Back to Journals » International Journal of Nanomedicine » Volume 9 » Issue 1
Preparation, characterization, and optimization of primaquine-loaded solid lipid nanoparticles
Authors Omwoyo W, Ogutu B, Oloo F, Swai H ![]() , Kalombo L, Melariri P, Mahanga G, Gathirwa JW
, Kalombo L, Melariri P, Mahanga G, Gathirwa JW
Received 17 February 2014
Accepted for publication 29 March 2014
Published 11 August 2014 Volume 2014:9(1) Pages 3865—3874
DOI https://doi.org/10.2147/IJN.S62630
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Wesley Nyaigoti Omwoyo,1,2 Bernhards Ogutu,3,4 Florence Oloo,3,5 Hulda Swai,6 Lonji Kalombo,6 Paula Melariri,6 Geoffrey Maroa Mahanga,2 Jeremiah Waweru Gathirwa3,4
1Department of Chemistry, Maasai Mara University, Narok, Kenya; 2Department of Chemistry, Jaramogi Oginga Odinga University of Science and Technology, Bondo, Kenya; 3Center for Research in Therapeutic Sciences, Strathmore University, Nairobi, Kenya; 4Kenya Medical Research Institute, Nairobi, Kenya; 5Department of Chemical Sciences and Technology, Technical University of Kenya, Nairobi, Kenya; 6Department of Polymers and Composites, Council for Scientific and Industrial Research, Pretoria, South Africa
Abstract: Primaquine (PQ) is one of the most widely used antimalarial drugs and is the only available drug that combats the relapsing form of malaria. PQ use in higher doses is limited by severe tissue toxicity including hematological- and gastrointestinal-related side effects. Nanoformulation of drugs in an appropriate drug carrier system has been extensively studied and shown to have the potential to improve bioavailability, thereby enhancing activity, reducing dose frequency, and subsequently reducing toxicity. The aim of this work was to design, synthesize, and characterize PQ-loaded solid lipid nanoparticles (SLNs) (PQ-SLNs) as a potential drug-delivery system. SLNs were prepared by a modified solvent emulsification evaporation method based on a water-in-oil-in-water (w/o/w) double emulsion. The mean particle size, zeta potential, drug loading, and encapsulation efficiency of the PQ-SLNs were 236 nm, +23 mV, 14%, and 75%, respectively. The zeta potential of the SLNs changed dramatically, from -6.54 mV to +23.0 mV, by binding positively charged chitosan as surface modifier. A spherical morphology of PQ-SLNs was seen by scanning electron microscope. In vitro, release profile depicted a steady drug release over 72 hours. Differential scanning calorimeter thermograms demonstrated presence of drug in drug-loaded nanoparticles along with disappearance of decomposition exotherms, suggesting increased physical stability of drug in prepared formulations. Negligible changes in characteristic peaks of drug in Fourier transform infrared spectra indicated absence of any interaction among the various components entrapped in the nanoparticle formulation. The nanoformulated PQ was 20% more effective as compared with conventional oral dose when tested in Plasmodium berghei-infected Swiss albino mice. This study demonstrated an efficient method of forming a nanomedicine delivery system for antimalarial drugs.
Keywords: double emulsion, nanomedicine drug-delivery system, antimalarial, nanotechnology
Introduction
Malaria continues to be a huge social, economic, and health problem, particularly in tropical countries. The disease kills between one and three million people, the majority of whom are young children.1 This death toll exceeds the mortality rate from AIDS (acquired immunodeficiency syndrome). Efforts to control malaria have included attempts in the development of effective vaccines, eradication of mosquito vectors, and development of new drugs. Malaria parasites have demonstrated some level of resistance to almost every antimalarial drug available,2 with some of the drugs in use having serious side effects. The failure of mosquito eradication, drug resistance, and the nonavailability of an effective antimalarial vaccine herald the need for alternative antiplasmodial therapy, a search for novel antimalarial compounds, or improvement of the existing antimalarials.3
Primaquine (PQ) is one of the most widely used antimalarial drugs and is the only available drug to date for combating the relapsing form of malaria, especially in the case of Plasmodium vivax and P. ovale.4 PQ has a unique and powerful role in the prevention and cure of malaria. Although its mechanism of action is not yet fully understood,5 it is thought to interfere with the cellular respiration of the parasite by generating oxygen free radicals and deregulating the electron transport.6 PQ is a tissue schizonticide 8-aminoquinoline group of drug that destroys exoerythrocytes and hypnozoites in the liver,7 thus preventing relapse and recrudescence. However, the drug has serious side effects including nausea, vomiting, stomach cramps, and hemolytic anemia.8 This prohibits its use in key groups, such as pregnant women.5 The PQ dose-limiting side effects including acute hemolytic anemia in individuals with glucose-6-phosphate dehydrogenase deficiency (G6PD deficiency), methemoglobinemia, leukocytopenia, leukocytosis, gastrointestinal disturbances, and abdominal cramps9 are partly due to the nonspecific targeting and short half-life that necessitates frequent dosing. For instance, for P. vivax, the dosage is 30 mg daily for 14 days, while for P. ovale, the dosage is 15 mg daily for 14 days.4 The drug is also a casual prophylactic, especially for travelers to endemic areas,10 but the dose frequency is also relatively frequent as the dosage involves 30 mg once daily, starting the day before travel and continues up to 7 days after returning. PQ oral bioavailability is also limited due to pre-systemic metabolism and excretion.11 With all this in mind, there is a need to reformulate this drug in order to enhance efficacy, reduce dose frequency, and subsequently reduce toxicity. A promising strategy to overcome these problems involves the development of a suitable drug-carrier system. Nanomedicine drug-delivery systems present the ability to enhance the therapeutic properties of current antimalarials. Nanomedicine drug-delivery systems including liposomes, dendrimers, lipids, pheroids, solid lipid nanoparticles (SLNs), polymeric nanoparticles, and nanocapsules have been developed in the effort to overcome delivery barriers.12
