Back to Journals » International Journal of Nanomedicine » Volume 15
Preparation, Characterization, and Pharmacokinetic Study of a Novel Long-Acting Targeted Paclitaxel Liposome with Antitumor Activity
Authors Han B, Yang Y, Chen J, Tang H, Sun Y, Zhang Z, Wang Z, Li Y, Li Y, Luan X, Li Q ![]() , Ren Z
, Ren Z ![]() , Zhou X, Cong D, Liu Z, Meng Q, Sun F, Pei J
, Zhou X, Cong D, Liu Z, Meng Q, Sun F, Pei J ![]()
Received 26 August 2019
Accepted for publication 15 December 2019
Published 24 January 2020 Volume 2020:15 Pages 553—571
DOI https://doi.org/10.2147/IJN.S228715
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Linlin Sun
Bing Han,1,* Yue Yang,1,2,* Jinglin Chen,1 Huan Tang,1 Yuxin Sun,1 Zheng Zhang,1 Zeng Wang,1 Yan Li,1 Yao Li,1 Xue Luan,1 Qianwen Li,1 Zhihui Ren,1 Xiaowei Zhou,1 Dengli Cong,1 Zhiyi Liu,1 Qin Meng,1 Fei Sun,1 Jin Pei1
1Department of Biopharmacy, School of Pharmaceutical Sciences, Jilin University, ChangChun, People’s Republic of China; 2Department of Pharmacy, Ministry of Health Service, Chinese PLA General Hospital, Beijing, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Jin Pei
Tel +86 431 85619725
Email [email protected]
Background: Breast cancer is the leading cause of cancer death in women. Chemotherapy to inhibit the proliferation of cancer cells is considered to be the most important therapeutic strategy. The development of long-circulating PEG and targeting liposomes is a major advance in drug delivery. However, the techniques used in liposome preparation mainly involve conventional liposomes, which have a short half-life, high concentrations in the liver and spleen reticuloendothelial system, and no active targeting.
Methods: Four kinds of paclitaxel liposomes were prepared and characterized by various analytical techniques. The long-term targeting effect of liposomes was verified by fluorescence detection methods in vivo and in vitro. Pharmacokinetic and acute toxicity tests were conducted in ICR mice to evaluate the safety of different paclitaxel preparations. The antitumor activity of ES-SSL-PTX was investigated in detail using in vitro and in vivo human breast cancer MCF-7 cell models.
Results: ER-targeting liposomes had a particle size of 137.93± 1.22 nm and an acceptable encapsulation efficiency of 88.07± 1.25%. The liposome preparation is best stored at 4°C, and is stable for up to 48 hrs. Cytotoxicity test on MCF-7 cells demonstrated the stronger cytotoxic activity of liposomes in comparison to free paclitaxel. We used the near-infrared fluorescence imaging technique to confirm that ES-SSL-PTX was effectively targeted and could quickly and specifically identify the tumor site. Pharmacokinetics and acute toxicity in vivo experiments were carried out. The results showed that ES-SSL-PTX could significantly prolong the half-life of the drug, increase its circulation time in vivo, improve its bioavailability and reduce its toxicity and side effects. ES-SSL-PTX can significantly improve the pharmacokinetic properties of paclitaxel, avoid allergic reaction of the original solvent, increase antitumor efficacy and reduce drug toxicity and side effects.
Conclusion: ES-SSL-PTX has great potential for improving the treatment of breast cancer, thereby improving patient prognosis and quality of life.
Keywords: breast cancer, estrogen receptors, paclitaxel, long-acting liposomes, targeted drug delivery
Introduction
Cancer is a complex and highly heterogeneous disease. Globally, breast cancer is the most common type of cancer and results in the most fatalities.1 Breast cancer is chemosensitive;2 thus, chemotherapy to inhibit the proliferation of primary cancer cells is considered to be the most important therapeutic strategy.3 Taxanes are one of the most effective classes of anticancer drugs in the clinic.
Paclitaxel (PTX) is a first-line treatment for breast cancer.4–7 Polyoxyethylene castor oil and absolute ethanol are used clinically (50:50, v/v) as a vehicle for intravenous PTX injections.8,9 The solvent significantly increases the solubility of PTX, however, it has been reported to cause allergic reactions, neurotoxicity, nephrotoxicity, and other toxic side effects,10–12 greatly limiting the clinical application of PTX. Additionally, like other broad-spectrum anticancer drugs, PTX has several limitations that seriously affect its clinical efficacy; it is not selectively distributed in vivo, has a short circulation half-life, and causes damage to normal cells while killing cancer cells.
The use of liposomes as drug carriers for chemotherapy was first proposed in 1974 by Gregoriadiset al13 and liposomes are regarded as good candidates because of their safety, size controllability, and capability for easy functionalization.14 Cisplatin and other chemotherapeutic drug-liposome preparations are approved by the FDA for clinical use in tumor therapy.15 The study of polyethylene glycol (PEG) long-circulating liposomes began in 1990.16 Because binding PEG to proteins was demonstrated to prolong their half-life in vivo, researchers have tried to determine if PEG conjugation could also extend the half-life of liposomes.17,18 The development of long-circulating PEG liposomes is a major advance in drug delivery, particularly tumor drug delivery. Kim et al showed that PEG-modified liposomes have enhanced anticancer effects and in vivo stability.19 The PEG chain bound to the lipid bilayer membrane enables the liposome to persist longer because the hydrophilic long chain of PEG can form a barrier on the surface of the liposome. This reduces the mutual polymerization between the liposomes and increases their stability. The PEG-modified liposomes have spatial stability and reduced adsorption by opsonized proteins in the blood. In addition, PEG can help liposomes escape capture by the reticuloendothelial system, greatly improving the stability of the liposomes in the vascular system, thereby reducing the distribution in important tissues and organs, and reducing drug toxicity.20 However, the techniques used in liposome preparation mainly involve conventional liposomes, which have a short in vivo half-life, high concentrations in the liver and spleen reticuloendothelial system, and no active targeting.21,22
Estrogen receptors (ERs) are powerful targets for receptor-mediated delivery of PTX to breast cancer cells. ERs are members of the nuclear receptor superfamily that mediate the multidirectional effects of estrogen and have a wide range of roles in various developmental and physiological processes.23–25 ERs were first identified in the late 1960s based on their ability to bind radiolabeled estrogens.26–28 Over the following decade, several different methods were developed to detect their expression in clinical breast cancer samples, and they became widely used as a predictor of hormone stress and the clinical invasiveness of tumors.29 Studies have shown that in breast cancer ERs are overexpressed by 60–80%.30 Estrone (ES)-conjugated liposomes loaded with doxorubicin (DOX) potentiated the delivery of DOX to cancer cells overexpressing ERs.31,32 A biodistribution test in female rats showed that the concentration of ES-conjugated liposomes in ER-positive cells (such as from the breast and uterus) was significantly increased; the concentration of DOX in the breast was 13.9 and 11.05 times higher than that of free DOX or DOX loaded in untargeted liposomes, respectively. Additionally, the concentration of DOX in myocardial tissue was significantly lower than that of free DOX.23 These studies suggest that ERs can be used to target breast cancer.
Here, we synthesized ER-targeting molecules and loaded them into conventional liposomes to develop and characterize a new, high-efficiency, low-toxicity liposome nanopreparation with long-term circulation and tumor-targeting characteristics. We also explored its ability to target breast cancer cells. We investigated the antitumor effect of this new PTX liposome on breast cancer in vitro and in vivo. Data from pharmacokinetic studies provides important evidence for the further reduced toxicity of PTX, which will help to improve the drug treatment index and compliance of patients with clinical cancer, in addition to facilitating its use by clinical staff.
Materials and Methods
Materials
The following reagents, drugs, and chemicals were used in this study: Paclitaxel (PTX, Dalian Meilun Biotechnology Co., Ltd.), Soy lecithin (PC, Tianjin Guangfu Fine Chemical Research Institute), Cholesterol (Chol, Shanghai Huishi Biochemical Reagent Co., Ltd.), distearoylphosphatidylethanolamine-oxypolyethylene glycol 2000 (DSPE-PEG2000, Shanghai Xibao Biotechnology Co., Ltd.), distearoylphosphatidylethanolamine-polyethylene glycol 2000-estrone (DSPE-PEG2000-ES, laboratory synthesis), chloroform (Beijing Chemical Plant), sodium hydroxide (Beijing Chemical Plant), sodium diethyldithiocarbamate (DDTC, Tianjin Tiantai Fine Chemicals Co., Ltd.), Rhodamine B (Tianjin Guangfu Fine Chemical Research Institute), DiR (Beijing Fanbo Biochemical Company), Thiazole Blue (MTT, MYM Biotechnology Co., Ltd.), and Hoechst 33342 (Sigma). All the reagents were of analytical or HPLC grade.
Cells
Human breast cancer MCF-7 cells (purchased from Wuhan Biotechnology Co., Ltd.) were cultured in DMEM high-glucose medium containing 10% fetal bovine serum (FBS) and cyan/streptomycin (0.1 and 0.06 mg/mL, respectively). The cells were cultured in a cell culture incubator at 37°C containing 5% CO2 humidified air.
