prepared by a double-emulsion (water/oil/water) method with subsequent solvent evaporation.41 BSA was used as a model protein. Briefly, BSA or FITC-BSA (8 mg) was first dissolved in water (1 mL) and then added to a solution of 150 mg of PLGA 50:50 (7,000–17,000 dalton MW) dissolved in chloroform (6 mL). The mixture was sonicated for 45 seconds using a Branson Sonifier 250 fitted with a Branson tapered microtip (3 mm) (Branson Sonifier® Cell Disrupters; Branson Electronics, VWR, Missisauga, ON, Canada) to form the first water-in-oil emulsion. The power of the tip sonicator was set at 60% duty cycle and was operated at maximum power (200 W). This water-in-oil emulsion was added to a solution of 2.5% (v/v) PVA (6,000 dalton MW) dissolved in water (50 mL) to form a water-in-oil-in-water double-emulsion. Sonication of the double-emulsion was performed at room temperature for 5 minutes to reduce droplet size to the nanosize range followed by evaporation of the organic solvent chloroform under vacuum to form PLGA-BSA and PLGA-FITC-BSA NPs. Again the power was 200 W with a 60% duty cycle. The PLGA NPs were then washed several times by centrifugation (14, 000 rpm, 4°C, 45 minutes) to remove the unbound protein and adsorbed PVA from the surface of the NPs and resuspended in a D-glucose (dextrose) solution. The NPs were then flash frozen with liquid nitrogen, freeze dried, and stored at −20°C until their use. Unloaded PLGA NPs were prepared using the same method except BSA was not added to the water during formation of the first water-in-oil emulsion. For the in vitro and in vivo studies, NPs were reconstituted in sterile water, and the suspensions were filtered through a sterile membrane with a pore size of 0.45 μm (EMD Millipore) and then kept at 4°C until their use. Freshly prepared NP batches were used unless otherwise noted.
Physicochemical characterization
The morphology of the NPs was analyzed using a Philips CM20 FEG transmission electron microscope operating at 200 kV. Images were obtained by drop-coating the PLGA NPs suspension onto 400 mesh copper grids with a precoated layer of holey carbon. Particle size and size distribution were measured by dynamic light scattering (DLS) using a Malvern Zetasizer Nano-ZS (Malvern Instruments, Malvern, UK). The width of the DLS hydrodynamic diameter distributions is indicated by the polydispersity index. Zeta potential was determined using the same instrument. NP suspensions were vortexed before analysis, and each sample was measured in triplicates.
Encapsulation efficiency
The amount of BSA and FITC-BSA contained in the NPs was spectrophotometrically determined directly after the extraction of the protein from the NPs. Briefly, the PLGA-BSA and PLGA-FITC-BSA NPs were treated with a solution of 2 M NaOH during 2 hours to allow degradation of the polymeric matrix and release of encapsulated BSA or FITC-BSA, respectively, from the NPs. The suspension was then centrifuged at 15,000 rpm for 15 minutes. Supernatant was collected, and the amount of protein released from the NPs was determined using a Bradford Protein Assay kit according to the manufacturer’s instructions. Absorbance was measured at 595 nm by a Synergy Mx multimode microplate reader (Biotek, Winooski, VT, USA) with the Gen5 Data Analysis Software. The amount of unbound BSA or FITC-BSA found in the supernatant was also measured after each of the three washes during the synthesis of the PLGA-BSA and PLGA-FITC-BSA NPs and quantified using the Bradford Protein Assay. The percentage of the encapsulation efficiency (EE%) was calculated according to the following equation:
EE% = (amount of protein released from the NPs/[amount of unbound BSA in the supernatant during the washing steps + amount of protein released from the NPs]) ×100 | (1) |
Loading of BSA was calculated using the following formula:
Loading% = (amount of protein/[amount of protein + amount of polymeric matrix]) ×100 | (2) |
Cell culture
THP-1 monocyte/macrophage cell line was obtained from the American Type Culture Collection43,44 and were maintained in RPMI 1640 supplemented with 10% heat-inactivated fetal bovine serum (FBS), 50 μg/mL gentamicin, and 2 mM L-glutamine at 37°C, 5% CO2. MDMs were obtained by adding PMA to the culture medium at a final concentration of 10 ng/mL. Cells were incubated for 4–6 days to allow the monocytes to differentiate into macrophages.
Cellular uptake and internalization
In order to evaluate the cellular uptake and internalization of BSA-loaded PLGA NPs, we used FITC-labeled BSA to prepare the NPs. For flow cytometry analysis, THP-1 cells were seeded in 24-well tissue culture plates at a density of 2×105 cells per well and allowed to differentiate. After 4 days, MDMs were washed and fresh culture medium containing various concentrations of PLGA-FITC-BSA NPs was added to the wells. Cells were incubated for 3 hours and then washed with PBS. Cellular uptake of fluorescent PLGA-FITC-BSA NPs was analyzed by flow cytometry (FACSCalibur™; BD Biosciences, San Jose, CA, USA). To further confirm cellular uptake of the PLGA-FITC-BSA NPs and study their internalization into MDMs, confocal microscopy analyses were performed. Cells were seeded in glass flat-bottomed 96-well tissue culture plates for confocal microscopy (MatTek, Ashland, MA, USA) at a density of 2×104 cells per well with culture medium containing PMA to allow differentiation. After 6 days of incubation, differentiated THP-1 cells were washed with PBS and fresh culture medium containing 100 μg/mL of fluorescent PLGA-FITC- BSA NPs or free FITC-BSA in solution was added to the wells. In control wells, only culture medium was added to the cells. Cells were incubated at 37°C for 3 hours, then washed with PBS, and fresh culture medium without NPs was added to each well. Uptake and intracellular localization of PLGA-FITC-BSA NPs or free FITC-BSA in viable MDM cells was visualized using laser scanning confocal microscopy (LSM700; Carl Zeiss Canada, Toronto, ON, Canada).
Cell-biocompatibility assay
THP-1 cells were seeded in flat-bottomed 96-well plates (Corning) at a density of 2×104 cells per well and allowed to differentiate. After 4 days, MDMs were washed with PBS and fresh culture medium containing various concentrations of either unloaded PLGA or PLGA-BSA NPs was added to the cells. After 48 hours of exposure to the NPs, cell viability was determined by the CellTiter-Blue® Assay (Promega Corporation) based on the ability of living cells to convert a redox dye (resazurin) into a fluorescent end product (resorufin) and quantified by the ratio to the control. Untreated cells served as 100% cell viability.
Neutrophil apoptosis assay
Blood donations were obtained from informed and consenting individuals and the study protocol was approved by the INRS–Institut Armand-Frappier Research Centre Ethics Committee. Freshly isolated human PMNs from healthy donors were isolated by dextran sedimentation followed by centrifugation over Ficoll-Hypaque (Pharmacia Biotech Inc., Quebec, Canada) as previously described.45–47 Purity (>98%) was verified by cytology from cytocentrifuged preparations stained with Diff-Quick staining (Thermo Fisher Scientific). Cell viability was monitored by trypan blue exclusion before and after each treatment and was always ≥99%. PMNs were cultured in RPMI 1640-HEPES-P/S, supplemented with 10% heat-inactivated autologous serum, and 10×106 cells/mL were treated with or without increasing concentrations of NPs for 24 hours. Control cultures were incubated with buffer (untreated control), 65 ng/mL granulocyte-macrophage colony-stimulating factor (GM-CSF, negative control), or plant lectin Viscum album agglutinin 1 (positive control). Apoptosis was assessed by cytology as described earlier.45,46 Cells were examined by light microscopy at 400× final magnification, and apoptotic neutrophils were defined as cells containing one or more characteristic, darkly stained pyknotic nuclei.