One of the strategies that can be employed to improve PQ is reformulation into nanoparticles. Nanoparticles have been investigated for many applications due to the enhanced material properties that result from a reduction in particle dimensions. The most active area of research is in the pharmaceutical industry, where nanoparticles have the potential to provide drug-delivery vehicles.13 Some advantages of nanoformulated drugs include maintaining drug therapeutic concentrations and circulatory time at target sites; protection from premature degradation in the gastrointestinal tract; improved pharmacokinetics, solubility, bioavailability, and stability; reduced toxicity; reduction in dose and dose frequency; enhancement of patient compliance; and prevention, reduction, or delay of onset of resistance.14
The main aim of using nanocarriers as drug-delivery systems is to promote drug protection against extracellular degradation, to improve selectivity in relation to the target, and to reduce dose frequency as well as duration of the treatment via enhancing the pharmacokinetic profile of the drug.15–17 Nanoformulation of drugs has made an impact in therapies for diseases like cancer.14 For example, Doxil® (Alza Corp., Mountain View, CA, USA) is a nanomedicine formulation of the anthracycline drug doxorubicin that is currently in use for cancer treatment. The four first-line antituberculosis drugs rifampicin, isoniazid, ethambutol, and pyrazinamide have been nanoformulated in polymeric nanoparticles.18,19 In contrast, nanotechnology has not been widely applied to transform therapies for poverty-related diseases such as malaria. Nevertheless, albumin and gelatin nanoparticles of PQ have been synthesized and shown to reduce toxicity in mice.20 Increased life-span index related to PQ-loaded nanoparticles was demonstrated after administration of free PQ diphosphate, free poly(diethyl methylidenemalonate) (poly[DEMM]) nanoparticles, and PQ-loaded poly(DEMM) nanoparticles as a single intraperitoneal injection to P. berghei-infected mice.21 In yet another study in mice, PQ-loaded liposomes were shown to protect nontarget tissues such as lung, kidneys, heart, and brain from PQ accumulation, thereby reducing the toxicity of PQ.22,23
The current study envisaged that reformulating PQ would enhance its efficacy and half-life, which may impact on its dosing regimen by enabling lower dosage and longer frequency. These will lead to reduced toxicity and better patient compliance. The strategy employed toward this was through the synthesis of PQ-loaded SLNs (PQ-SLNs). SLNs introduced in 1991 represent an alternative carrier system to traditional colloidal carriers such as emulsions, liposomes, and polymeric micro- and nanoparticles.24 SLNs are submicron colloidal carriers ranging from 50 nm to 1,000 nm. The system consists of spherical solid lipid particles in the nanometer ranges, which are dispersed in water or in aqueous surfactant solution. The solid core contains the drug dissolved or dispersed in the solid high melting fat matrix.25 The general ingredients include solid lipid(s), emulsifier(s), and water. A major advantage of SLNs is the fact that the lipid matrix is made from physiological lipids, which decreases the danger of acute and chronic toxicity.26 The solvent emulsification-evaporation method was selected for the formation of the nanoparticles. This method has emerged as a superior technique for preparing SLNs. A key step involves dispersions by precipitation in oil-in-water (o/w) emulsions. The lipophilic material is dissolved in a water-immiscible organic solvent that is emulsified in an aqueous phase. Upon evaporation of the solvent, nanoparticle dispersion is formed by precipitation of the lipid in the aqueous medium.27 Reproducibility of this method is reported to be high.28 The procedure used in forming the PQ-SLNs is outlined below.
Materials and methods
Materials
All materials, reagents, chemicals, and PQ base used in the research were supplied by our partners at the Center for Excellence in Nanomedicine at the Council for Scientific and Industrial Research (CSIR), Pretoria, South Africa. The stearic acid (SA), chitosan low-viscous, polyvinyl alcohol (PVA) of molecular weight 13,000–23,000 and partially hydrolyzed (87%–89%), D-lactose monohydrate, sulfanoyl, and ethyl acetate (EtOAc) were purchased from Sigma-Aldrich (Johannesburg, South Africa), and Pluronic® F127 Prill from BASF Corporation (Mount Olive, NJ, USA). All other chemical products were commercially available and of analytical grade. In this experimental design, stearic acid was the matrix; PVA and pluronic were surfactants and thus stabilized the emulsion, and chitosan was a mucoadhesive helping to increase circulation time in the intestines to enable most of the nanoparticles to be absorbed. Lactose enhanced particle size reduction, since it is a binder, while sulfanoyl was used as an antifoaming agent.
Preparation of SLNs
Nanoparticles were prepared using a modified double-emulsion solvent evaporation technique. Aqueous 2 mL of 2% PVA was stirred for a short period with 100 mg PQ using a magnetic stirrer to form the first aqueous phase containing the drug. The organic phase was made by dissolving 50 mg of stearic acid in 10 mL of EtOAc. The aqueous phase was dispersed in the organic phase by means of a high-speed homogenizer (Silverson L4R; Silverson Machines Limited, Buckinghamshire, UK), with a speed varying between 3,000 and 6,000 rpm for 3–6 minutes. The resulting water-in-oil emulsion was transferred into a specific volume of an aqueous solution made by mixing solutions of 10 mL of 2% PVA (weight/volume [w/v]), 5 mL of 0.2% (w/v), chitosan low-viscous, and 5 mL of 5% (w/v) D-lactose monohydrate. A drop of sulfanoyl was added to the resultant water-in-oil-in-water (w/o/w) emulsion, and the mixture was further emulsified for 5 minutes by homogenization at 8,000 rpm. The double emulsion (w/o/w) obtained was directly fed into a benchtop Buchi mini-spray dryer (Model B-290; BÜCHI Labortechnik AG, Flawil, Switzerland) and spray-dried at a temperature ranging between 80°C and 110°C, with an atomizing pressure varying between 5 and 8 bars. Nanoparticles obtained were characterized for particle size, polydispersity index (PDI), zeta potential, encapsulation efficiency (EE%) and drug loading (DL%). Experimental procedures above were repeated with varied homogenization speed and time, varied concentrations of the surfactant, change of surfactant, the effect of internal phase to external phase ratio, and the effect of chitosan and lactose to the emulsion. Scale-up experiments were also performed to determine the large scale application of this method.