Animals
Female BALB/c nude mice, aged 4 to 5 weeks old and weighing 20 ± 2 g, were purchased from Beijing Vital River Laboratory Animal Technology Co., Ltd. (animal license number: SCXK Beijing 2009–0004 (Beijing, China)). Female ICR mice, aged 6–8 weeks and weighing 30±2 g, were purchased from the Experimental Animal Center of the Basic Medical College of Jilin University. All mice were reared indoors with a 12 hr alternating light-dark cycle. All operation procedures were in accordance with the requirement of the ethical approval by the Ethics Committee of the School of Pharmaceuticals Sciences, Jilin University (Approval No. 20120010). All the animal studies have been conducted strictly under the guidelines of “National Animal Management Regulations of China”, and approved by the Animal Ethics Committee of the School of Pharmaceuticals Sciences, Jilin University.
Synthesis of Estrogen Receptor-Targeting Fragments
The synthesis of estrogen receptor (ER)-targeting fragments had two steps. First, estrone succinic acid monoester was synthesized from the starting materials estrone (ES) and succinic anhydride under the catalysis of DMAP. Next, estrone succinic acid monoester and DSPE-PEG2000-NH2 were used to synthesize the estrogen receptor-targeting fragment DSPE-PEG2000-ES in the presence of DCC (Figure 1).
 |
Figure 1 The synthetic route of targeting fragment DSPE-PEG2000-ES. |
Liposome Preparation
Preparation of PTX Liposomes
Four PTX liposome preparations33 were generated by the membrane hydration method: common PTX liposomes (L-PTX; soy lecithin phospholipid: cholesterol: PTX, molar ratio of 9:6:1), targeted PTX liposomes (ES-L-PTX; soy lecithin phospholipid: cholesterol: PTX: ES-PEG2000-DSPE, molar ratio of 9:6:1:0.075); long-acting PTX liposomes (SSL-PTX; soy lecithin phospholipid: cholesterol: PTX: mPEG2000-DSPE, molar ratio of 9:6:1:0.75), and targeted long-acting PTX liposomes (ES-SSL-PTX; soy lecithin phospholipid: cholesterol: PTX: mPEG2000-DSPE: ES-PEG2000-DSPE, molar ratio of 9:6:1:0.75:0.075). Soy lecithin, cholesterol, PTX, mPEG2000-DSPE and ES-PEG2000-DSPE were weighed and added to a rotary evaporation flask according to the above molar ratios and dissolved in chloroform. The lipid film was formed by rotary evaporation under vacuum conditions using a rotary evaporator at a rotational speed of 90 to 120 rpm, ensuring that no chloroform solvent remained. Phosphate-buffered saline (PBS) was added to fully hydrate the film at room temperature. A vortexer was used to disperse the lipid film evenly. After the liposome suspension was sonicated in an ice water bath, the liposomes were passed through a 220 nm polycarbonate membrane 5 times and an 80 nm polycarbonate membrane 5 times using a high-pressure filtration extruder. As a result, a PTX liposome solution with a uniform particle size was obtained.
Preparation of Rhodamine B Liposomes and DiR Liposomes
Four rhodamine B (RhB) liposome formulations were prepared by membrane hydration.34 Precision weighed phosphatidylcholine (PC), cholesterol (Chol), mPEG2000-DSPE, and RhB (molar ratio of 11:9:1:0.67) were added to a rotary evaporation flask and dissolved in chloroform. The liposome suspension preparation was performed as described in section of preparation of PTX liposomes. The ES-PEG2000-DSPE target fragment was incubated with the obtained liposome suspension in a 37°C water bath at a ratio of 1:200 (fragment to total lipid) to obtain RhB long-acting targeted liposomes (ES-SSL-RhB). The RhB long-acting liposomes (SSL-RhB) were prepared in the same manner as ES-SSL-RhB, but without the addition of the targeting fragment ES-PEG2000-DSPE. The RhB-targeted liposomes (ES-L-RhB) were prepared using the same method used for ES-SSL-RhB, but the long-acting fragment, mPEG2000-DSPE, was not added. The RhB common liposome (L-RhB) was prepared using the same method as ES-L-RhB, but the targeting fragment, ES-PEG2000-DSPE, was not added.
DiR long-acting targeted liposomes (ES-SSL-DiR; PC, Chol, mPEG2000-DSPE, ES-PEG2000-DSPE, and DiR at a molar ratio of 11:9:1:0.1:0.02) and DiR long-acting liposomes (SSL-DiR; PC, Chol, mPEG2000-DSPE, and DiR at a molar ratio of 11:9:1:0.02) were prepared using the membrane hydration method,34 as described in section of preparation of PTX liposomes.
Liposome Characterization
Encapsulation Efficiency (EE) and Drug-Loading Capacity (DL) Determination
Due to PTX’s poor solubility in water, free PTX35 that was not loaded into the liposomes could be removed by low-speed centrifugation. To break the liposomes, methanol was added to 50 μL of PTX liposome solution, followed by sonication for 5 min. The total PTX content was determined by HPLC. 0.1 mL of PTX liposomes were mixed with an appropriate amount of PBS solution and centrifuged at 1000 rpm for 10 min. The insoluble free PTX precipitated and the supernatant contained homogeneous PTX liposomes. The supernatant was collected, de-emulsified by adding methanol solution, and sonicated for 5 min to completely break the liposomes. The PTX content was determined by HPLC, which allowed us to determine the mass of PTX loaded into the liposomes.

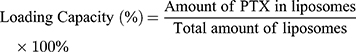
The chromatographic conditions were based on previously published methods36 and described below. Column: Shimadzu ODS C-18 column (250 mm × 4.6 mm, 5 μm), column temperature: 25°C, mobile phase: acetonitrile-water (60:40, v/v), detection wavelength: 227 nm, flow rate: 1.0 mL/min, injection amount: 10 μL.
Determination of Liposome Particle Size, Polydispersity Index (PDI) and Zeta Potential
Each type of PTX liposome solution (1mL) was placed in a sample cell, and the particle size, polydispersity coefficient (representing the uniformity of particle size distribution) and zeta potential were measured at 25°C using the Malvern Nano-ZS90 dynamic light scattering particle size analyzer.
Observation of Liposome Morphology
The ES-SSL-PTX liposome solution, described above, was diluted 3 times with PBS buffer at room temperature. The sample was carefully dropped onto a pure carbon film copper mesh, which gave the droplets a hemispherical liquid surface. After drying, the solution was dyed using phosphotungstic acid solution;37,38 the dyeing agent was blotted around the copper mesh using filter paper. Ultrapure water was then added dropwise to the copper mesh, followed by blotting dry with filter paper. After air-drying, the sample imaged using a transmission electron microscope (TEM) (JEM-2100F).
Liposomal Stability
The four liposome preparations were stored at both 4°C and room temperature, and the appropriate amount was collected at 0, 1, 2, 4, 8, 24, and 48 h. Free PTX was removed by centrifugation as described in section of encapsulation efficiency and drug-loading capacity determination and characterized by HPLC. The PTX concentration in the supernatant liposomes was recorded as “Cn” (where n is each time point).
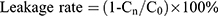
Cytotoxicity Test
The toxicity of different liposome preparations on MCF-7 cells was determined using thiazolyl blue colorimetry (MTT colorimetry). Cell suspensions were prepared by trypsinizing MCF-7 cells in the logarithmic growth phase. Cells were then counted and seeded into 96-well plates at a density of 1×104 cells/well. Cells were grown to confluency at 5% CO2, and 37°C for 24 h. Seven different concentrations (0.1, 1, 10, 100, 500, 1000, and 5000 ng/mL) of free PTX, L-PTX, ES-L-PTX, SSL-PTX and ES-SSL-PTX diluted with DMEM medium were added to the cultured cells. After continuous culture for 24, 48, and 72 h, 20 µL of MTT solution (5 mg/mL) were added to each well. After 4 h, the incubation was terminated. The culture medium was carefully aspirated and 150 μL of dimethyl sulfoxide was added. The mixture was shaken at low speed. After 10 mins, the crystals were sufficiently dissolved. The absorbance value (OD) of each well was measured at a wavelength of 492 nm with a microplate reader. The cell survival rate curve was plotted and the IC50 value of each group was calculated by the following equation:
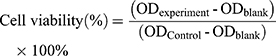
ODblank is the OD of the blank well containing only medium; ODControl is the OD of the control cell well; and ODexperiment is the OD of the experimental well.
Determination of Cell Intake and Endocytosis Mechanisms
Fluorescence microscopy was used to observe the uptake of rhodamine liposomes by MCF-7 cells. MCF-7 cells were seeded 2×105 cells/well in 24-well plates and incubated at 5% CO2 and 37°C for 24 h until 60–80% confluence was reached. Media containing L-RhB, ES-L-RhB, SSL-RhB, and ES-SSL-RhB was separately added to a final concentration of 5 μg/mL RhB, and the cells were further incubated for 1, 2, 3, or 4 h, respectively. After incubation, the media was aspirated and the cells were washed three times with PBS buffer. Cell nuclei were then stained with Hoechest 33342 nuclear stain and observed using a fluorescence microscope.34
To verify the targeting of ES-SSL-RhB, MCF-7 cells were incubated for 0.5 hrs with excess free ES (at a final concentration of 5×10−4 M) before the addition of ES-SSL-RhB solution, blocking the surface estrogen receptors on the MCF-7 cells. After washing the cells three times to remove excess ES, the ES-SSL-RhB solution was added. The MCF-7 cells were then incubated at 37°C for 2 h and observed using a fluorescence microscope.