Animal study
CD-1 female mice (6-8 weeks of age) from Charles River Laboratories (St-Constant, QC, Canada) after a period of acclimation of 1 week were anesthetized with isofurane at days 0 and 3 to create an air pouch by injecting 3 mL of sterile air subcutaneously in the back (with a 26-gauge needle) as previously described.45,48–50 At day 6, 1 mL of water or various concentrations of NPs or free BSA was injected into the air pouch. After 6 hours, mice were sacrificed by CO2 asphyxiation, the pouches were washed three times (once with 1 mL and then twice with 2 mL of Hank’s Balanced Salt Solution containing 10 mM EDTA), and the exudates were centrifuged. Cells were resuspended at a density of 0.5×106 cells/mL, spread onto microscope slides, and stained with Hema-Stain for identification/quantification of leukocyte cell subpopulations. Experiments were performed as per protocols approved by Animal Use and Care Committees at INRS-Institut Armand-Frappier.
Statistics
Statistical analysis was performed using Sigma Stat for Windows (version 3.0) for the Student’s t-test or SigmaPlot (version 11.0) for the ANOVA analysis. A P-value of <0.05 was considered as statistically significant. All data are presented as mean ± standard error of the mean (SEM) unless otherwise noted.
Results
Morphology and physicochemical properties of the nanoparticles
Using the double-emulsion solvent evaporation method, we were able to successfully prepare spherical NPs as shown in the electron microscope image (Figure 1A). Particle size analysis and zeta potential of BSA-loaded and unloaded PLGA NPs were determined by DLS in different solvents and summarized in Table 1. The mean particle size of the PLGA-BSA NPs in water was 186±17 nm compared with 160±8 nm for unloaded PLGA NPs, indicating the presence of the protein. In cell culture medium supplemented with 10% serum, we observed a reduction in the size of the PLGA-BSA NPs to 126±2 nm, whereas the size of the unloaded control PLGA NPs increased to 195±18 nm. Surface charge was negative in all conditions, but a significant increase in zeta potential was observed when NPs were dispersed in saline buffer or cell culture medium as compared with water. As summarized in Table 1, PLGA-BSA NPs had a zeta potential of −12.3±2.5 mV in water compared with −30.8±3.2 for unloaded PLGA NPs. In saline or RPMI, zeta potential increased to reach up to −4.6 mV for PLGA-BSA and −2.3 mV for PLGA NPs. To note, addition of serum in RPMI did not affect the surface charge of the NPs. The polydispersity index was 0.041 and 0.195 for unloaded PLGA NPs (Figure 1B) and PLGA-BSA NPs (Figure 1C), respectively. The DLS results indicated a narrow size distribution and a good colloidal stability of the NPs (Figure 1B and C). The results were reproducible in multiple batches. The size of FITC-BSA-loaded PLGA NPs was 207 nm, and the zeta potential of the NPs in water was −18 mV (data not shown).
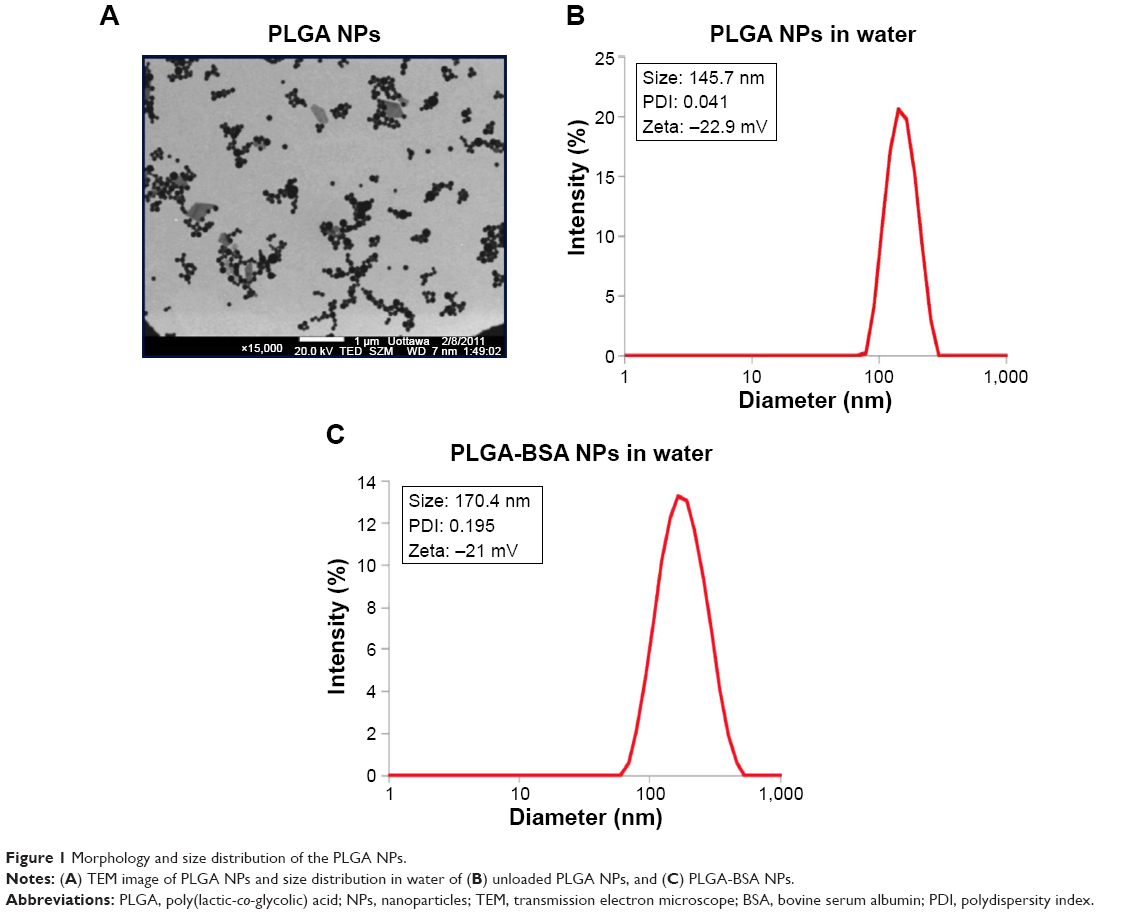 | Figure 1 Morphology and size distribution of the PLGA NPs. |
 | Table 1 Particle size and zeta potential in different solvents |
EE% of the BSA protein and stability of nanoparticles
EE% of BSA was evaluated at 38%±5% from four different experiments done in triplicates. BSA content in the NPs and free BSA in supernatants were determined by the Bradford method as described in the Materials and methods section. The percentage of loading of BSA in the NPs was 3.85%. EE% of FITC-BSA was evaluated at 27.6%±1.1% from triplicate measurements. To assess the stability of the NPs, fluorescein-labeled BSA-loaded PLGA NPs were prepared, stored at 4°C, and monitored for up to 1 month. THP-1 MDMs were treated with 100 μg/mL of PLGA-FITC-BSA NPs for 3 hours, and then cell-associated fluorescence was analyzed by flow cytometry. As shown in Figure S1, fluorescence was detected up to the end of the experiment (28 days). Weaker fluorescence intensity was observed at days 21 and 28; however, the difference between cell-associated fluorescence from day 0 up to day 28 was not found to be statistically significant. These results indicate that the NPs are stable at least for 1 month.
Cellular uptake and internalization
In order to evaluate cellular uptake, THP-1 MDM cells were treated with different concentrations of PLGA-FITC-BSA NPs for 3 hours, and fluorescence intensity of the resulting labeled cells was measured by flow cytometry. Results are shown in Figure 2. Flow cytometry data demonstrated that PLGA-FITC-BSA NPs are rapidly and efficiently taken up by THP-1 MDMs (Figure 2A). A significant dose-dependent increase in fluorescence was observed from 20 up to 200 μg/mL and then increased more slowly up to the highest concentration tested, 1,000 μg/mL (Figure 2B). We next compared the cellular uptake of PLGA-FITC-BSA with free FITC-BSA. THP-1 MDM cells were exposed to different concentrations of free FITC-BSA or PLGA-FITC-BSA for 3 hours, and then the fluorescence was measured by flow cytometry. Data are shown in Figure 3. In contrast to Figure 2 in which the concentration represented the amount of NPs, in this study, the data are expressed in terms of the amount of BSA. As shown in Figure 3, cell-associated fluorescence increased up to fivefold when the protein was incorporated into PLGA NPs compared with free delivery, demonstrating that uptake of BSA is more efficient when loaded into PLGA NPs.