Particle size, size distribution, and zeta potential
Particle size and size distribution were measured by dynamic laser scattering or photon correlation spectroscopy using a Malvern Zetasizer Nano ZS (Malvern Instruments, Malvern, UK). For each sample, 1–3 mg of nanoparticles was suspended in distilled water, then vortexed and/or sonicated for a few minutes. Each sample was measured in triplicate. Zeta potential was also determined using the same instrument.
EE% and DL%
EE% was determined using both the direct and indirect method. In the indirect method, 20 mg of the prepared nanoparticles were dispersed in 10 mL of water and vortexed in falcon tubes until fully dispersed. The resultant solution was then ultra-centrifuged at 15,000 rpm for 20 minutes at 4°C. The supernatant was then taken for ultraviolet-visible (UV-VIS) analysis at wavelengths between 400 nm and 200 nm. For the direct method, the precipitate was taken and dissolved in a specified amount of EtOAc. Water was then added to the solution and left overnight. The aqueous phase was then separated by means of a separating funnel and analyzed for PQ concentration. The concentration of the drug was determined by means of a standard curve as extracted from UV-VIS spectrometry analysis by using different known concentrations of the drug. The EE% and DL% were calculated using the formulas below:
EE% = (drug in precipitate/total added drug) ×100 | (1) |
DL% = (drug in precipitate/drug in precipitate + added excipients) ×100 | (2) |
where “drug in precipitate” = total drug added − free drug after ultra-centrifugation (indirect method) and “added excipients” = lipids + surfactant mixtures + other ingredients used.
Thermal properties
Thermogravimetric analysis (TGA) was used to measure the physical and chemical changes of the nanoparticles as a function of temperature. A sample of PQ-free drug, empty nanoparticles, and drug-loaded nanoparticles were analyzed using TGA (TGA-601; PerkinElmer Inc., Waltham, MA, USA) connected to an inert nitrogen gas flow and at a heating rate of 10°C/minute.
Polymorphism analysis
Differential scanning calorimetry (DSC) was performed using model Q200 equipped with an automated computer-controlled refrigerated cooling system (RSC-90) and TZero capabilities (TA Instruments, New Castle, DE, USA). DSC thermograms were collected using a sample weight of 3–5 mg powder and placed in TZero alodine-coated aluminum DSC pans, which were then hermetically sealed with a TZero hermetic sealer (TA Instruments). Phase transition temperatures – melting point – of the samples were measured under a 50 mL/minute dry ultra-high purity nitrogen gas (Scott-Gross, Winchester, KY, USA) purge in DSC. The samples were heated at 10°C/minute from 10°C to 200°C. At least four melting scans were carried out to ensure melting point temperature reproducibility. Measured DSC data were analyzed using a coupled DSC Q200-1740 data station. DSC can be used to determine the nature and speciation of crystallinity within nanoparticles through the measurement of glass and melting point temperatures and their associated enthalpies.
Surface morphology
Scanning electron microscopy was used to provide a way to directly observe the morphological appearance of the nanoparticles. The particles were first coated in gold to minimize the effect of heat during high-power magnification and were then run through a scanning electron microscope machine (SU1510 model; Hitachi Ltd., Tokyo, Japan) where pictures of the nanoparticles were taken. Fourier transform infrared spectroscopy (FTIR) was also used to determine the functional groups present on the surface of the nanoparticle.
Storage stability of SLNs
Physical stability of SLNs during prolonged storage in room temperature (22°C–28°C) was determined routinely by monitoring changes in zeta potential, particle size, and PDI as a function of time. External parameters such as temperature and light are of primary importance for long-term stability. Zeta potential should in general remain high for a dispersion to remain physically stable.
In vitro release experiment
In vitro release studies were performed using the method described by Mühlen and Mehnert,29 with modifications. Twenty milligrams of the spray-dried nanoparticles were weighed into 13 different falcon tubes and dispersed in 10 mL of distilled water. The dispersion was vortexed for 10 minutes to ensure uniformity. The dispersion was then ultra-centrifuged at 15,000 rpm for 20 minutes at 4°C. The supernatant was then discarded as a means of washing the nanoparticles. The precipitates in the falcon tubes were further dispersed in a buffer solution of pH 7.0 and then placed in a shaker, with bath water maintained at 37°C to mimic body conditions. The supernatant from each tube was taken for UV-VIS analysis at wavelengths between 400 nm and 200 nm after 0, 6, 12, 18, 24, 30, 36, 42, 48, 54, 60, 66, and 72 minutes respectively. The analysis gave the amount of drug released from the nanoparticles with time.
In vivo 4-day suppressive test
Prior to commencement, ethics approval to carry out the study was given by the Kenya Medical Research Institute Animal Care and Use Committee. In vivo antimalarial efficacy was evaluated based on a documented 4-day suppressive test.30 Swiss albino mice were infected intraperitoneally with blood containing 2×107 parasitized P. berghei ANKA red blood cells contained in 0.2 mL inoculums on day zero. Five mice were used per group. Two hours post-infection, the test animals were orally treated with two dose levels of either PQ-SLNs or PQ free drug at 5 and 2 mg/kg/day. Negative control received vehicle used to suspend samples, which was composed of 7% Tween-80 and 3% ethanol in water. Drug administration was repeated 24, 48, and 72 hours post-infection. On day 4 (96 hours post-infection), blood smears were taken by making a thin film from a tail snip of each mouse, fixed in methanol and stained with 10% Giemsa for 20 minutes. Blood smears were used to estimate the level of parasitemia. Percentage chemosuppression (parasite reduction) was calculated as described in the literature.31
Results and discussion
Role of different excipients to basic characteristics of formulations
Various parameters were examined to optimize nanoparticle size, charge, and DL%. Particles of size <500 nm were targeted, as particles of this size are known to be absorbed in the gastrointestinal tract intact. Considering that PQ targets liver stages of malaria parasites, the targeted size was thus found to be ideal. Table 1 gives results obtained under varied parameters.