To investigate the endocytosis of ES-SSL-RhB, MCF-7 cells were seeded 2×105 cells/well in 24-well plates and incubated at 5% CO2 and 37°C for 24 h, at which point 100% confluence was achieved. Each well was then incubated with sucrose (final concentration 0.45 M), genistein (final concentration 0.2 M), amiloride hydrochloride (final concentration 0.01 M), and PBS buffer for 0.5 h. Media was then aspirated and each well was incubated for 2 hrs with medium containing 5 μg/mL ES-SSL-RhB.
In vivo Targeting Study
Healthy female BALB/c nude mice (aged 5–6 weeks, weighing 18–22 g) were inoculated with 1×107 MCF-7 cells in the second pair of right breast fat pads and used for in vivo imaging experiments. The tumor volume was measured:
Tumor volume (mm3) = (a × b2)/2, where “a” is the longest diameter of the tumor, and “b” is the shortest diameter of the tumor.
After the average tumor volume reached 100 mm3, the tumor-bearing mice were randomly divided into two groups. DiR liposomes (SSL-DiR or ES-SSL-DiR), at a dose of 100 ng/g, were injected into the tail veins of the MCF-7 cell-bearing mice. At 1, 2, 6, 12, and 24 h after injection of DiR liposomes, the corresponding tumor-bearing mice were given an intraperitoneal injection of 10% chloral hydrate at a dose of 0.004 mL/g. The mice were then imaged using a live imaging system (excitation wavelength: 710 nm, emission wavelength: 780 nm) (IVIS SPECTRUM, USA). The mice were sacrificed and the hearts, livers, spleens, lungs, kidneys and tumors were taken out, observed fluorescence imaging results. All operation procedures were in accordance with the requirement of the ethical approval.
Pharmacokinetics
Eight-week-old female ICR mice were weighed and received tail vein injections of: PTX (at a standard dose of 10 mg/kg), L-PTX, and ES-SSL-PTX. Blood was collected from the eye socket of mice into tubes containing heparin sodium at 5 min, 10 min, 15 min, 30 min, 1 h, 2 h, 4 h, 6 h, and 8 h (n=3 per group, total of 9 groups) from the PTX injected group. Collection times for the L-PTX group were 5 min, 10 min, 15 min, 30 min, 1 h, 2 h, 4 h, 6 h, 8 h, 12 h, and 24 h (n=3 per group, total of 11 groups), and collection times for the ES-SSL-PTX group were 5 min, 10 min, 15 min, 30 min, 1 h, 2 h, 4 h, 6 h, 8 h, 12 h, 24 h, 36 h, and 48 h (n=3 per group, total of 13 groups). Blood was centrifuged at 3000 rpm for 10 min in a low-temperature centrifuge, and stored at −20°C. A total of 200 μL of plasma was transferred into a 5 mL centrifuge tube. 10 µL of 100 μg/mL norethisterone internal standard and 2 mL of methyl tert-butyl ether were added and the mixture was vortexed for 2 min. The supernatant was taken after centrifugation and air-dried with nitrogen at 40°C. For detection, 200 μL of mobile phase (acetonitrile: water = 60:40) was added until dissolved and the sample was vortexed for 1 min. 20 μL was collected for injection under the HPLC conditions described in section of encapsulation efficiency and drug-loading capacity determination. Normal organs (including heart, liver, spleen, lung and kidney) were taken out, washed with cold saline, dried over filter paper, weighed and stored at −20 °C before analysis. The organs were homogenized in normal saline solution. The processing and detection methods were the same as above. All operation procedures were in accordance with the requirement of the ethical approval.
In vivo Antitumor Assessment
The in vivo antitumor effects of PTX liposomes were evaluated using tumor-bearing nude mice. Female BALB/c nude mice weighing 16–18 g were inoculated with 1×107/0.2 mL MCF-7 cell suspension into the second pair of breast fat pads. The mice were randomly divided into 7 groups, with 6 mice in each group: (1) saline group (model group), (2) blank liposome group, (3) PTX free drug group, (4) PTX liposome group, (5) long-acting PTX liposome group, (6) targeted PTX liposome group, and (7) long-acting targeted PTX liposome group. PTX was administered to each group at a dose of 5 mg/kg body weight via tail vein injection once every 2 days for 4 consecutive administrations. The mice were monitored each day, and body weight was measured. The tumor volume was measured twice a week, and the survival time was recorded at 34 days. In the model group, the tumor diameter reached 15 mm. The mice were then sacrificed, blood collection from mouse orbit. The tumors of mice were photographed, and a tumor growth curve was plotted. All operation procedures were in accordance with the requirement of the ethical approval.
Acute Toxicity Test
160 ICR mice (an equal number of males and females, weighing 24–30 g) were divided into the following 4 groups randomly according to gender and body weight: PTX injection group (50 mice), L-PTX group (50 mice), ES-SSL-PTX group (50 mice), and normal saline group (10 mice).
50 ICR mice were treated with PTX injection. These mice were randomly divided into 5 groups (10 mice per group) which received the following doses: 20 mg/kg, 25 mg/kg, 30 mg/kg, 35 mg/kg, and 40 mg/kg. The mice were weighed and the drug was administered via tail vein injection.
There were 50 L-PTX ICR mice that were randomly divided into 5 groups (10 mice per group) receiving the following doses: 80 mg/kg, 100 mg/kg, 130 mg/kg, 160 mg/kg, and 200 mg/kg (the dose ratio between doses was 1:0.6). The mice were weighed and the drug was administered via tail vein injection. Administration of drug to ES-SSL-PTX ICR mice was performed in the same manner as with L-PTX ICR mice.
10 ICR mice (an equal number of males and females, weighing 24–30 g) were treated with saline as a control group. The mice were weighed and 0.4 mL of physiological saline was injected into the tail vein.
The mice were monitored the day after administration, and the time of death or symptom onset was recorded. The mice were observed every day to carefully track lethality, and the surviving mice were weighed. Observation continued for 14 days in order to determine if there was any longer term toxicity. On the 14th day after drug administration (AAD14), whole blood was collected from the eyeball of the surviving mice for measurement of hematological parameters using a whole blood analyzer. The heart, liver, spleen, lung, and kidney were dissected and weighed. All operation procedures were in accordance with the requirement of the ethical approval.
Statistical Analysis
Statistical analysis was performed using GraphPad Prism 5.0 software. Intergroup t-test comparisons were performed using the mean ± standard deviation (x ± s), and P < 0.05 was considered statistically significant.
Results and Discussion
Liposome Characterization
Particle Size, PDI, Zeta Potential, and TEM
The particle sizes, PDIs, and zeta potentials of various PTX liposomes were shown in Table 1, Figure 2A–D. The particle size distribution of the four preparations was uniform and normalized to a single-peak distribution. The PDI ranged from 0.1 to 0.2, and the surface liposome particle size distribution range was narrow. The average particle size of L-PTX was 119.10 nm, and the average particle size of ES-L-PTX was 125.57 nm. Introduction of the targeting fragment, ES-PEG2000-DSPE, slightly increased the average particle size. The average particle sizes of SSL-PTX and ES-SSL-PTX were 137.93 nm and 135.93 nm, respectively. The increase in particle size compared with L-PTX and ES-L-PTX is due to the introduction of the long-acting fragment, mPEG2000-DSPE; the PEG chain forms a protective layer on the surface of the liposomes, which increases their particle size.
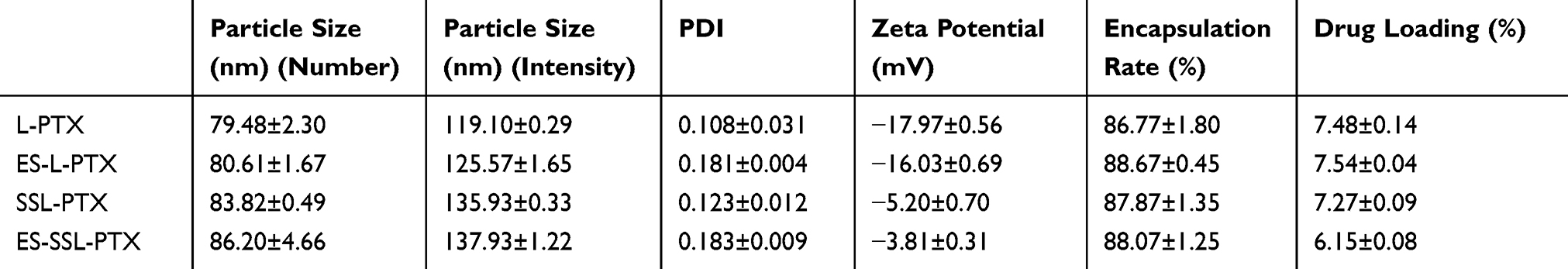 |
Table 1 Physicochemical Characteristics of PTX Liposomes |
The zeta potentials of L-PTX, ES-L-PTX, SSL-PTX, and ES-SSL-PTX were all negatively charged, thus, electrostatic repulsion is preventing the particles from accumulating, a beneficial feature for preserving liposomes. The L-PTX potential was −17.97 mV, and the ES-L-PTX potential was −16.03 mV. Introduction of the targeting fragment, ES-PEG2000-DSPE, had no significant effect on the zeta potential. The zeta potentials of SSL-PTX and ES-SSL-PTX were −5.20 mV and −3.81 mV, respectively; the introduction of the long-acting fragment reduced the zeta potential.