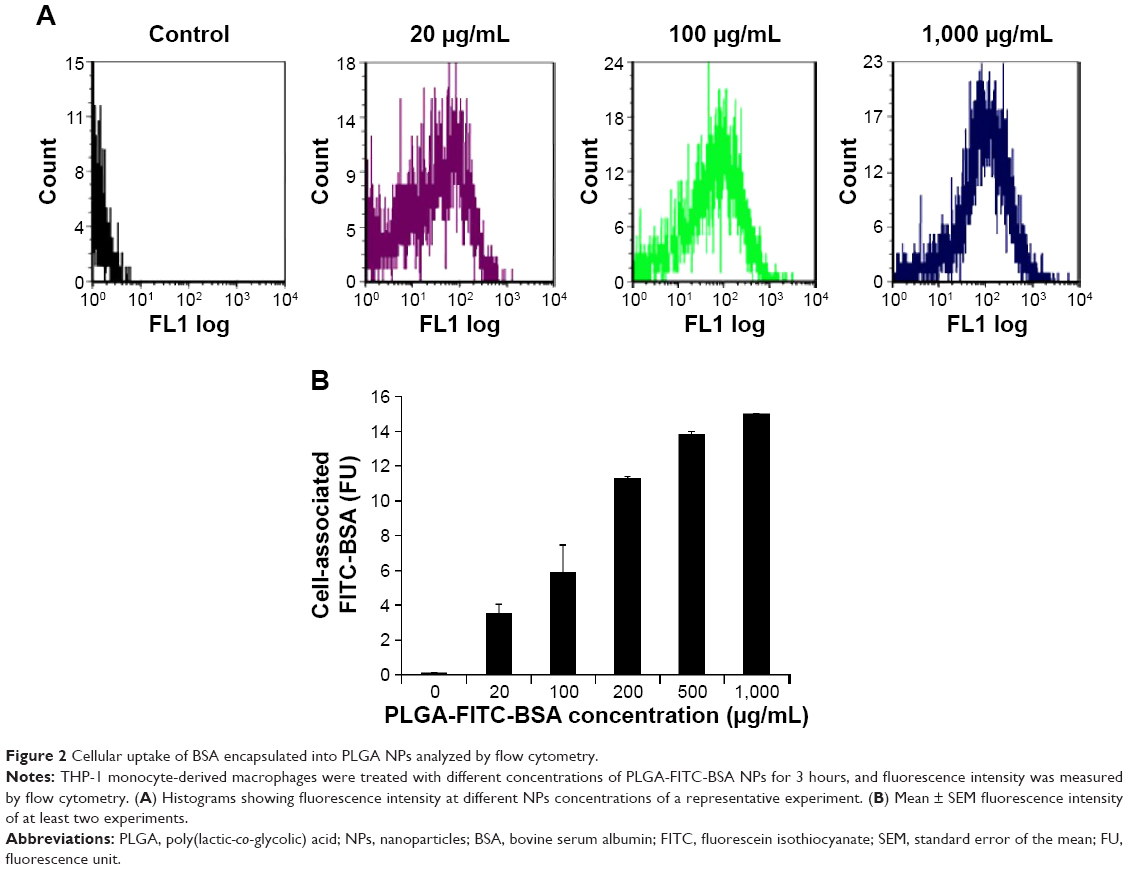 | Figure 2 Cellular uptake of BSA encapsulated into PLGA NPs analyzed by flow cytometry. |
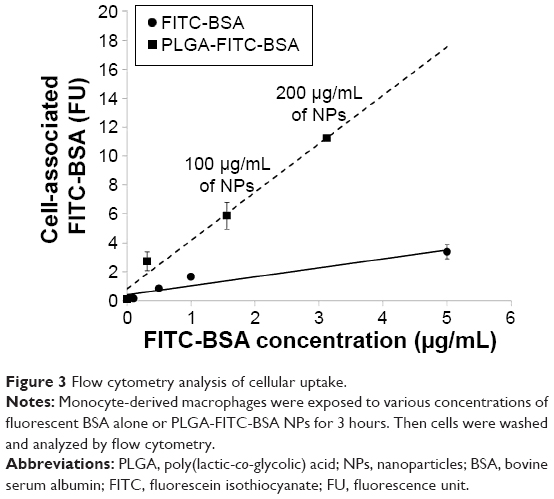 | Figure 3 Flow cytometry analysis of cellular uptake. |
Flow cytometry analysis cannot determine if the PLGA NPs are inside the cells or simply on the cell surface. Therefore, to further investigate whether the NPs was indeed taken up and internalized within the cells, we performed confocal laser scanning microscopy studies. The data highlighted in Figure 4 are representative images of live cells incubated with PLGA-FITC-BSA NPs or free FITC-BSA for 3 hours. While in untreated control cultures (Figure 4A), no fluorescence was detected. In cells treated with PLGA-FITC-BSA NPs, fluorescence intensity increased with the concentration of NPs (Figure 4C–F). At the lowest dose tested, 20 μg/mL, a high level of cellular uptake was already observed (Figure 4C), and at 100 μg/mL, almost all cells have had taken up the BSA-loaded PLGA NPs (Figure 4D). These results support the flow cytometry data and demonstrate that the PLGA-FITC-BSA NPs were efficiently and rapidly taken up by the cells. In addition, the confocal analysis allowed us to visualize the intracellular distribution of the fluorescent BSA either free or encapsulated into the PLGA-FITC-BSA NPs. Z-stack analysis (data not shown) and Figure 4C–F show that the fluorescence is distributed inside the cells, confirming the internalization of the NPs. By comparing the cellular uptake of free FITC-BSA (Figure 4B) with the uptake of PLGA-FITC-BSA (Figure 4C–F), we again observed a higher rate of delivery when FITC-BSA was incorporated into PLGA NPs, confirming the flow cytometry data (Figure 3).
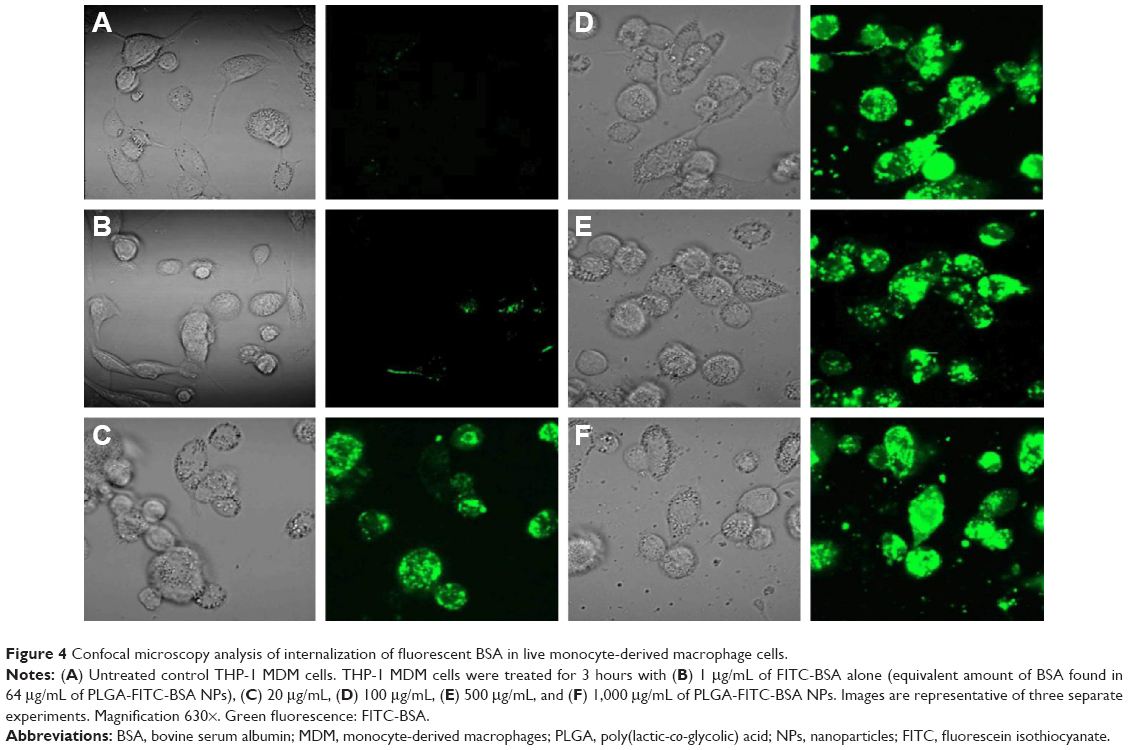 | Figure 4 Confocal microscopy analysis of internalization of fluorescent BSA in live monocyte-derived macrophage cells. |
Safety evaluation
Cytotoxicity
Next, we ran a series of experiments to define the safety and inflammatory properties of the NPs. First, we evaluated the effects on THP-1 MDMs on enzymatic activity after 48 hours of exposure to increased concentrations of unloaded PLGA or PLGA-BSA NPs using the CellTiter-Blue® colorimetric cell viability assay. As illustrated in Figure 5, PLGA NPs (Figure 5A) did not affect cell viability of MDMs up to the maximum concentration tested, 500 μg/mL. Interestingly, when the cells were treated with low doses (5 or 50 μg/mL) of PLGA-BSA NPs, we observed an increase in the number of cells (Figure 5B). This increase was not observed with unloaded PLGA NPs (Figure 5A). At higher concentrations of PLGA-BSA NPs, cell viability was not affected (Figure 5B). PLGA NPs were also evaluated for their cytotoxicity on the TZM-bl epithelial cell line model, and again, no effect on cell viability was observed up to the highest concentration used in these cells, 4,000 μg/mL (data not shown).