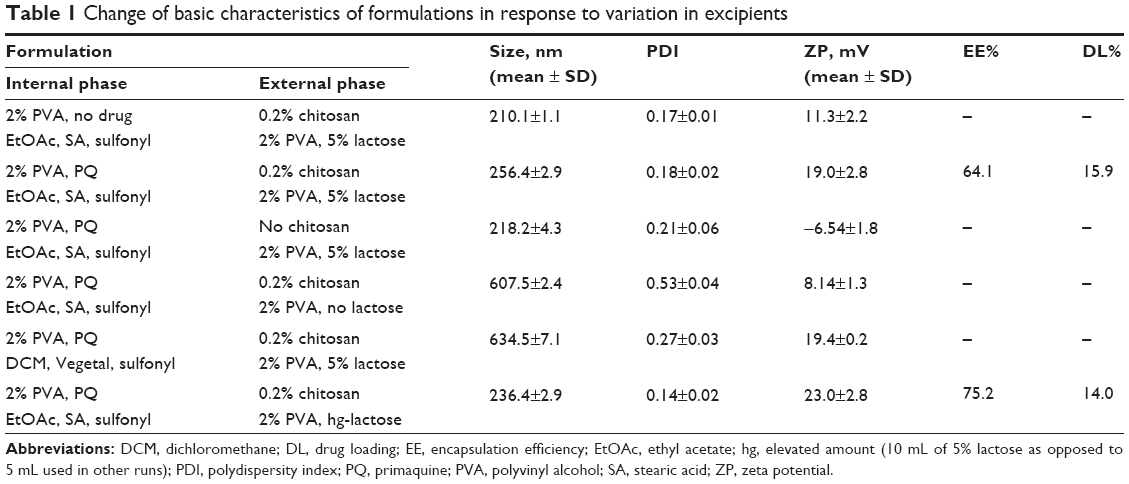 | Table 1 Change of basic characteristics of formulations in response to variation in excipients |
Empty nanoparticles formed at a size of 210 nm in run one. The size increased to 256 nm upon inclusion of 100 mg PQ. This colloidal system was stabilized by 2% PVA. To try to reduce the particle size further, chitosan was excluded in run three. The size reduced to 218 nm; however, the surface charge of the particles became negative. Considering that the gastrointestinal tract lining is negatively charged, a repulsion of the like charges would significantly reduce absorption of the negatively charged PQ-loaded nanoparticles. Chitosan a high positively charged molecule was thus found to be a very important additive for the purpose of introducing positive charge on the particle surface to increase absorption. In the absence of lactose (run four), particle sizes of 607 nm were obtained. Lactose acts as a binder by bringing together the matrix units, thereby reducing size to a tightly intertwined network of matrix and drug. Vegetal oil as a replacement of the SA as the solid lipid also gave significantly larger nanoparticles as shown in run five. Increasing amounts of lactose as the binder reduced the particle size to 236 nm, with a higher positive surface charge of 23.0 mV and an EE% of 75%, and thus was taken as the optimum condition. High amounts of lactose, however, negatively impacted on DL% capacity, which declined from 15.9% to 14% when the amount was doubled. The loading capacity is generally expressed as a percentage related to the lipid phase (matrix lipid + drug). Loading capacities of typically 1%–5% yielded an EE% of up to 50% for ubidecarenone,32 while for tetracaine and etomidate, loading capacities of 10%–20% have been reported.33–35 Other reports have shown loading capacities of 5%, 20%, and 20%–25% for retinol, coenzyme Q10, and cyclosporine, respectively.36,37 PQ-SLNs in this experiment depicted similar loading capacity. Factors determining the loading capacity of drug in lipid are the solubility of drug in lipid, miscibility of drug and lipid, chemical and physical structure of solid lipid matrix, and the polymorphic state of the lipid material.37 Prerequisite to obtain sufficient loading capacity is sufficiently high solubility of the drug in the lipid. The solubility in lipid is enhanced by addition of solubilizers.38
Effects of surfactant concentration and homogenization time on particle size
Effect of surfactant concentration and homogenization time on particle size, PDI, and zeta potential was investigated using a two-factorial completely randomized design, with surfactant concentration as the main factor and homogenization time as a subfactor. When surfactant concentration was gradually increased from 0.5% to 5%, particle size significantly (P≤0.05) reduced with an increase in surfactant concentration with smallest nanoparticles being obtained at a PVA concentration of 2%. Further increase in surfactant concentration beyond that point significantly (P≤0.05) increased size of nanoparticles. Similarly, increase in homogenization time significantly (P≤0.05) reduced the particle size, up to an optimum at 10 minutes.
There was a significant (P≤0.05) interaction between homogenization time and surfactant concentration, indicating that the choice of surfactant and surfactant concentration could affect homogenization time. Similar trends were observed for the PDI and zeta potential. The coefficients of variation were within the statistically acceptable limits.
Effect of internal to external ratio on the particle size
The ratio between the internal and external phases was also examined. As shown in Table 2, at equal volume of the two phases, the particle size was large. This could be attributed to the fact that no phase could comfortably disperse in the other, as an enclosure of a smaller phase by a larger phase is ordinarily mandatory. The more the amount of the external phase, the smaller the size of the resultant nanoparticle.
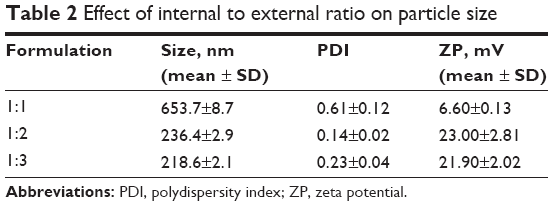 | Table 2 Effect of internal to external ratio on particle size |
Effect of inlet temperature during spray drying
Spray-drying conditions were found to impact on the nanoparticles characteristics. Table 3 gives various conditions that were applied during drying and the state of nanoparticles recovered. At 70°C inlet temperature, the powder recovered was not free-flowing, indicating that moisture content was high. Percentage recovery yield was highest at 65%, though this could be attributed to retained moisture. Particle size gradually decreased with increase in inlet temperature, and at 100°C, a free-flowing powder of nanoparticles was obtained. At 100°C, outlet temperature coming into contact with the nanoparticles was found to be 65°C, and at 110°C, outlet temperature was found to equal the melting point of the SA particle matrix (67°C–69°C); thus 100°C was taken as the optimum temperature to avoid melting the nanoparticle matrix.