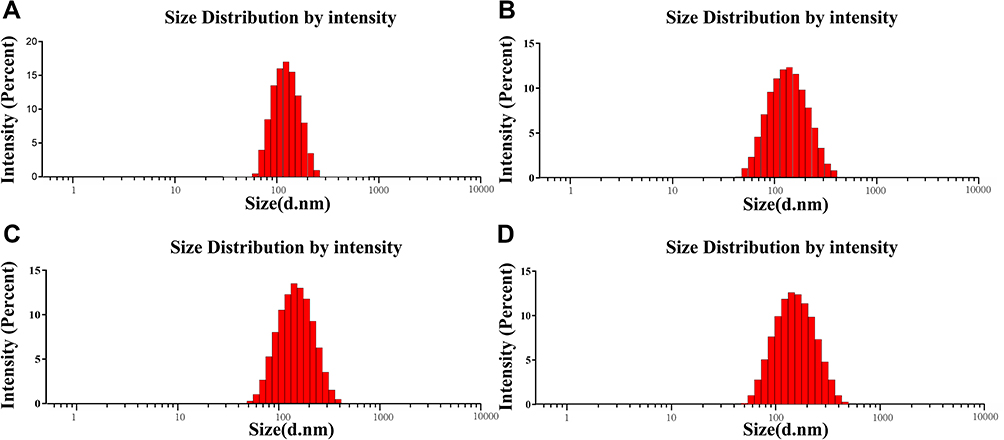 |
Figure 2 PTX liposome particle size distribution. Liposome particle sizes (L-PTX (A), ES-L-PTX (B), SSL-PTX (C), and ES-SSL-PTX (D), n=3 per group) were measured at 25°C using a Malvern Nano-ZS90 dynamic light scattering particle size analyzer. |
Encapsulation Efficiency and Drug Loading
As shown in Table 1, the encapsulation efficiency of each of the four liposome preparations (L-PTX, ES-L-PTX, SSL-PTX, and ES-SSL-PTX) was between 86.0% and 88.0%, and there was no difference in encapsulation efficiency among the preparations. When the targeting fragment, ES-PEG2000-DSPE, or the long-acting fragment, mPEG2000-DSPE, were introduced by the direct introduction method, (liposomes are formed together with phospholipids and cholesterol), there was no change in the encapsulation efficiency or the amount of drug loaded.
Liposomal Stability
As shown in Figure 3A, the leakage rate of the four PTX liposomes stored at room temperature for 48 hrs was approximately 20%, suggesting that the liposome preparations were relatively stable. After storage for 48 hrs at 4°C (Figure 3B), the leakage rate of the four liposomal formulations was less than 10%. Therefore, the liposome preparation is best stored at 4°C, and is stable for up to 48 hrs. The leakage rates of ES-SSL-PTX and SSL-PTX were lower than the leakage rates of ES-L-PTX and L-PTX, suggesting that the introduction of long-acting fragments could improve the stability of liposomes. The introduction of targeting fragments did not affect the liposome stability.
 |
Figure 3 PTX liposome stability. Stability curves (n = 3 per group) for each of the different PTX formulations at 25°C (A) and at 4°C (B). Liposomes were collected at 0, 1, 2, 4, 8, 24, and 48 h, and free PTX was removed by centrifugation. The PTX concentration in the liposomes was determined by HPLC. |
Cytotoxicity Test
As shown in Figure 4, the blank liposome group was not cytotoxic to MCF-7 cells, and all five PTX preparations had some concentration- and time-dependent inhibitory effects on MCF-7 cells. The toxicity of five PTX preparations on MCF-7 cells was similar after 24 hrs (Figure 4A), and none of them reached IC50. After prolonged incubation for 48 (Figure 4B) or 72 h (Figure 4C), the cell survival rate of each group decreased. The IC50 values of the five different PTX preparations were significantly different (Table 2). The toxicity of the five PTX preparations to MCF-7 cells was as follows, from strongest to weakest: ES-SSL-PTX, SSL-PTX, ES-L-PTX, L-PTX, and PTX. The IC50 values of the four PTX liposomal preparations and free PTX were significantly different at 72 h (P < 0.01). This is because the liposomes had high affinity with the cell membrane, and the PTX that was encapsulated in the liposomes entered the cells, exerting an antitumor effect. Over time, SSL-PTX and ES-SSL-PTX showed more cytotoxicity than ES-L-PTX and L-PTX. At 48 and 72 h, the IC50 values of SSL-PTX were significantly different from the IC50 values of L-PTX (P < 0.05) because the long-acting fragments included in SSL-PTX and ES-SSL-PTX lengthened their metabolism by the cells, which increased the accumulation of drug in tumor cells and enhanced cell inhibition. The inhibitory effect of ES-SSL-PTX on the cells was significantly higher than that of other preparations. The IC50 value of ES-SSL-PTX was the lowest, at 0.31±0.05 ng/mL. This was 36 times lower than the IC50 value of L-PTX, 19 times lower than that of ES-L-PTX, and 12 times lower than that of SSL-PTX, all of which were significantly different (P < 0.01). Because ES-SSL-PTX could specifically target the estrogen receptor on the surface of MCF-7 cells, it could be recognized and internalized more quickly. The acting time on tumor cells is prolonged, and the antitumor effect is significantly improved.
 |
Figure 4 MCF-7 cell survival. Cell survival curves after treating MCF-7 cells with different PTX preparations for 24 h (A), 48 h (B), and 72 h (C). *P < 0.05, **P < 0.01 compared with L-PTX. |
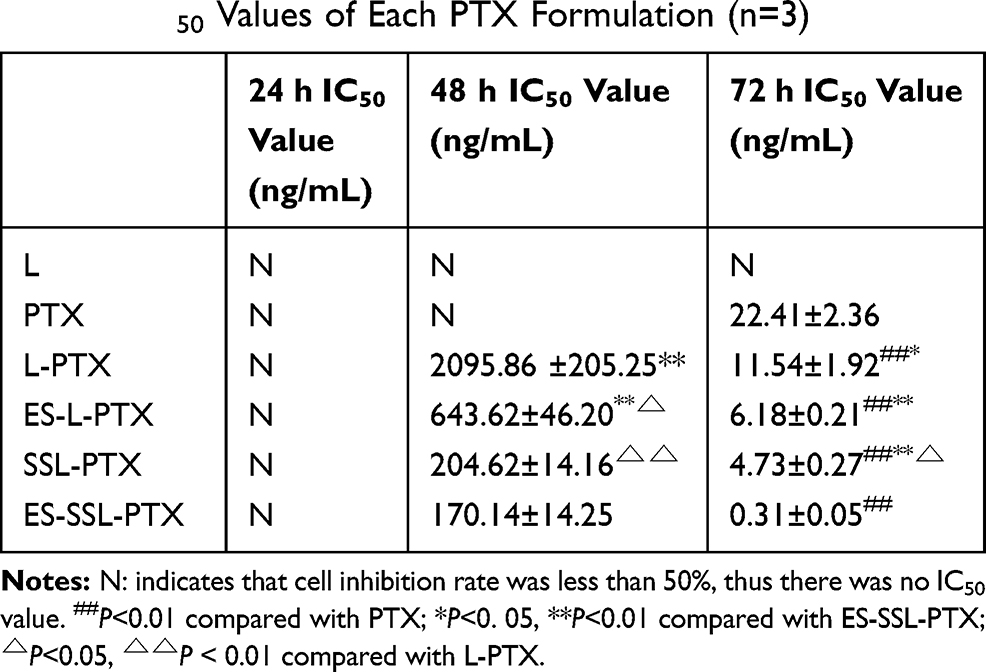 |
Table 2 IC50 Values of Each PTX Formulation (n=3) |
Cell Uptake Assay
As shown in Figure 5A and B, free RhB had the strongest fluorescence intensity at 1 and 2 hrs, and decreased at 3h and 4h, because it entered the cells by diffusion and metabolized quickly. The fluorescence intensity of L-RhB and ES-L-RhB both increased at first, but then decreased, which may be attributed to the cell metabolism effect. The fluorescence intensity of SSL-RhB and ES-SSL-RhB gradually increased from 1 to 4 h because the long-acting fragment, mPEG2000-DSPE, has a steric protection effect on liposomes, which prolongs their activity in cells.
Given the same incubation time, the fluorescence intensity of ES-L-RhB and ES-SSL-RhB was significantly higher than that of free RhB. Compared with L-RhB, the fluorescence intensity of ES-L-RhB was significantly higher from 2 to 4 h, the result of ES-SSL-RhB compared with SSL-RhB was the same as this. The estrone-targeting fragment, incorporated as a liposome surface modification, can specifically recognize and bind to estrogen receptors on the surface of MCF-7 cells, enter the cell through estrogen receptor-mediated endocytosis, and increase uptake of the liposomes.