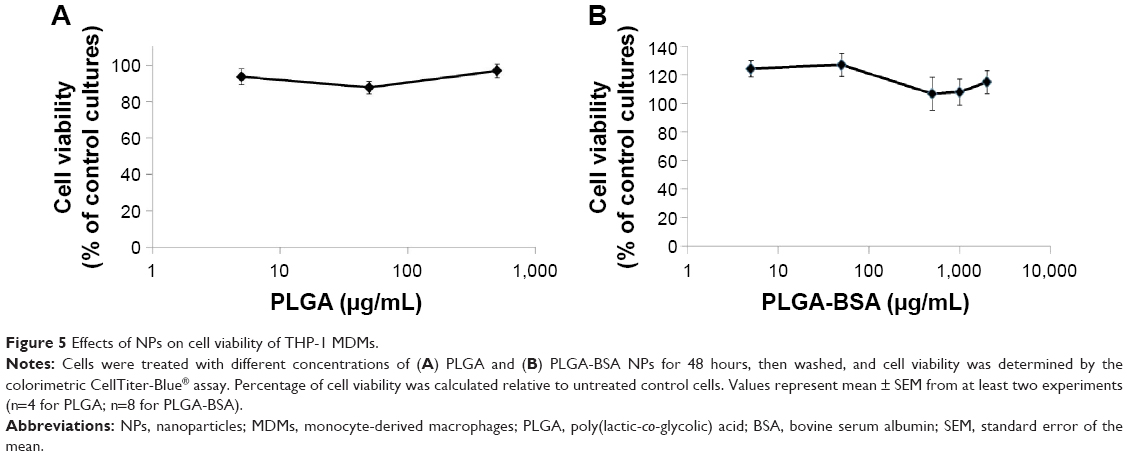 | Figure 5 Effects of NPs on cell viability of THP-1 MDMs. |
Inflammatory properties
The inflammatory profile of the PLGA NPs was performed with a neutrophil apoptosis assays. As illustrated in Figure 6, neither PLGA nor PLGA-BSA NPs affected apoptosis of human neutrophils up to the highest concentration tested (1,000 μg/mL). As expected, V. album agglutinin increased and GM-CSF inhibited neutrophil apoptosis when compared with the controls.51,52 To further investigate the pro-inflammatory property of the PLGA NPs, we also conducted in vivo assays using the murine air pouch model. This model has been commonly used to determine pro-inflammatory effects of molecules including the evaluation of acute pro-inflammatory activity of NPs.53–55 As illustrated in Figure 7, administration of 10, 100, 250, 500, or 1,000 μg/mL of unloaded PLGA NPs or 10, 100, 250, 500 μg/mL of PLGA-BSA NPs did not significantly increase the total number of attracted leukocytes into the air pouch 6 hours postinjection when compared with the control mice. However, administration of 1,000 μg/mL of PLGA-BSA NPs significantly increased leukocyte infiltration into the air pouch to the same extent of theTiO2 positive control NPs (Figure 7B). However, when BSA alone was administered into the air pouch at concentrations equivalent to the amount of BSA found in 1,000, 2,000, or 4,000 μg/mL of PLGA-BSA NPs, we did not observe increased leukocyte infiltration into the air pouch (Figure 7C). Cytology examinations revealed that 75% of the leukocytes attracted into the air pouch by 1,000 μg/mL of PLGA-BSA NPs were neutrophils compared with 56% in control mice or 93% when TiO2 NPs were injected (Figure 8B). The remainders of the leukocytes were mononuclear cells. Although the total number of leukocytes attracted into the air pouch did not increase when unloaded PLGA NPs were administered, the proportion of PMNs was slightly higher (72%) compared with the control mice (63%), but this difference was not found statistically significant (Figure 8A). Finally, BSA alone at any of the concentrations evaluated showed the same proportion of cell subpopulations as in the control mice (Figure 8C).