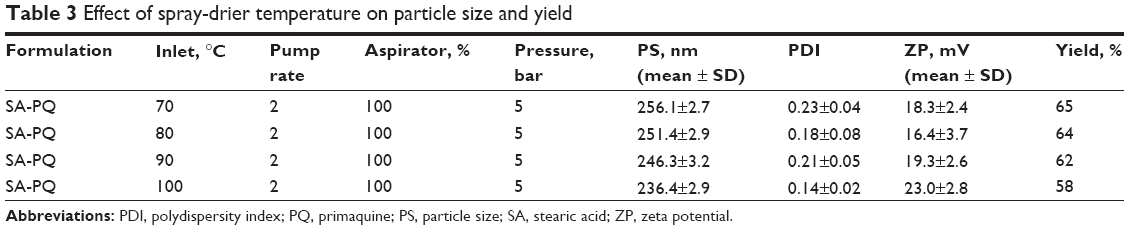 | Table 3 Effect of spray-drier temperature on particle size and yield |
TGA
TGA analysis gave the degradation points of the tested samples shown in Figure 1. The first depression in the curves was a measure of moisture content. From TGA analysis, sample moisture content was found to be 4%–5%. A moisture content of less than 5% is acceptable in the pharmaceutical industry. PQ-loaded nanoparticles gradually degraded at a temperature above 176°C. This was comparable to that of free PQ. A residue of between 32% and 35% was retained for both free PQ and the nanoformulated PQ, even when they were heated to 700°C, while SA degraded to a minimal amount of residue.
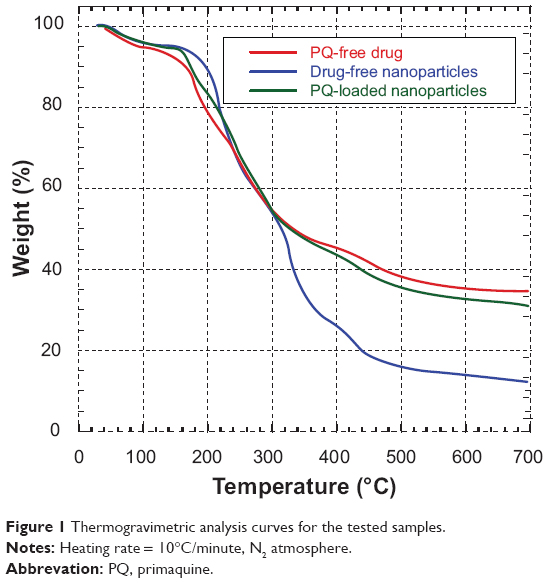 | Figure 1 Thermogravimetric analysis curves for the tested samples. |
Figure 2 gives DSC results of the nanoformulated PQ. DSC was performed to determine the crystallinity of drug in nanoparticles. Crystallinity in nanoparticles is an important consideration as it greatly affects the solubility and dissolution characteristics of the drug.39 To determine the crystallinity state of the PQ-SLNs, samples were gradually heated and then allowed to cool in the DSC instrument as explained previously, and the DSC thermograms then collected. The DSC curve of PQ-SLNs revealed an endothermic melting peak (maximum 67°C). There was a very slight shift in thermograms of PQ-loaded nanoparticles as compared with empty nanoparticles, probably occasioned by drug to polymer interactions. The shift of the endothermic peak of PQ in SLNs indicated that the drug was molecularly dispersed inside the matrix of stearic acid. PQ in crystalline form is known to melt sharply at temperatures of about 175°C–178°C. After nanoformulation, this peak disappeared, and the only peak observed was the one of matrix (stearic acid) at about 67°C. This is attributed to the fact that the drug changed its characteristics from crystalline form to amorphous form. When considering the crystallinity of polymeric nanoparticles, it is suggested that degradation occurs first in the amorphous regions of the particle, followed by a slower degradation in the crystalline regions. However, in SLNs, the opposite is observed. This observation thus suggests that the crystallinity affects the degradation rate and thus the drug-release kinetics.40 In general, amorphous (non-crystalline) forms of the particles may present faster dissolution rates compared with crystalline forms.40 Drug release is less rapid in amorphous nanoparticles than in crystalline particles. Lack of crystallinity suggests better drug dispersion and increased drug–matrix interactions, leading to the conclusion that if slow-release kinetic is required, reduced crystallinity is favored,39 as was achieved in our study.
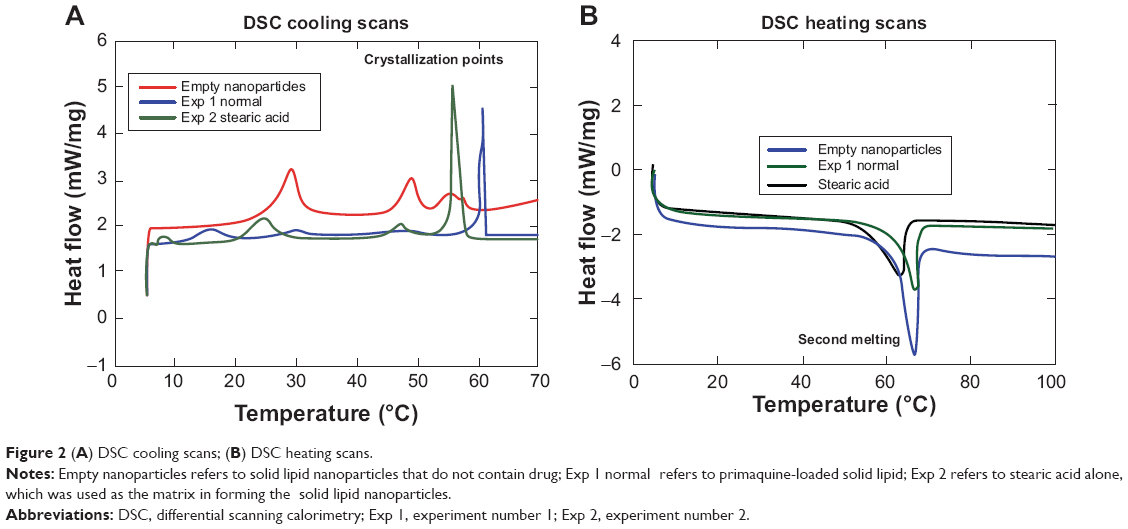 | Figure 2 (A) DSC cooling scans; (B) DSC heating scans. |
Morphological analysis
Figure 3 shows images from the scanning electron microscope of the nanoformulated PQ. PQ-SLNs obtained were seen to be spherical in shape. Despite gold coating, magnification of >×30,000 caused melting of particles, causing them to agglomerate, as could be seen from the images. This was expected, as stearic acid used as matrix melts at temperatures above 67°C.