As shown in Figure 6A–C, when free ES was added as a competitive inhibitor of ES-SSL-RhB, the fluorescence intensity in the cells was significantly reduced. Excessive free ES binds to estrogen receptors on the surface of MCF-7 cells, blocking the recognition and binding of the ES fragments on the surface of the liposomes (i.e., ES-SSL-RhB with estrogen receptors), thereby inhibiting the binding of ES-SSL-RhB to the receptors. Blocking the targeting of ES-SSL-RhB to the estrogen receptor greatly reduced the uptake of ES-SSL-RhB by MCF-7 cells.
 |
Figure 5 (A) Fluorescent images showing uptake of different liposome preparations by MCF-7 cells after 1–4 h of incubation. (B) The integrated fluorescent intensity of Figure 5A. ΔP<0.05, ΔΔP<0.01, ΔΔΔP<0.001, ΔΔΔΔP<0.0001 compared with RhB; *P<0.05, ***P<0.001, ****P<0.0001 compared with L-RhB; ###P<0.001 compared with SSL-RhB. |
 |
Figure 6 ES-SSL-RhB uptake by MCF-7 cells after addition of ES. (A) MCF-7 cells after incubation with ES-SSL-RhB solution for 2 h at 37°C. (B) Free ES was added to the MCF-7 cells for 0.5 h before adding ES-SSL-RhB solution (final concentration of 5×10−4 M) to block estrogen receptors on the surface of MCF-7 cells. After washing with medium three times to remove excess ES, ES-SSL-RhB solution was added, and MCF-7 cells were incubated at 37°C for 2 h. Cells were then imaged using a fluorescence microscope. (C) The integrated fluorescent intensity of Figure 6A and B. *P<0.05 compared with ES-SSL-RhB+ES. |
Mechanism of Endocytosis
Amiloride can inhibit cell macropinocytosis, as shown in Figure 7B, the fluorescence intensity was significantly reduced after the addition of amiloride (as compared to cells without amiloride, Figure 7A and E). This indicates that the uptake of ES-SSL-RhB by the cells was reduced, which suggests that ES-SSL-RhB enters MCF-7 cells mainly via macropinocytosis. Genistein inhibits tyrosine kinase activation, which thereby inhibits caveolin-mediated endocytosis. As shown in Figure 7C and E, after adding genistein, the fluorescence intensity was reduced after adding genistein, indicating that the uptake of ES-SSL-RhB by MCF-7 cells was reduced. This demonstrates that ES-SSL-RhB enters MCF-7 cells via caveolin-dependent endocytosis. Sucrose can inhibit endocytosis by capturing or agglomerating clathrin, thereby inhibiting clathrin-dependent endocytosis. As shown in Figure 7D and E, the fluorescence intensity was decreased when sucrose was added, compared to samples without sucrose. The uptake of ES-SSL-RhB by MCF-7 cells was reduced by sucrose, but compared with amiloride, the inhibition was weaker, indicating that ES-SSL-RhB enters MCF-7 cells via clathrin-dependent endocytosis, but this is not the primary pathway.
 |
Figure 7 Inhibition of ES-SSL-RhB uptake. MCF-7 cells were treated with (A) PBS buffer, (B) amiloride hydrochloride (final concentration 0.01 M), (C) genistein (final concentration 0.2 M), or (D) sucrose (final concentration 0.45 M), for 0.5 h. Then, media containing 5 μg/mL ES-SSL-RhB were added and cells were incubated for an additional 2 h. (E) The integrated fluorescent intensity of Figure 7A–D. **P<0.01, ***P<0.001, ****P<0.0001 compared with control. |
In vivo Targeting Study
To detect the metabolic distribution of long-acting targeted liposomes in vivo, we used the near-infrared fluorescence imaging technique. PTX liposomes and long-acting targeted liposomes mainly accumulated in the liver, spleen, and kidneys. As shown in Figure 8, after administration of SSL-DiR and ES-SSL-DiR, fluorescence intensity began to increase in the liver at 1 h, suggesting aggregation in the liver with time. ES-SSL-DiR began to accumulate in the tumor at 6 h, and peaked at 12 h. Figure 9 showed the fluorescence imaging results in ex vivo, which more clearly demonstrated the distribution of liposomes in various organs. SSL-DiR was notably concentrated in the spleen and liver reticuloendothelial system from 1 to 24 h and accumulated at the tumor site at 24 h. ES-SSL-DiR accumulated at the tumor site within 1 h. The accumulation at the tumor site peaked at 12 h and was significantly higher than the fluorescence intensity of SSL-DiR at the tumor site. This confirmed that ES-SSL-DiR was effectively targeted and could quickly and specifically identify the tumor site. Increased drug accumulation at the tumor site was important for improving drug efficacy.
 |
Figure 8 In vivo imaging of MCF-7 tumor-bearing mice. SSL-DiR and ES-SSL-DiR (at a dose of 100 ng/g of DiR) were administered to mice via tail vein injection. At 1, 2, 6, 12, and 24 h, one mouse per group was anesthetized by intraperitoneal injection of 10% chloral hydrate at a dose of 0.004 mL/g. Fully anesthetized mice were imaged using the in vivo imaging system. |
 |
Figure 9 Imaging organs from MCF-7 tumor-bearing mice. The mice described in Figure 8 were sacrificed after being imaged, and their hearts (lower left), livers (lower right), spleens (upper right), lungs (lower middle), kidneys (upper left), and tumors (middle) were dissected out and imaged ex vivo using the in vivo imaging system. |
Pharmacokinetics
As shown in Figure 10A, the free PTX was rapidly removed from circulation, and was undetectable in plasma after ~8 h. The blood concentration of PTX in liposomal formulations was significantly increased compared to that of free PTX. Most importantly, the plasma concentration of ES-SSL-PTX at each time point was significantly higher than that of the L-PTX group. The PTX could be detected till the time of 48 hrs after the administration in ES-SSL-PTX group.
In addition, the elimination half-lives of PTX, L-PTX, and ES-SSL-PTX were 1.79, 2.26, and 20.98 hrs, respectively (Table 3). The elimination half-life of ES-SSL-PTX was significantly prolonged; it was 11.1 and 9.3 times higher than that of free PTX and L-PTX, respectively. The area under the curve (AUC0-∞) of ES-SSL-PTX was 107.69 mg/L×h, indicating a 5.2-fold and 3.8-fold higher bioavailability than free PTX and L-PTX, respectively. In addition, the clearance rate (CL) of ES-SSL-PTX was significantly reduced, and the mean residence time (MRT) was significantly prolonged. This is related to the presence of the long-acting PEG molecules in liposomes. The PEG chain forms a barrier on the surface of the liposomes, which reduces their adsorption by opsonin in the blood. It can also help the liposomes escape capture by the reticuloendothelial system, greatly improving their stability in the circulatory system. The results of our pharmacokinetic experiments show that ES-SSL-PTX can significantly prolong the drug’s blood residence time, maintaining a higher blood concentration for a long time and improving bioavailability.
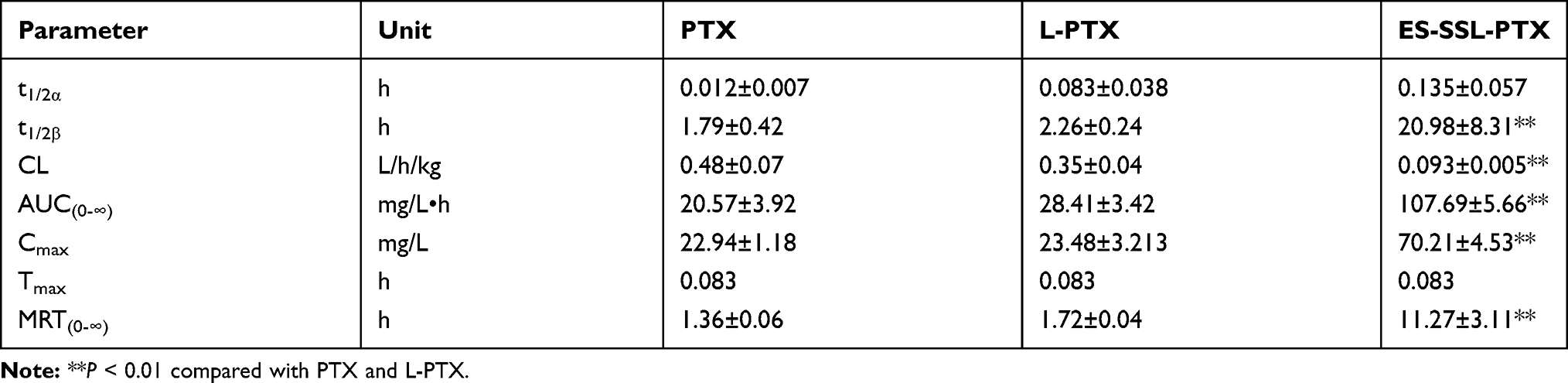 |
Table 3 Pharmacokinetic Parameters of PTX Preparations |
Concentration of PTX was determined in mice various tissues including heart, liver, spleen, lung and kidney at 0.5 and 8h after a single dose intravenous administration of PTX liposomal and injectable formulation and bio-distribution study results are shown in Figure 10B, C and Table 4. Notably, PTX concentration in spleen and liver of ES-SSL-PTX group was much higher than PTX group, it may be that the amount of drug entering the systemic circulation of ES-SSL-PTX was significantly higher than that of L-PTX and PTX injections. Furthermore, the PTX concentration in mice other tissues including heart, kidney, lung, treated mice at 8 h almost maintained at almost an equal low level compared with that at 0.5 h, showing reduced the distribution of drugs in normal tissues and organs and reduced drug toxicity.