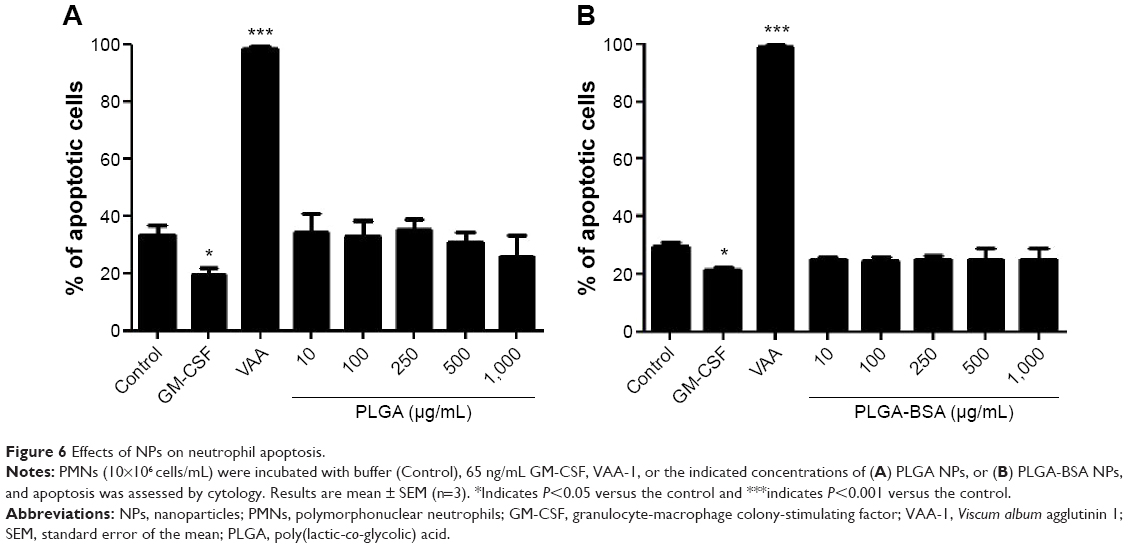 | Figure 6 Effects of NPs on neutrophil apoptosis. |
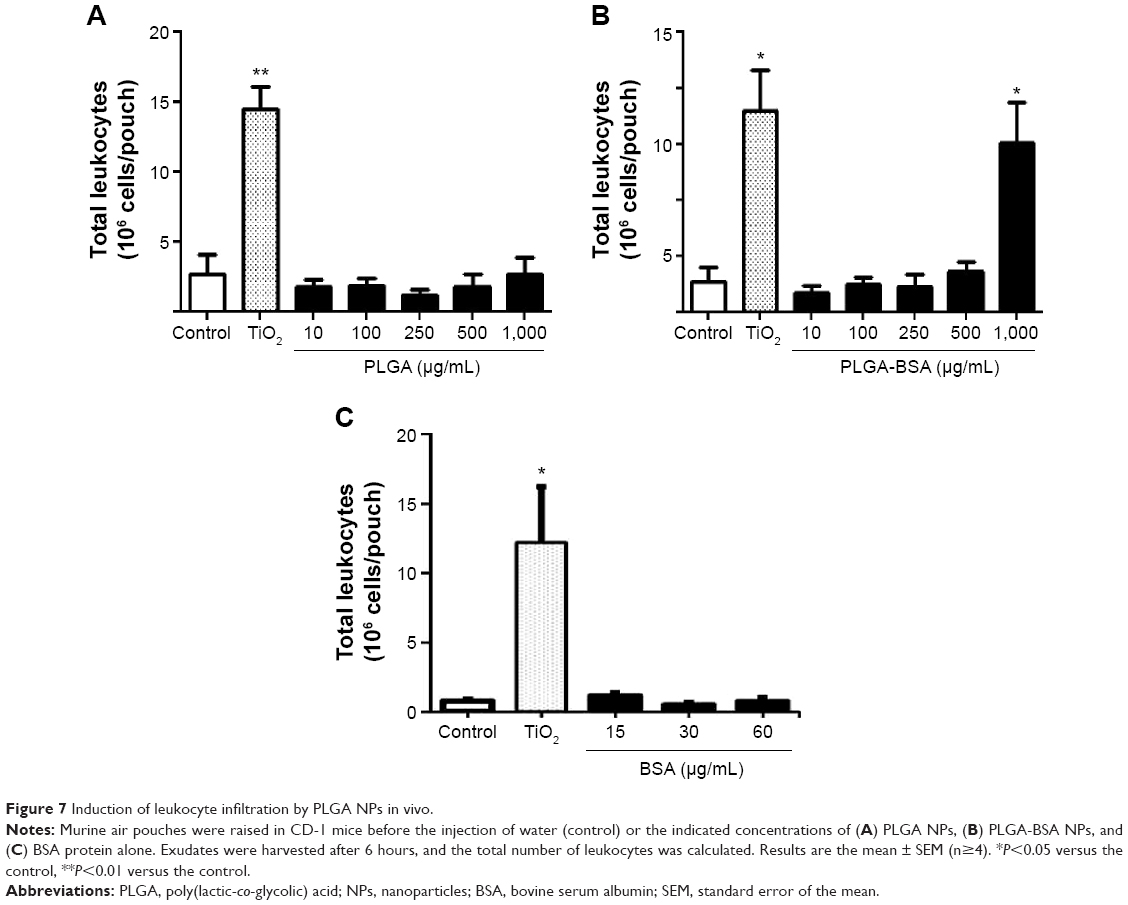 | Figure 7 Induction of leukocyte infiltration by PLGA NPs in vivo. |
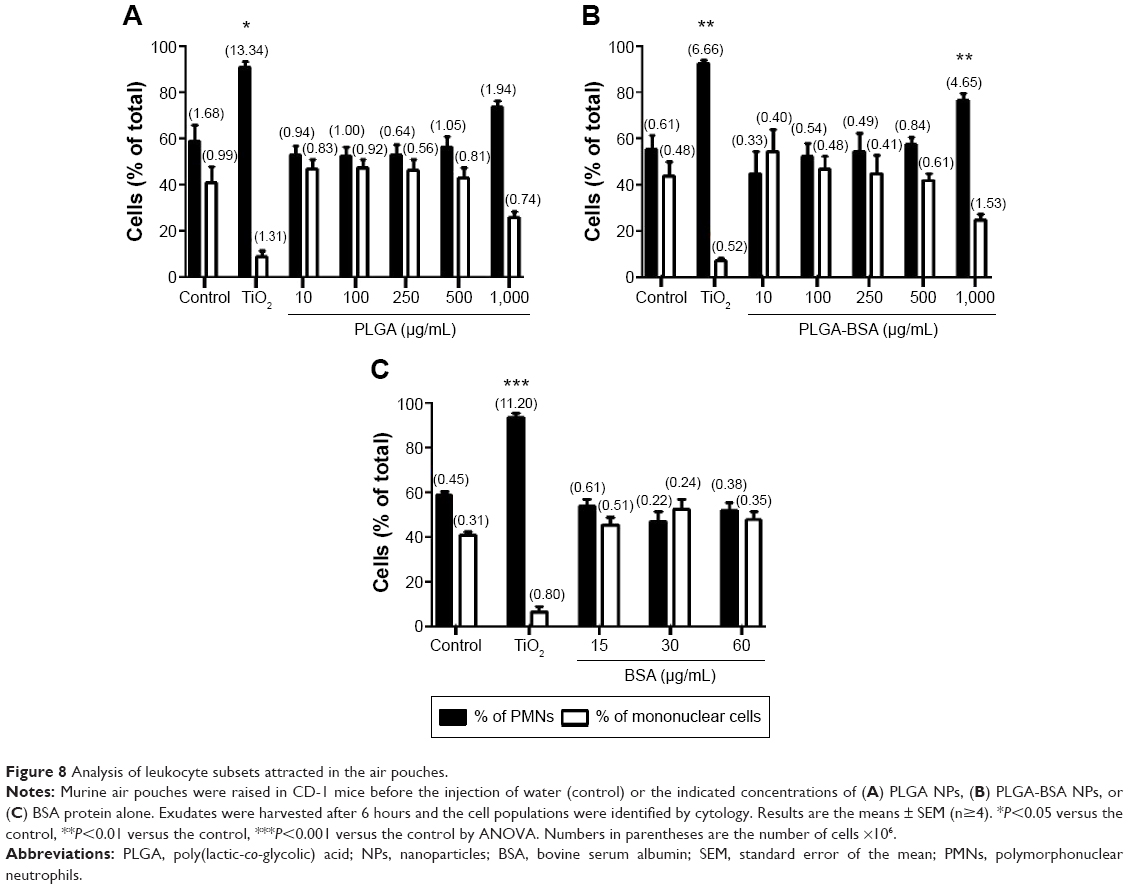 | Figure 8 Analysis of leukocyte subsets attracted in the air pouches. |
Discussion
Like CD4+ T cells, monocyte/macrophage lineage cells are regarded as the early targets for HIV-1 infection. Because macrophages are widely distributed and present in many tissues of the body, they not only play important roles in disease control and progression but can also contribute to virus dissemination throughout the body including in the central nervous system. Therefore, HIV-1-infected macrophages might play a pivotal role in virus transmission, the establishment of latent reservoirs, and AIDS pathogenesis. As such, eliminating or preventing viral infection in macrophages is a key element to achieve viral eradication. Previous studies have demonstrated the potential of PLGA-based NPs to deliver drugs in vitro and in vivo, but the toxicity profile of PLGA NPs have not yet been fully investigated. Therefore, the objective of the present study was to further characterize PLGA-based NPs for the delivery of therapeutic proteins into human macrophages with an emphasis on a better characterization of their inflammatory profile, which is currently poorly documented. We have prepared PLGA NPs containing BSA that remain stable in cell culture medium supplemented with 10% serum with a homogenous size of 126 nm and a negative surface charge (zeta potential =−5.61 mV). In addition, these PLGA-BSA NPs were found to be stable at 4°C up to at least 1 month in solution. Specifically, the size of PLGA NP increases from 156 to 167 nm, and zeta potential decreases from −33.3 to −31.5 mV, respectively, after storage for 2 months in water (data not shown). However, further experiments are required in order to confirm their stability for future use in clinical application. Indeed, the change in fluorescence observed could be assumed to be dependent from loss of albumin stabilization or change in surface characteristics of NPs, which have not been investigated in this study. Most importantly, these PLGA NPs did not agglomerate in serum-containing culture medium and remained well below 200 nm in diameter. This was crucial for the present study because we wanted to evaluate the NPs in vitro and in vivo, so their properties in biological media had to be characterized.