 | Figure 3 Scanning electron microscopy images of drug-loaded solid lipid nanoparticles at ×20,000 (A) and ×50,000 (B) magnification. |
The FTIR chromatogram in Figure 4 shows peaks of the drug formulations. The level of drug enclosure in the nanoparticles or absence in the case of the drug-free nanoparticles could be deduced from the chromatograms. Of particular interest was the NH2 bending at 1,614 cm−1 and aromatic C=C stretching at around 1,532 cm−1 and 1,467 cm−1. As seen in Figure 4, free drug had more pronounced peaks in those particular absorbance regions, but in the case of the nanoparticles with 5 mL of lactose, the peaks appeared reduced. When 15 mL of lactose was used as a structural binder, the peaks reduced much further, indicating successful encapsulation in the solid lipid matrix as has been similarly observed in other studies.41–43
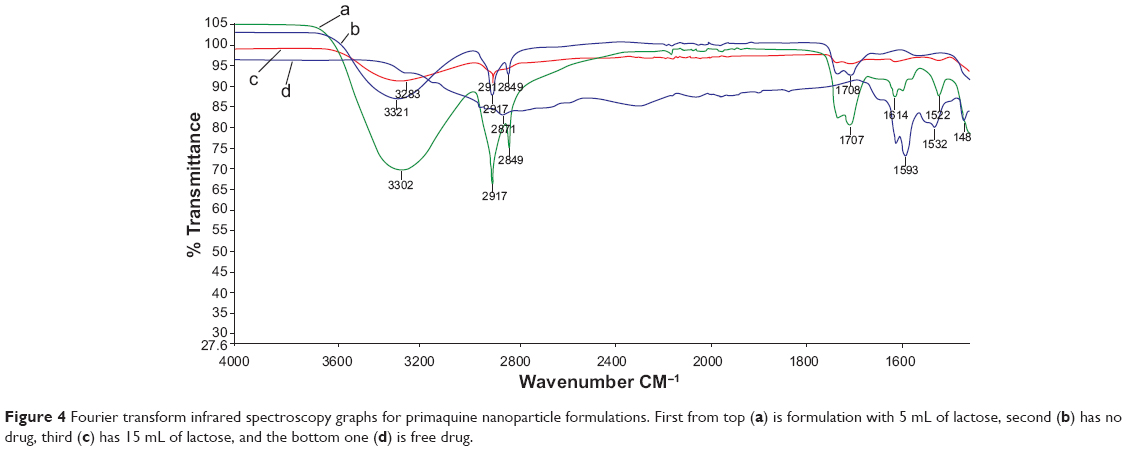 | Figure 4 Fourier transform infrared spectroscopy graphs for primaquine nanoparticle formulations. First from top (a) is formulation with 5 ml of lactose, second (b) has no drug, third (c) has 15 ml of lactose, and the bottom one (d) is free drug. |
Stability of the SLNs with time
The nanoparticles showed that they were stable in reference to particle size, zeta potential, and PDI. Changes in nanoparticle characteristics with respect to time are given in Table 4. It was observed that drug-loaded particles maintained their characteristics when stored at room temperature. Similar observations have been made previously, where it was noted that topotecan-loaded lipid nanoparticles are stable between temperatures of 4°C and 50°C over a period of 90 days, but temperatures above this cause the nanoparticles to agglomerate.38 It is thus advisable to store the PQ-SLNs at room temperature, as they did not show any effects.
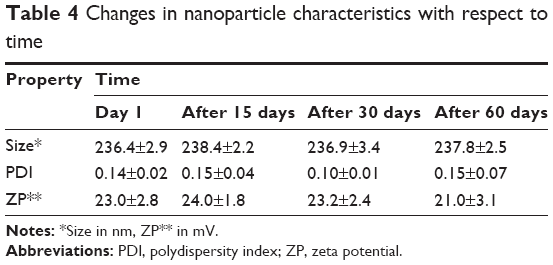 | Table 4 Changes in nanoparticle characteristics with respect to time |
In vitro release studies
There are distinctly less data available about drug release,29 especially information about the release mechanisms. Most of the data about in vitro drug-release mechanisms have been generated by studying the model drugs tetracaine, etomidate, and prednisolone.29,44 A major problem during the work with lipid nanopellets has been the burst release observed with these systems. When not washed properly, PQ-SLNs could show a burst release due to unencapsulated drug. A prolonged drug release has been obtained when studying the incorporation of prednisolone.37 This demonstrated the principal suitability of the SLN system for prolonged drug release.29 More importantly, one can modify the release profiles as a function of lipid matrix, surfactant concentration, and production parameters such as temperature,29 although this was not investigated in this experiment. In vitro drug release was achieved for up to 72 hours, as shown in Figure 5. The profiles can be modulated showing prolonged release without any burst at all through proper washing of the nanoparticles, but also generating systems with burst can be exploited to deliver an initial high dose when desired.37
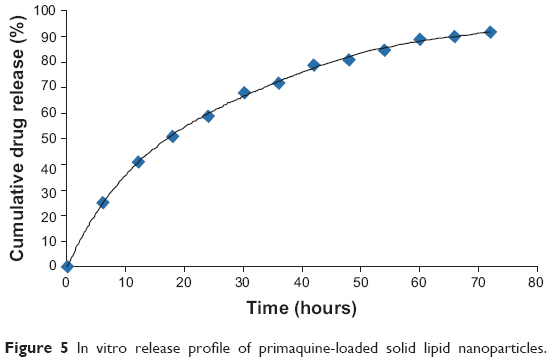 | Figure 5 In vitro release profile of primaquine-loaded solid lipid nanoparticles. |
In vivo antimalarial efficacy
Suppressive efficacy against the malaria parasite was evaluated by comparing the percentage reduction of parasitemia (chemosuppression) between the treated and untreated groups. Results are shown in Table 5. Average parasitemia in both nanoformulated and free PQ drug were significantly lower (P≤0.05) than that of the untreated group for both test concentrations. There was no significant difference in parasitemia levels and survival time between the unloaded nanoparticles and the untreated control group, indicating that the empty nanoparticles and the excipients therein did not exert any antimalarial activity. When mice were treated with PQ-SLNs at a dose of 2 mg/kg/day, chemosuppression of 93.5% was observed. In comparison, only 71.9% chemosuppression was observed when mice were treated with free PQ at a similar dose. This indicated that nanoformulation of PQ increased its efficacy by more than 20%. The mean survival time of mice treated with the nanoformulated PQ was similarly enhanced when compared with the group of mice that received the conventional dose of PQ. In a previous study, it was observed that formulating PQ into the nanoemulsion shows effective antimalarial activity against P. berghei infection in Swiss albino mice at 25% lower dose level compared with the conventional oral dose.11