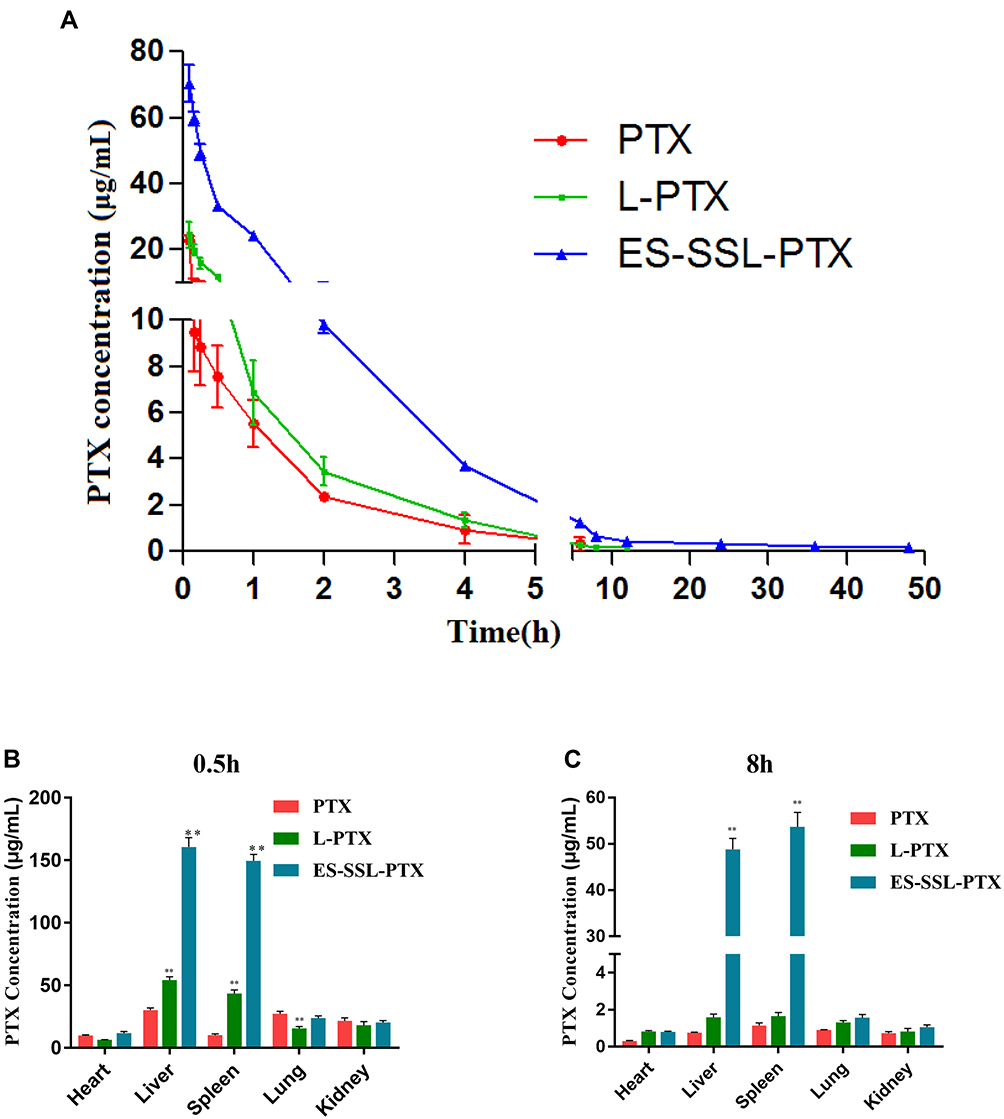 |
Figure 10 Pharmacokinetics and biodistribution studies of PTX liposomes. (A) Blood concentration curves. Eight-week-old female ICR mice were weighed and received a standard 10 mg/kg dose of PTX only (PTX), PTX liposomes (L-PTX), or long-acting PTX liposomes (ES-SSL-PTX) via tail vein injection. Blood was collected (n=3 per time point) at 5 min, 10 min, 15 min, 30 min, 1 h, 2 h, 4 h, 6 h, and 8 h from all groups, as well as at 12 h and 24 h from the L-PTX and ES-SSL-PTX groups, and at 36 h and 48 h for the ES-SSL-PTX group. (B) PTX accumulation in main organs of MCF-7 tumor-bearing mice at 0.5 h and (C) 8 h after administration with different PTX formulations (dose: 10 mg/kg PTX) (TWO-WAY ANOVA). Data are presented as mean ± SD (n = 3). **P <0.01 compared with PTX. |
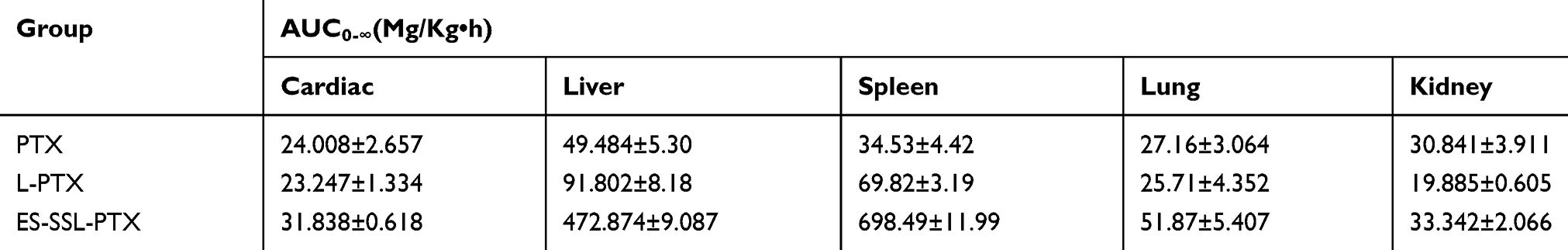 |
Table 4 Area Under the Curve of PTX Preparations in Different Tissues (n=3) |
Antitumor Efficacy in vivo
Each group displayed a different increase in tumor volume after the start of drug administration (Figure 11). Tumor volumes in the model group and the blank liposome group increased more rapidly than other groups; they reached 1000 mm3 after 18 days of administration and 4500 mm3 after 34 days of administration. Tumor growth was slowed in each PTX group, and effect was most significant in the ES-SSL-PTX group. After 18 days of administration, the tumor volume was only 160 mm3 and was maintained thereafter at 300 mm3. After 34 days, the average tumor volume in this group was only 420 mm3, compared to 2300 mm3 in the free PTX group and 1700 mm3 in the L-PTX group. Thus, ES-SSL-PTX effectively inhibited MCF-7 tumor growth in mice, and their efficacy was significantly better than PTX alone or the common PTX liposome preparation (Figure 12).
There was a small increase in body weight in tumor-bearing mice after administration of different PTX preparations (Figure 13). In the free PTX group, mice lost weight 8–24 days after administration and then recovered to pre-administration levels. In contrast, the ES-SSL-PTX group showed a steady rise in weight. There were no obvious side effects of the long-acting PTX liposomes at the therapeutic dose, suggesting that they are safer than free PTX.
 |
Figure 11 Tumor volume changes. Nude female BALB/c mice weighing 16–18 g were inoculated with MCF-7 cell suspension (1×107/0.2 cells/mL) into the second pair of breast fat pads (n=6). After the average tumor volume reached approximately 50 mm3, mice were randomly divided into 7 groups (6 mice in each group): (1) saline group (model group), (2) blank liposome group, (3) free PTX group (PTX), (4) PTX liposome group (L-PTX), (5) long-acting PTX liposome group (SSL-PTX), (6) targeted PTX liposome group (ES-L-PTX), and (7) long-acting targeted PTX liposome group (ES-SSL-PTX). Mice received tail vein injections of 5 mg/kg body weight (dose based on PTX content). The drug was administered once every 2 days for 4 consecutive doses, and the tumor volume was measured twice a week. *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001 compared with ES-SSL-PTX. |
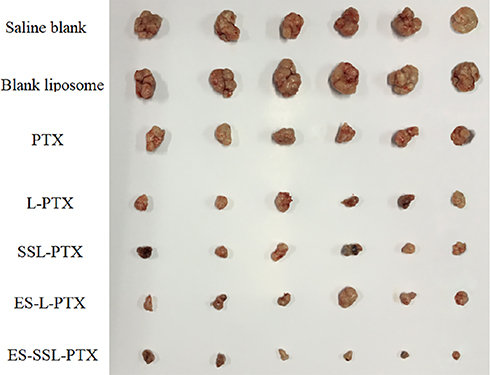 |
Figure 12 Mouse tumors. After the tumors in the model group reached 15 mm, the mice in each group were sacrificed, and the tumor tissues were collected and imaged. |
 |
Figure 13 Changes in mouse body weight (n=6 per group). |
In vivo Toxicity
The survival of the mice after administration of PTX, L-PTX, and ES-SSL-PTX are shown in Tables 5–7. The LD50 values of PTX, L-PTX, and ES-SSL-PTX administration in ICR mice were 36.15 mg/kg, 134.46 mg/kg, and 166.39 mg/kg, respectively. The LD50 of the targeted long-acting PTX liposomes was 4.6 times and 1.24 times higher than that of PTX alone and common PTX liposomes, respectively. There was no significant difference between males and females.