Macrophages obtained by differentiation of THP-1 monocytes were chosen as the in vitro cell model because these have been shown to be a relevant in vitro human macrophage models43,44 and have been used successfully to investigate the toxicity of NPs.56–58 The use of FITC-labeled BSA allowed us to detect and localize the protein either free or encapsulated into PLGA NPs and monitor its uptake and delivery process in THP-1 MDM cells. Flow cytometry analysis demonstrated that BSA-loaded PLGA NPs are easily taken up by human MDMs and that BSA delivery in this cell model is increased fivefold when the protein is encapsulated into PLGA NPs. Our results are in agreement with a previous study showing a similar increase range of cell uptake of PLGA-formulated antituberculosis drugs in human MDMs.40 The authors observed a three- and tenfold increase in human MDMs delivery of a hydrophobic isoniazid derivative and rifampicin, respectively, when compared with free drugs. Using live-cell confocal microscopy, we further demonstrated that PLGA-FITC-BSA NPs are rapidly taken up and that higher uptake of BSA is achieved when delivered by PLGA nanocarriers compared with its free form in solution. We also confirmed their internalization in human MDMs as they were found inside the cells within 3 hours. This study demonstrates and supports previous studies showing the rapid uptake of PLGA NPs by macrophages. The focus of this study was not to investigate the uptake mechanism in human MDMs, but it has been previously demonstrated that PLGA NPs are phagocytosed by macrophages within 30 minutes, and electron microscopy studies have shown that they are found within the cytoplasm of human MDMs 45 minutes following treatment. Therefore, as shown in this study and previous studies, the use of polymeric-based nanocarriers is a promising strategy to deliver therapeutic agents in human MDMs and to target pathogens that proliferate inside macrophages including visceral leishmaniasis, malaria, and tuberculosis.29,38–40
It is well known that PLGA-based material is suitable for therapeutic application since it is biodegradable and show low toxic effects. The results of our cytotoxicity assays support previous studies showing that PLGA NPs do not significantly affect human MDM cell viability.38,39,56–58 In the present study, PLGA NPs were not found to be cytotoxic for neither TZM-bl nor human MDMs when cells were exposed for 48 hours at concentrations up to 4,000 or 500 μg/mL, respectively. In 2013, Shibata et al reported no significant cytotoxicity up to 28 days of treatment with 500 μg/mL of PLGA NPs using three cell models including the human monocyte U937 cell model.39 Therefore, we and others have demonstrated that PLGA NPs do not affect cell activity even at concentrations well above therapeutically relevant doses (100 μg/mL and below), confirming their potential as delivery nanocarriers for therapeutic applications. Interestingly, in this study, we observed that cells treated with PLGA-BSA NPs showed an increase in cell proliferation. We also observed an increase when cells were treated with BSA alone at equivalent amount found in the PLGA NPs but to a lesser extent than the PLGA-BSA NPs (data not shown). This might be explained in part by the fact that cells were exposed to higher concentrations of BSA when it was delivered by PLGA NPs compared with free BSA in solution, confirming again higher delivery of BSA when the protein is encapsulated. More experiments are needed though to confirm this.
One of the major toxic effects of NPs frequently reported in the literature is inflammation.57,59–61 Because PMNs are key players in innate immunity and inflammation,62,63 it is important to evaluate the effects of NPs on the response and functions of human neutrophils. Therefore, in this study, we wanted to further investigate the toxicological profile of PLGA NPs by evaluating their effects on human PMNs. To do so, we performed in vitro apoptosis neutrophil assays. The resolution of inflammation is known to occur by the elimination of apoptotic PMNs, so the modulation of neutrophil apoptosis may indicate pro-inflammatory (inhibition of apoptosis) or anti-inflammatory (induction of apoptosis) properties depending on whether the agent inhibits or induces apoptosis, respectively. To determine whether PLGA NPs had pro- or anti-inflammatory effects, we investigated their ability to modulate human primary PMNs apoptosis in vitro. To validate the assay, control cultures were exposed to either the cell growth factor GM-CSF known to act as an inhibitor of apoptosis or to the plant lectin V. album agglutinin 1, a potent pro-apoptotic molecule that we used in this study as an inducer of neutrophil apoptosis. In contrast to other types of NPs such as ZnO or TiO2 that have been found to modulate PMN apoptosis,47,64 in this study, PLGA NPs were not found to have inflammatory activities. The NPs did not significantly inhibit or induce PMN apoptosis in vitro. Of note, this is the first study to report the lack of inflammatory effect of PLGA NPs using the neutrophil apoptosis assay. Based on these results, one can speculate that these PLGA NPs would not disturb normal homeostasis in vivo by modifying the life span of PMNs. Hence, they should not induce accumulation of PMNs in tissues by inhibiting apoptosis and thus induce inflammation in vivo. To confirm this and further investigate the inflammatory properties of the PLGA NPs, we used the murine air pouch model to determine whether our PLGA NPs caused leukocyte infiltration in vivo as in our previous studies with TiO2 NPs.54 We and others have previously used this in vivo model to classify the degree of pro-inflammatory potential of NPs, but it is the first time that the inflammatory effects of PLGA NPs are evaluated using this animal model. In this study, we did not observe an increase in leukocyte infiltration into the air pouch when PLGA NPs were injected up to the highest concentration tested (1,000 μg/mL) demonstrating their lack of inflammatory effect. Furthermore, when PLGA NPs were loaded with BSA, a significant increase in the number of leukocytes recruited into the air pouch was noted when compared with equivalent amount of BSA in solution, suggesting that encapsulation of BSA into the PLGA NPs might have protected it from degradation for a longer period of time allowing leukocyte recruitment. More experiments are needed to confirm that this was the case. Nevertheless, these results show the potential of PLGA nanocarrier to transport and deliver a protein in vivo and confirm its low toxicity profile. Finally, in agreement with the low pro-inflammatory properties of the PLGA NPs observed in the neutrophil apoptosis assay and in the air pouch murine model, we found that they did not significantly increase cytokine secretion of interleukin (IL)-1β, IL-6, IL-10, and TNF-α in differentiated THP-1 cells up to the highest concentration tested (400 μg/mL) 48 hour post-treatment as determined by a luminex multiplex assay (Table S1). Further evaluation of the effect of these PLGA NPs on the activation of the inflammasome is necessary to confirm these preliminary observations.
Conclusion
PLGA NPs loaded with BSA protein were successfully synthesized and rapidly internalized in macrophages. Encapsulation in PLGA NPs significantly increased cellular uptake of the BSA protein in macrophages compared with the free protein in solution. In addition, the PLGA NPs did not cause cytotoxicity and showed low inflammatory properties. Indeed, they did not modulate apoptosis in vitro in the neutrophil apoptosis assay and did not induce leukocyte infiltration in vivo in the murine air pouch model. Altogether, the results of this study further demonstrate the safety and efficiency of PLGA NPs to deliver therapeutic molecules in human MDMs. Further study is necessary to determine if these NPs could be a promising strategy for the prevention and eradication of HIV-1 infection.
Acknowledgments
This work was supported by the Public Health Agency of Canada and the National Research Council of Canada. Denis Girard was supported partly by Natural Sciences and Engineering Research Council of Canada (NSERC) and Institut de recherche Robert-Sauvé en santé et en sécurité du travail (IRSST). David Gonçalves was supported by a PhD NSERC scholarship award. AS Guedj has received a scholarship from the Université Montpellier II for her research training in Canada. We are grateful to Michèle Bergeron and Xuefen Yang for their technical help with the flow cytometry analysis and Sylvie Faucher for her help with the luminex bead assay.