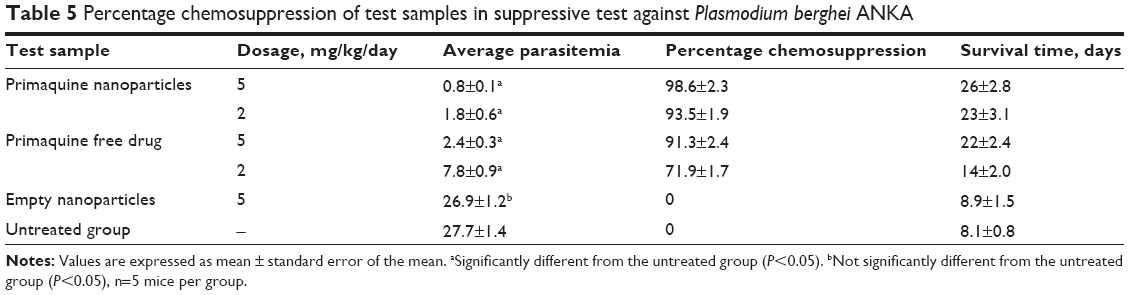 | Table 5 Percentage chemosuppression of test samples in suppressive test against Plasmodium berghei ANKA |
Conclusion
PQ-SLNs were successfully formed using a modified solvent emulsification evaporation method based on a w/o/w double emulsion. The particle size was small enough to allow direct absorption in the gastrointestinal tract. This coupled with a moderately positive surface charge is envisaged to allow for high absorption that could trigger an elevated concentration of PQ base in the liver. Since the drug is highly effective against hypnozoites/liver stages of all malaria species, the formulation will be highly efficacious, even at reduced dosages. A prolonged drug-release profile was observed which may positively impact on dosage frequency and consequently result in reduced toxicity. Efficacy evaluation in mice showed that the nanoformulated PQ was 20% more effective as compared with the conventional oral dose, indicating the great potential of improving antimalarial efficacy of drugs by nanoformulating them into SLNs.
Acknowledgments
This study was sponsored by the Consortium for National Health Research, grant number RCDG-2012-008. Partial funding was received from the National Commission for Science, Technology and Innovation, grant number NCST/RCD/ST & i/KE-SA/3RDCALL/02. National Research Foundation South Africa provided funding for our partners at CSIR. The authors sincerely thank the funding organizations for their relentless support. We also wish to express our gratitude to the CSIR, Kenya Medical Research Institute, and Institute for Primate Research for providing necessary facilities to develop the research study.
Disclosure
The authors report no conflicts of interest in this work.
References
Snow RW, Guerra CA, Noor AM, Myint HY, Hay SI. The global distribution of clinical episodes of Plasmodium falciparum malaria. Nature. 2005;434:214–217. | ||
Bloland PB. Drug resistance in malaria. Available from: http://www.who.int/csr/resources/publications/drugresist/malaria.pdf. Accessed July 16, 2014. | ||
World Health Organization. Global Plan for Artemisinin Resistance Containment. Geneva: World Health Organization; 2011. Available from: http://www.who.int/malaria/publications/atoz/artemisinin_resistance_containment_2011.pdf. Accessed April 13, 2014. | ||
Baird JK, Hoffman SL. Primaquine therapy for malaria. Clin Infect Dis. 2004;39:1336–1345. | ||
Fernando SD, Rodrigo C, Rajapakse S. Chemoprophylaxis in malaria: drugs, evidence of efficacy and costs. Asian Pac J Trop Med. 2011;4(4):330–336. | ||
Hill DR, Baird JK, Parise ME, Lewis LS, Ryan ET, Magill AJ. Primaquine: report from CDC expert meeting on malaria chemoprophylaxis I. Am J Trop Med Hyg. 2006;75:402–415. | ||
Fernando D, Rodrigo C, Rajapakse S. Primaquine in vivax malaria: an update and review on management issues. Malar J. 2011;10:351. | ||
Kilawa W, Ntoumi F. Malaria, a search agenda for the eradication era. Lancet. 2009;374:1480–1482. | ||
Baird JK, Rieckmann KH. Can primaquine therapy for vivax malaria be improved? Trends Parasitol. 2003;19:115–120. | ||
Alving A, Anold J, Hockwald R. Prevention of curative action of primaquine in vivax malaria by quinine and chloroquine. J Lab Clin Med. 1985;4:301–306. | ||
Singh KK, Vingkar SK. Formulation, antimalarial activity and biodistribution of oral lipid nanoemulsion of primaquine. Int J Pharm. 2008;347:136–143. | ||
Allen T, Cullis P. Drug delivery systems: entering the mainstream. Science. 2004;303:1818–1822. | ||
Peters W, Robinson B, Tovey G, Rossier J, Jefford C. The chemotherapy of rodent malaria. L. The activities of some synthetic 1,2,4-trioxanes against chloroquine-sensitive and chloroquine resistant parasites. Part 3: Observations on ‘Fenzan-50F’, a difluorinated 3,3 spirocyclopentane 1,2,4- trioxane. Ann Trop Med Parasitol. 1993;87:111–123. | ||
Malam Y, Loizidou M, Seifalian A. Liposomes and nanoparticles: nanosized vehicles for drug delivery in cancer. Trends Pharmacol Sci. 2009;30(11):592–599. | ||
Vauthier C, Couvreur P. Nanomedicines: a new approach for the treatment of serious diseases. J Biomed Nanotechnol. 2007;3:223–234. | ||
Barratt G. Colloidal drug carriers: achievements and perspectives. Cell Mol Life Sci. 2003;60:21–37. | ||
Devalapally H, Chakilam A, Amiji MM. Role of nanotechnology in pharmaceutical product development. J Pharm Sci. 2007;96:2547–2565. | ||
Semete B, Booysen L, Lemmer Y, et al. In vivo evaluation of the biodistribution and safety of PLGA nanoparticles as drug delivery systems. Nanomedicine. 2010;6(5):662–671. | ||