 |
Table 5 Acute Toxicity of PTX Injection |
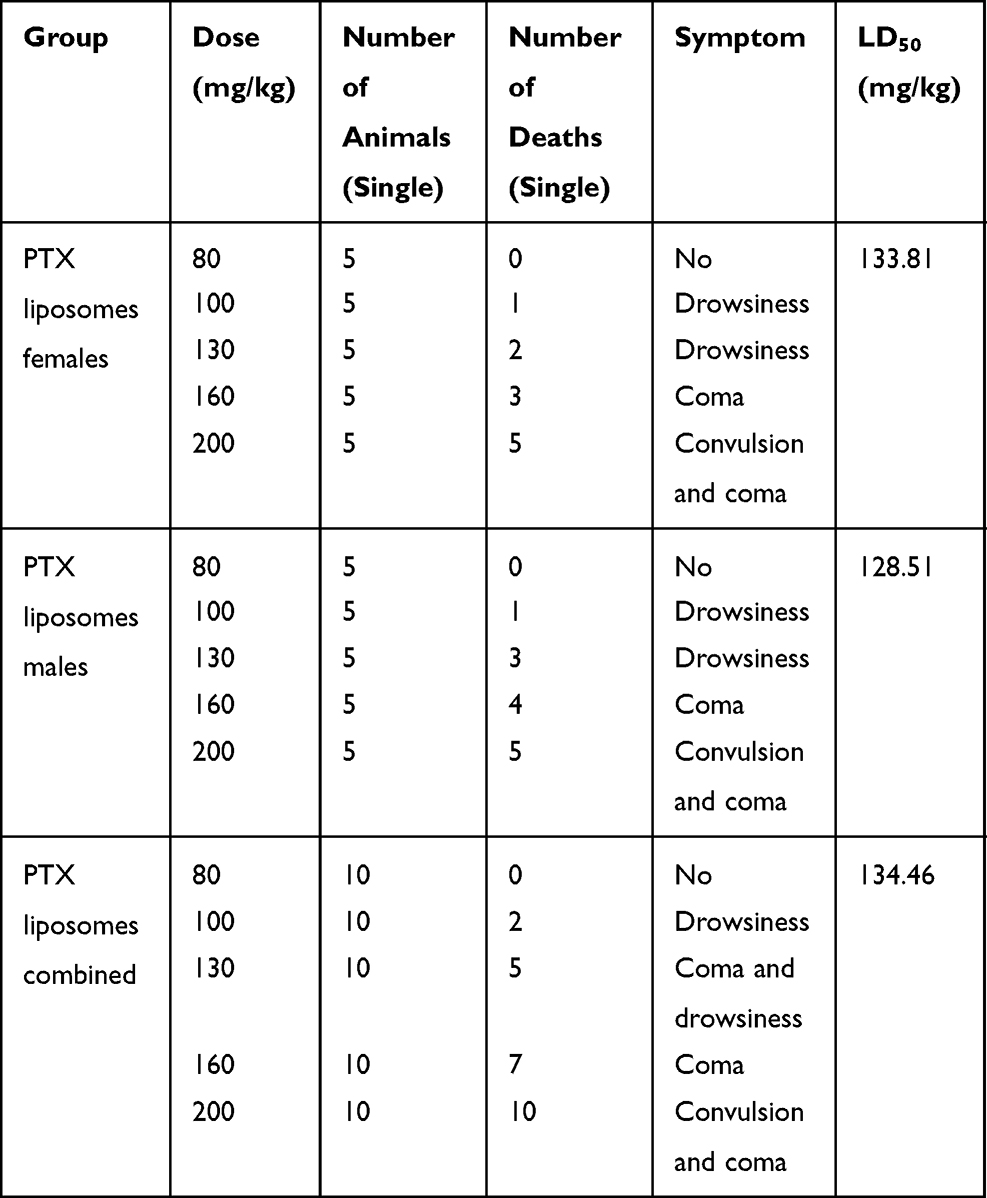 |
Table 6 Acute Toxicity of Common PTX Liposomes |
 |
Table 7 Acute Toxicity of Long-Acting PTX Liposomes |
Mice injected with different doses of PTX formulations had no obvious symptoms at 20 and 25 mg/kg doses. Mice dosed at 30 and 35 mg/kg showed lethargy and death on the first day after administration (AAD1). At a dose of 40 mg/kg, the mice immediately developed seizures and coma, and died 1 h after administration. Some mice recovered on AAD1 or AAD2 but were still lethargic. These mice fully returned to normal on AAD3. Mice injected with L-PTX and ES-SSL-PTX displayed similar symptoms: no obvious symptoms at 80 mg/kg, lethargy lasting until AAD2 or AAD3 at 100 and 120 mg/kg, and coma lasting one day followed by lethargy for two days at 160 mg/kg. Death was also observed at doses ranging from 80–160 mg/kg. Death occurred on AAD1. After administration of a 200 mg/kg dose, we immediately observed convulsions and coma; 1 h after administration, 8 mice in the L-PTX group had died, and 2 more died on AAD1. Seven mice in the ES-SSL-PTX group died 1 hr after administration, and 1 died on AAD1. Two mice in this group survived, recovering from coma on AAD1 and fully recovering by AAD2. The result of toxicity study showed the LD50 value of PTX, L-PTX and ES-SSL-PTX were 36.15, 134.46 and 166.39 mg/kg, respectively. Compared with that of PTX and L-PTX, the acute toxicity of ES-SSL-PTX was significantly lower which indicating that our ES-SSL-PTX had low toxicity and high safety.
Conclusions
In this study, estrogen receptors, which are highly expressed in breast cancer cells, were used to target long-acting liposomes for drug delivery. We successfully prepared a long-acting estrogen-receptor-targeting liposome formulation containing estrone-targeting fragments and PEG long-acting fragments. The formulation had good encapsulation efficiency and stability. In vivo and in vitro targeting studies, as well as anti-tumor studies and pharmacokinetic experiments, demonstrated that the long-acting targeted PTX liposome formulation (ES-SSL-PTX) selectively targeted breast cancer cells, prolonged drug action time, increased drug uptake by tumor cells, and increased drug accumulation in tumor sites. The formulation significantly improved PTX antitumor efficacy and reduced its side effects. Thus, ES-SSL-PTX has great potential for improving the treatment of breast cancer, thereby improving patient prognosis and quality of life.
Abbreviations
PTX, paclitaxel; PEG, polyethylene glycol; ERs, Estrogen receptors; ES, Estrone; DOX, doxorubicin; EE, Encapsulation efficiency; DL, drug-loading capacity; PDI, polydispersity index; TEM, transmission electron microscope; L-PTX, common PTX liposomes; ES-L-PTX, targeted PTX lipid plasmin; SSL-PTX, long-acting PTX liposomes or sterically stabilized liposomes;ES-SSL-PTX, targeted long-acting PTX liposomes; PBS, Phosphate-buffered saline; RhB, rhodamine B; PC, phosphatidylcholine; Chol, cholesterol; ES-SSL-RhB, RhB long-acting targeted liposomes; SSL-RhB, RhB long-acting liposomes; ES-L-RhB; RhB-targeted liposomes L-RhB, RhB common liposomes; SSL-DiR, DiR long-acting liposomes; CDI, N, N’-carbonyl diimidazole; DiR, (1,1′-dioctadecyl-3,3,3′,3′-tetramethylindotricarbo-cyanine iodide); mPEG, methoxy poly (ethylene glycol); SSL-PTX, long-acting PTX liposomes.