Author contributions
Lavigne was responsible for the study design and overall data collection, analysis, and writing paper. Kell was responsible for experimental design for the synthesis and characterization of the nanoparticles and contributed to the editing of the paper. Girard was responsible for the experimental design of the in vitro and in vivo inflammatory studies and reviewed the paper. Guedj, Stals, Gonçalves, and Barnes were responsible for carrying out experiments and contributed to the analysis of the results. All authors approved the final version to be published and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
Wong JK, Hezareh M, Günthard HF, et al. Recovery of replication-competent HIV despite prolonged suppression of plasma viremia. Science. 1997;278:1291–1295. | ||
Davey RT Jr, Bhat N, Yoder C, et al. HIV-1 and T cell dynamics after interruption of highly active antiretroviral therapy (HAART) in patients with a history of sustained viral suppression. Proc Natl Acad Sci U S A. 1999;96:15109–15114. | ||
Palmer S, Josefsson L, Coffin JM. HIV reservoirs and the possibility of a cure for HIV infection. J Intern Med. 2011;270:550–560. | ||
Finzi D, Hermankova M, Pierson T, et al. Identification of a reservoir for HIV-1 in patients on highly active antiretroviral therapy. Science. 1997;278:1295–1300. | ||
Chun TW, Fauci AS. HIV reservoirs: pathogenesis and obstacles to viral eradication and cure. AIDS. 2012;26:1261–1268. | ||
Chan CN, Dietrich I, Hosie MJ, Willett BJ. Recent developments in human immunodeficiency virus-1 latency research. J Gen Virol. 2013;94:917–932. | ||
Coleman CM, Wu L. HIV interactions with monocytes and dendritic cells: viral latency and reservoirs. Retrovirology. 2009;6:51. | ||
Gavegnano C, Schinazi RF. Antiretrovial therapy in macrophages: implication for HIV eradication. Antivir Chem Chemother. 2009;20:63–78. | ||
Le Douce V, Herbein G, Rohr O, Schwartz C. Molecular mechanisms of HIV-1 persistence in the monocyte-macrophage lineage. Retrovirology. 2010;7:32. | ||
Best BM, Letendre SL, Koopmans P, et al; CHARTER Study Group. Low cerebrospinal fluid concentrations of the nucleotide HIV reverse transcriptase inhibitor, tenofovir. J Acquir Immune Defic Syndr. 2012;59:376–381. | ||
Gartner S, Markovits P, Markovitz DM, Kaplan MH, Gallo RC, Popovic M. The role of mononuclear phagocytes in HTLVIII/LAV infection. Science. 1986;233:215–219. | ||
Verani A, Gras G, Pancino G. Macrophages and HIV-1: dangerous liaisons. Mol Immunol. 2005;42:195–212. | ||
Venzke S, Keppler OT. Role of macrophages in HIV infection and persistence. Expert Rev Clin Immunol. 2006;2:613–626. | ||
Koppensteiner H, Brack-Werner R, Schindler M. Macrophages and their relevance in Human Immunodeficiency Virus Type I infection. Retrovirology. 2012;9:82. | ||
Kumar A, Abbas W, Herbein G. HIV-1 latency in monocytes/macrophages. Viruses. 2014;6:1837–1860. | ||
Campbell JH, Hearps AC, Martin GE, et al. The importance of monocytes and macrophages in HIV pathogenesis, treatment, and cure. I. 2014;28:2175–2187. | ||
Eisele E, Siliciano RF. Redefining the viral reservoirs that prevent HIV-1 eradication. Immunity. 2012;37:377–388. | ||
Jorajuria S, Dereuddre-Bosquet N, Becher F, et al. ATP binding cassette multidrug transporters limit the anti-HIV activity of zidovudine and indinavir in infected human macrophages. Antivir Ther. 2004;9:519–528. | ||
Amiji MM, Vyas TK, Shah LK. Role of nanotechnology in HIV/AIDS treatment: potential to overcome the viral reservoir challenge. Discov Med. 2006;6:157–162. | ||
das Neves J, Amiji MM, Bahia MF, Sarmento B. Nanotechnology-based systems for the treatment and prevention of HIV/AIDS. Adv Drug Deliv Rev. 2010;62:458–477. | ||
van’t Klooster G, Hoeben E, Borghys H, et al. Pharmacokinetics and disposition of rilpivirine (TMC278) nanosuspension as a long-acting injectable antiretroviral formulation. Antimicrob Agents Chemother. 2010;54:2042–2050. | ||
Wong HL, Chattopadhyay N, Wu XY, Bendayan R. Nanotechnology applications for improved delivery of antiretroviral drugs to the brain. Adv Drug Deliv Rev. 2010;62:503–517. | ||
Roy U, McMillan J, Alnouti Y, et al. Pharmacodynamic and antiretroviral activities of combination nanoformulated antiretrovirals in HIV-1-infected human peripheral blood lymphocyte: reconstituted mice. J Infect Dis. 2012;206:1577–1588. | ||
Lenjisa JL, Woldu MA, Satessa GD. New hope for eradication of HIV from the body: the role of polymeric nanomedicines in HIV/AIDS pharmacotherapy. J Nanobiotechnology. 2014;12:9. | ||
Mahajan SD, Aalinkeel R, Law WC, et al. Anti-HIV-1 nanotherapeutics: promises and challenges for the future. Int J Nanomedicine. 2012;7:5301–5314. | ||
Cory T, Schacker TW, Stevenson M, Fletcher CV. Overcoming pharmalogical sanctuaries. Curr Opin HIV AIDS. 2013;8:190–195. | ||
Lavigne C, Slater K, Gajanayaka N, et al. Influence of lipoplex surface charge on siRNA: application to the in vitro downregulation of CXCR4 HIV-1 co-receptor. Expert Opin Biol Ther. 2013;13:973–985. | ||
Kell AJ, Slater K, Barnes ML, Lavigne C. Functionalized silica nanoparticles stable in serum-containing medium efficiently deliver siRNA targeting HIV-1 co-receptor CXCR4 in mammalian cells. Int J Nano Biomater. 2012;4:223–242. | ||
Destache CJ, Belgum T, Goede M, Shibata A, Belshan MA. Antiretroviral release from poly(DL-lactide-co-glycolide) nanoparticles in mice. J Antimicrob Chemother. 2010;65:2183–2187. | ||
Lü JM, Wang X, Marin-Muller C, et al. Current advances in research and clinical applications of PLGA-based nanotechnology. Expert Rev Mol Diagn. 2009;9:325–341. | ||
Semete B, Booysen L, Lemmer Y, et al. In vivo evaluation of the biodistribution and safety of PLGA nanoparticles as drug delivery systems. Nanomedicine. 2010;6:662–671. | ||
Danhier F, Ansorena E, Silva JM, Coco R, Le Breton A, Préat V. PLGA-based nanoparticles: an overview of biomedical applications. J Control Release. 2012;161:505–522. | ||