Semete B, Booysen LIJ, Kalombo L, et al. In vivo uptake and acute immune response to orally administered chitosan and PEG coated PLGA nanoparticles. Toxicol Appl Pharmacol. 2010;249(2):158–165. | ||
Bruce-Chwatt L. Essential Malariology. New York: John Wiley and Sons; 1980:46–59. | ||
Povinelli L, Monson TA, Fox BC, Parise ME, Morrisey JM, Vaidya AB. Plasmodium vivax malaria in spite of atovaquone/proguanil (malarone) prophylaxis. J Travel Med. 2003;10:353–355. | ||
Pirson P, Steiger R, Trouet A. The disposition of free and liposomally encapsulated antimalarial primaquine in mice. Biochem Pharmacol. 1982;31:3501–3507. | ||
Arica B, Ozer AY, Ercan MT, Hincal AA. Characterization, in vitro and in vivo studies on primaquine diphosphate liposomes. J Microencapsul. 1995;12:469–485. | ||
Ekambaram P, Abdul HS, Priyanka K. Solid lipid nanoparticles: a review. Sci Rev Chem Commun. 2012;2(1):80–102. | ||
Shah C, Shah V, Upadhyay U. Solid lipid nanoparticles: a review. Curr Pharm Res. 2011;1(4):351–368. | ||
Mehnert W, Mäder K. Solid lipid nanoparticles: production, characterization and applications. Adv Drug Deliv Rev. 2001;47:165–196. | ||
Sjostrom B, Bergenstahl B. Preparation of submicron particles in lecithin-stabilized o/w emulsions. I. Model drug studies of the precipitation of cholesteryl acetate. Int J Pharm. 1992;88:53–62. | ||
Siekmann B, Westesen K. Investigations on solid lipid nanoparticles prepared by precipitation in o/w emulsions. Eur J Pharm Biopharm. 1996;43:104–109. | ||
Mühlen A, Mehnert W. Drug release and release mechanism of prednisolone loaded solid lipid nanoparticles. Pharmazie. 1998;53:552–555. | ||
Peters W, Portus JH, Robinson BL. The chemotherapy of rodent malaria XXII. The value of drug resistant strains of Plasmodium berghei in screening for schizontocidal activity. Ann Trop Med Parasitol. 1975;69:155–171. | ||
Tona L, Mesia K, Ngimbi NP, et al. In-vivo antimalarial activity of Cassia occidentalis, Morinda morindoides and Phyllanthus niruri. Ann Trop Med Parasitol. 2001;95:47–57. | ||
Westesen K, Bunjes H, Koch MHJ. Physicochemical characterization of lipid nanoparticles and evaluation of their drug loading capacity and sustained release potential. J Control Release. 1997;48:223–236. | ||
Iscan YY, Hekimoglu S, Kas S, Hincal AA. Formulation and characterization of solid lipid nanoparticles for skin delivery, conference ‘lipid and surfactant dispersed systems’. Moscow: Proceedings Book. 1999:163–166. | ||
Schwarz C, Freitas C, Mehnert W, Müller RH. Sterilisation and physical stability of drug-free and etomidate-loaded solid lipid nanoparticles. Proc Int Symp Control Release Bioact Mater. 1995;22:766–767. | ||
Schwarz C. Feste Lipidnanopartikel: Herstellung, Charakterisierung, Arzneistof nkorporation under freisetzung, Sterilisation und Lyophilisation [doctoral thesis]. Berlin: Free University of Berlin; 1995. | ||
Penkler L, Müller RH, Runge SA, Ravelli V. Inventors; Pharmatec International S.R.L., assignee. Pharmaceutical cyclosporin formulation with improved biopharmaceutical properties, improved physical quality and greater stability, and method for producing said formulation. United States patent US6551619 B1. 2003 Apr 22. | ||
Müller RH, Mäder K, Gohla S. Solid lipid nanoparticles (SLN) for controlled drug delivery – a review of the state of the art. Eur J Pharm Biopharm. 2000;50(1):161–177. | ||
Souza LG, Silva EJ, Martins ALL, et al. Development of topotecan loaded lipid nanoparticles for chemical stabilization and prolonged release. Eur J Pharm Biopharm. 2011;79:189–196. | ||
Mahato RI. Pharmaceutical Dosage Forms and Drug Delivery. Boca Raton: CRC Press; 2007. | ||
Izumikawa S, Yoshioka S, Aso Y, Takeda Y. Preparation of poly(l-lactide) microspheres of different crystalline morphology and effect of crystalline morphology on drug release rate. J Control Release. 1991;15(2):133–140. | ||
Gupta N, Rajera R, Nagpal M, Arora S. Primaquine loaded chitosan nanoparticles for liver targeting. Pharm Nanotechnol. 2013;1:35–43. | ||
Dudhani AR, Kosaraju SL. Bioadhesive chitosan nanoparticles: preparation and characterization. Carbohydr Polym. 2010;81:243–251. | ||
Li P, Wang Y, Peng Z, et al. Development of chitosan nanoparticles as drug delivery systems for 5-fluorouracil and leucovorin blends. Carbohydr Polym. 2011;85:698–704. | ||
Mehnert W, Zur Mühlen A, Dingler A, Weyhers H, Müller RH. Solid lipid nanoparticles – ein neuartiger Wirkstoff-Carrier für Kosmetika und Pharmazeutika. II. Mitteilung: Wirkstoff Inkorporation, Freisetzung und Sterilisierbarkeit. Pharm Ind. 1997;59:511–514. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.