Acknowledgements
This work was funded by the Science and Technology Development Project form Jilin Science and Technology Department (20130727054YY, 20160209013YY, and 20180311072YY), and Jilin Province Development and Reform Commission (2014N149).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Ramadass SK, Anantharaman NV, Subramanian S, Sivasubramanian S, Madhan B. Paclitaxel/epigallocatechin gallate coloaded liposome: a synergistic delivery to control the invasiveness of MDA-MB-231 breast cancer cells. Colloids Surf B Biointerfaces. 2015;125:65–72. doi:10.1016/j.colsurfb.2014.11.005
2. King KM, Lupichuk S, Baig L, et al. Optimal use of taxanes in metastatic breast cancer. Curr Oncol. 2009;16(3):8–20. doi:10.3747/co.v16i3.377
3. Bedard PL, Di Leo A, Piccart-Gebhart MJ. Taxanes: optimizing adjuvant chemotherapy for early-stage breast cancer. Nat Rev Clin Oncol. 2010;7(1):22–36. doi:10.1038/nrclinonc.2009.186
4. Xiang J, Wu B, Zhou Z, et al. Synthesis and evaluation of a paclitaxel-binding polymeric micelle for efficient breast cancer therapy. Sci China Life Sci. 2018;61(4):436–447. doi:10.1007/s11427-017-9274-9
5. Liu Y, Xu Z, Zhang Z, Wen G, Sun J, Han F. Efficacy and safety of TE/TEC/intensive paclitaxel neoadjuvant chemotherapy for the treatment of breast cancer. Oncol Lett. 2019;17(1):907–912. doi:10.3892/ol.2018.9658
6. Fu S, Chen X, Lo HW, Lin J. Combined bazedoxifene and paclitaxel treatments inhibit cell viability, cell migration, colony formation, and tumor growth and induce apoptosis in breast cancer. Cancer Lett. 2019;448:11–19. doi:10.1016/j.canlet.2019.01.026
7. Merchan JR, Jayaram DR, Supko JG, He X, Bubley GJ, Sukhatme VP. Increased endothelial uptake of paclitaxel as a potential mechanism for its antiangiogenic effects: potentiation by Cox-2 inhibition. Int J Cancer. 2005;113(3):490–498. doi:10.1002/(ISSN)1097-0215
8. Du X, Khan AR, Fu M, Ji J, Yu A, Zhai G. Current development in the formulations of non-injection administration of paclitaxel. Int J Pharm. 2018;542(1–2):242–252. doi:10.1016/j.ijpharm.2018.03.030
9. Dian LH, Hu YJ, Lin JY, et al. Fabrication of paclitaxel hybrid nanomicelles to treat resistant breast cancer via oral administration. Int J Nanomedicine. 2018;13:719–731. doi:10.2147/IJN.S150140
10. Barbosa MV, Monteiro LO, Carneiro G, et al. Experimental design of a liposomal lipid system: a potential strategy for paclitaxel-based breast cancer treatment. Colloids Surf B Biointerfaces. 2015;136:553–561. doi:10.1016/j.colsurfb.2015.09.055
11. Takashima T, Kawajiri H, Nishimori T, et al. Safety and efficacy of low-dose nanoparticle albumin-bound paclitaxel for HER2-negative metastatic breast cancer. Anticancer Res. 2018;38(1):379–383. doi:10.21873/anticanres.12233
12. Alemi A, Zavar Reza J, Haghiralsadat F, et al. Paclitaxel and curcumin coadministration in novel cationic PEGylated niosomal formulations exhibit enhanced synergistic antitumor efficacy. J Nanobiotechnology. 2018;16(1):28. doi:10.1186/s12951-018-0351-4
13. Gregoriadis G, Wills EJ, Swain CP, Tavill AS. Drug-carrier potential of liposomes in cancer chemotherapy. Lancet. 1974;1(7870):1313–1316. doi:10.1016/S0140-6736(74)90682-5
14. Yuba E. Liposome-based immunity-inducing systems for cancer immunotherapy. Mol Immunol. 2018;98:8–12. doi:10.1016/j.molimm.2017.11.001
15. Malam Y, Loizidou M, Seifalian AM. Liposomes and nanoparticles: nanosized vehicles for drug delivery in cancer. Trends Pharmacol Sci. 2009;30(11):592–599. doi:10.1016/j.tips.2009.08.004
16. Silvander M, Johnsson M, Edwards K. Effects of PEG-lipids on permeability of phosphatidylcholine/cholesterol liposomes in buffer and in human serum. Chem Phys Lipids. 1998;97(1):15–26. doi:10.1016/S0009-3084(98)00088-7
17. Klibanov AL, Maruyama K, Torchilin VP, Huang L. Amphipathic polyethyleneglycols effectively prolong the circulation time of liposomes. FEBS Lett. 1990;268(1):235–237. doi:10.1016/0014-5793(90)81016-H
18. Shen Z, Ye H, Kroger M, Li Y. Aggregation of polyethylene glycol polymers suppresses receptor-mediated endocytosis of PEGylated liposomes. Nanoscale. 2018;10(9):4545–4560. doi:10.1039/C7NR09011K
19. Kim CE, Lim SK, Kim JS. In vivo antitumor effect of cromolyn in PEGylated liposomes for pancreatic cancer. J Control Release. 2012;157(2):190–195. doi:10.1016/j.jconrel.2011.09.066
20. Gomez-Vallejo V, Puigivila M, Plaza-Garcia S, et al. PEG-copolymer-coated iron oxide nanoparticles that avoid the reticuloendothelial system and act as kidney MRI contrast agents. Nanoscale. 2018;10(29):14153–14164. doi:10.1039/C8NR03084G
21. Allen TM, Hansen C. Pharmacokinetics of stealth versus conventional liposomes: effect of dose. Biochim Biophys Acta. 1991;1068(2):133–141. doi:10.1016/0005-2736(91)90201-I
22. Xu X, Wang L, Xu HQ, Huang XE, Qian YD, Xiang J. Clinical comparison between paclitaxel liposome (Lipusu(R)) and paclitaxel for treatment of patients with metastatic gastric cancer. Asian Pac J Cancer Prev. 2013;14(4):2591–2594. doi:10.7314/APJCP.2013.14.4.2591
23. Kastrati I, Semina S, Gordon B, Smart E. Insights into how phosphorylation of estrogen receptor at serine 305 modulates tamoxifen activity in breast cancer. Mol Cell Endocrinol. 2019;483:97–101. doi:10.1016/j.mce.2019.01.014
24. Wu H, Li J, Guo E, Luo S, Wang G. MiR-410 acts as a tumor suppressor in estrogen receptor-positive breast cancer cells by directly targeting ERLIN2 via the ERS pathway. Cell Physiol Biochem. 2018;48(2):461–474. doi:10.1159/000491777
25. Chen YZ, Kim Y, Soliman HH, Ying G, Lee JK. Single drug biomarker prediction for ER- breast cancer outcome from chemotherapy. Endocr Relat Cancer. 2018;25(6):595–605. doi:10.1530/ERC-17-0495
26. Jensen EV, DeSombre ER. Mechanism of action of the female sex hormones. Annu Rev Biochem. 1972;41:203–230. doi:10.1146/annurev.bi.41.070172.001223
27. Gorski J, Toft D, Shyamala G, Smith D, Notides A. Hormone receptors: studies on the interaction of estrogen with the uterus. Recent Prog Horm Res. 1968;24:45–80. doi:10.1016/b978-1-4831-9827-9.50008-3
28. Madhu Krishna B, Chaudhary S, Mishra DR, et al. Estrogen receptor alpha dependent regulation of estrogen related receptor beta and its role in cell cycle in breast cancer. BMC Cancer. 2018;18(1):607. doi:10.1186/s12885-018-4528-x
29. Osborne CK, Yochmowitz MG, Knight WA
30. Osborne CK. Steroid hormone receptors in breast cancer management. Breast Cancer Res Treat. 1998;51(3):227–238. doi:10.1023/A:1006132427948
31. Rai S, Paliwal R, Vaidya B, et al. Targeted delivery of doxorubicin via estrone-appended liposomes. J Drug Target. 2008;16(6):455–463. doi:10.1080/10611860802088481
32. Aghanejad A, Babamiri H, Adibkia K, Barar J, Omidi Y. Mucin-1 aptamer-armed superparamagnetic iron oxide nanoparticles for targeted delivery of doxorubicin to breast cancer cells. BioImpacts: BI. 2018;8(2):117–127. doi:10.15171/bi.2018.14
33. Chen L, Liu Y, Wang W, Liu K. Effect of integrin receptor-targeted liposomal paclitaxel for hepatocellular carcinoma targeting and therapy. Oncol Lett. 2015;10(1):77–84. doi:10.3892/ol.2015.3242
34. Xiang Y, Liang L, Wang X, Wang J, Zhang X, Zhang Q. Chloride channel-mediated brain glioma targeting of chlorotoxin-modified doxorubicine-loaded liposomes. J Control Release. 2011;152(3):402–410. doi:10.1016/j.jconrel.2011.03.014
35. Luo LM, Huang Y, Zhao BX, et al. Anti-tumor and anti-angiogenic effect of metronomic cyclic NGR-modified liposomes containing paclitaxel. Biomaterials. 2013;34(4):1102–1114. doi:10.1016/j.biomaterials.2012.10.029
36. Zhao H, Wang JC, Sun QS, Luo CL, Zhang Q. RGD-based strategies for improving antitumor activity of paclitaxel-loaded liposomes in nude mice xenografted with human ovarian cancer. J Drug Target. 2009;17(1):10–18. doi:10.1080/10611860802368966
37. Dai W, Yang F, Ma L, et al. Combined mTOR inhibitor rapamycin and doxorubicin-loaded cyclic octapeptide modified liposomes for targeting integrin alpha3 in triple-negative breast cancer. Biomaterials. 2014;35(20):5347–5358. doi:10.1016/j.biomaterials.2014.03.036
38. Du D, Chang N, Sun S, et al. The role of glucose transporters in the distribution of p-aminophenyl-alpha-d-mannopyranoside modified liposomes within mice brain. J Control Release. 2014;182:99–110. doi:10.1016/j.jconrel.2014.03.006
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.