Desai N. Challenges in development of nanoparticle-based therapeutics. AAPS J. 2012;14:282–295. | ||
Moghimi SM, Parhamifar L, Ahmadvand D, et al. Particulate systems for targeting of macrophages: basic and therapeutic concepts. J Innate Immun. 2012;4:509–528. | ||
Kumari A, Yadav SK, Yadav SC. Biodegradable polymeric nanoparticles based drug delivery systems. Colloids Surf B Biointerfaces. 2010;75:1–18. | ||
Thomas C, Rawat A, Hope-Weeks L, Ashan F. Aerosolized PLA and PLGA nanoparticles enhance humoral, mucosal and cytokine responses to hepatitis B vaccine. Mol Pharm. 2011;8:405–415. | ||
Chitkara D, Kumar N. BSA-PLGA-based core-shell nanoparticles as carrier system for water-soluble drugs. Pharm Res. 2013;30:2396–2409. | ||
Destache CJ, Belgum T, Christensen K, et al. Combination antiretroviral drugs in PLGA nanoparticle for HIV-1. BI. 2009;9:198. | ||
Shibata A, McMullen E, Pham A, et al. Polymeric nanoparticles containing combination antiretroviral drugs for HIV type 1 treatment. AIDS Res Hum Retroviruses. 2013;29:746–754. | ||
Edagwa BJ, Guo D, Puligujja P, JoEllyn HC. Long-acting antituberculous therapeutic nanoparticles target macrophage endosomes. FASEB J. 2014;28:5071–5082. | ||
Panyam J, Zhou WZ, Prabha S, Sahoo SK, Labhasetwar V. Rapid endo-lysosomal escape of poly(DL-lactide-coglycolide) nanoparticles: implications for drug and gene delivery. FASEB J. 2002;16:1217–1226. | ||
Kumar R, Sahoo GC, Pandey K, Das V, Das P. Study the effects of PLGA-PEG encapsulated amphotericin B nanoparticle drug delivery system against Leishmania donovani. Drug Deliv. 2015;22(3):383–388. | ||
Tsuchiya S, Yamabe M, Yamaguchi Y, Kobayashi Y, Konno T, Tada K. Establishment and characterization of a human acute monocytic leukemia cell line (THP-1). Int J Cancer. 1980;26:171–176. | ||
Tsuchiya S, Kobayashi Y, Goto Y, et al. Induction of maturation in cultured human monocytic leukemia cells by a phorbol diester. Cancer Res. 1982;42:1530–1536. | ||
Pelletier M, Bouchard A, Girard D. In vivo and in vitro roles of IL-21 in inflammation. J Immunol. 2004;173:7521–7530. | ||
Binet F, Cavalli H, Moisan E, Girard D. Arsenic trioxide (AT) is a novel human neutrophil pro-apoptotic agent: effects of catalase on AT-induced apoptosis, degradation of cytoskeletal proteins and de novo protein synthesis. Br J Haematol. 2006;132:349–358. | ||
Goncalves DM, Chiasson S, Girard D. Activation of human neutrophils by titanium dioxide (TiO2) nanoparticles. Toxicol In Vitro. 2010;24:1002–1008. | ||
Lavastre V, Cavalli H, Ratthe C, Girard D. Anti-inflammatory effect of Viscum album agglutinin-I (VAA-I): induction of apoptosis in activated neutrophils and inhibition of lipopolysaccharide-induced neutrophilic inflammation in vivo. Clin Exp Immunol. 2004;137:272–278. | ||
Pelletier M, Girard D. Biological functions of interleukin-21 and its role in inflammation. Scientific World Journal. 2007;7:1715–1735. | ||
Ratthe C, Ennaciri J, Garces Goncalves DM, Chiasson S, Girard D. Interleukin (IL)-4 induces leukocyte infiltration in vivo by an indirect mechanism. Mediators Inflamm. 2009;2009:193970. | ||
Lavastre V, Pelletier M, Saller R, Hostanska K, Girard D. Mechanisms involved in spontaneous and Viscum album agglutinin-I (VAA-I)-induced human neutrophil apoptosis: VAA-I accelerates the loss of the antiapoptotic Mcl-1 expression and the degradation of the cytoskeletal paxillin and vimentin proteins via caspases. J Immunol. 2002;168:1419–1427. | ||
Kobayashi SD, Voyich JM, Whitney AR, DeLeo FR. Spontaneous neutrophil apoptosis and regulation of cell survival by granulocyte macrophage-colony stimulating factor. J Leukoc Biol. 2005;78:1408–1418. | ||
Gonçalves DM, Girard D. Titanium dioxide (TiO2) nanoparticles induce neutrophil influx and local production of several pro-inflammatory mediators in vivo. Int Immunopharmacol. 2011;11:1109–1115. | ||
Gonçalves DM, Girard D. Evidence that polyhydroxylated C60 Fullerene (Fullerenol) nanoparticles amplify the effect of lipopolysaccharides to induce a rapid leukocyte infiltration in vivo. Chem Res Toxicol. 2013;26:1884–1892. | ||
Vandooren J, Berghmans N, Dillen C, et al. Intradermal air pouch leukocytosis as an in vivo test for nanoparticles. Int J Nanomedicine. 2013;8:4745–4756. | ||
Chellat F, Grandjean-Laquerriere A, Le Naour R, et al. Metalloproteinase and cytokine production by THP-1 macrophages following exposure to chitosan-DNA nanoparticles. Biomaterials. 2005;26:961–970. | ||
Chen HW, Su SF, Chien CT, et al. Titanium dioxide nanoparticles induce emphysema-like lung injury in mice. FASEB J. 2006;20:2393–2395. | ||
Grabowski N, Hillaireau H, Vergnaud J, et al. Surface coating mediates the toxicity of polymeric nanoparticles towards human-like macrophages. Int J Pharm. 2015;482(1–2):75–83. | ||
Hussain S, Vanoirbeek JA, Luyts K, et al. Lung exposure to nanoparticles modulates an asthmatic response in a mouse model. Eur Respir J. 2011;37:299–309. | ||
Srinivas A, Rao PJ, Selvam G, Murthy PB, Reddy PN. Acute inhalation toxicity of cerium oxide nanoparticles in rats. Toxicol Lett. 2011;23:23. | ||
Brandenberger C, Rowley NL, Jackson-Humbles DN, et al. Engineered silica nanoparticles act as adjuvants to enhance allergic airway disease in mice. Part Fibre Toxicol. 2013;10:26. | ||
Nathan C. Neutrophils and immunity: challenges and opportunities. Nat Rev Immunol. 2006;6:173–182. | ||
Wright HL, Moots RJ, Bucknall RC, Edwards SW. Neutrophil function in inflammation and inflammatory diseases. Rheumatology (Oxford). 2010;49:1618–1631. | ||
Goncalves DM, Girard D. Zinc oxide nanoparticles delay human neutrophil apoptosis by a de novo protein synthesis-dependent and reactive oxygen species-independent mechanism. Toxicol In Vitro. 2014;28:926–931. |
Supplementary materials
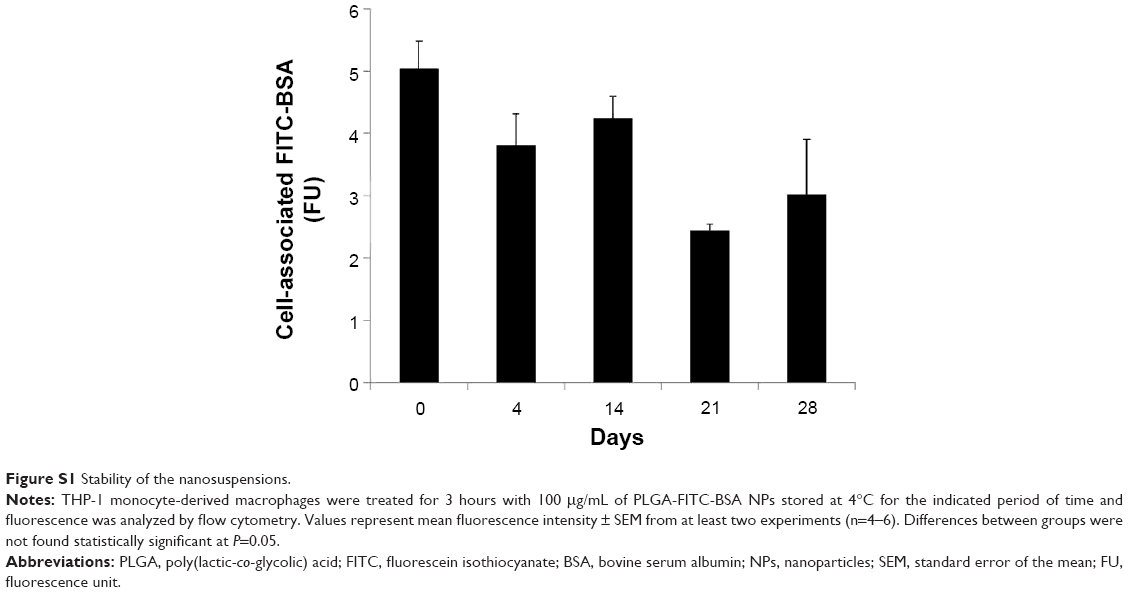 | Figure S1 Stability of the nanosuspensions. |
 | Table S1 Effects of PLGA nanoparticles on cytokine secretion |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.