Back to Journals » Drug Design, Development and Therapy » Volume 10
Preparation, characterization, and in vivo evaluation of intranasally administered liposomal formulation of donepezil
Authors Al-Asmari A, Ullah Z, Tariq M, Fatani A
Received 6 August 2015
Accepted for publication 1 December 2015
Published 12 January 2016 Volume 2016:10 Pages 205—215
DOI https://doi.org/10.2147/DDDT.S93937
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Prof. Dr. Wei Duan
Abdulrahman K Al Asmari,1 Zabih Ullah,1 Mohammad Tariq,1 Amal Fatani2
1Department of Research, Prince Sultan, Military Medical City, 2Department of Pharmacy, King Saud University, Riyadh, Saudi Arabia
Abstract: The adequate amount of drug delivery to the brain in neurological patients is a major problem faced by the physicians. Recent studies suggested that intranasal administration of liposomal formulation may improve the drug delivery to the brain. In the present study, an attempt was made to study the brain bioavailability of commonly used anti-Alzheimer drug donepezil (DNP) liposomal formulation by intranasal route in rats. We adopted the thin layer hydration technique for the preparation of liposomes by using cholesterol, polyethylene glycol, and 1,2-distearyl-sn-glycero-3-phosphocholine (DSPC). The prepared liposomes were characterized by determining particle size, shape, surface morphology, zeta potential, encapsulation efficiency, and in vitro release of DNP. The pharmacokinetic parameters of liposomal DNP in plasma and brain of rats were determined following oral and nasal administration. The results of this study showed that the DNP liposomal formulation was stable with a consistent size (102±3.3 nm) and shape. The prepared liposomes showed high encapsulation efficiency (84.91%±3.31%) and sustained-release behavior. The bioavailability of DNP in plasma and brain increased significantly (P<0.05) after administration of liposomal formulation by the intranasal route. Histopathological examination showed that the formulation was safe and free from toxicity. It can be concluded that the nasal administration of liposomal preparation may provide an efficient and reliable mode of drug delivery to the central nervous system.
Keywords: donepezil, intranasal, liposomes, bioavailability, blood–brain barrier
Introduction
Neuropsychiatric diseases are one of the leading causes of global disease burden, which significantly contribute to disability and adjusted life years.1 Treatment of central nervous system (CNS) diseases is particularly challenging because of a variety of formidable obstacles that often impede drug delivery to the brain and spinal cord.2 The blood–brain barrier (BBB), situated at the level of brain capillaries (comprising specialized cells like microglia endothelial cells, pericytes, and astrocytes), imposes major obstruction to the passage of active drug molecules from the vascular compartment to brain tissues.3 The cells of the BBB display few essential qualities, such as the presence of tight and compact junction, lack of aperture, and a reduced pinocytic behavior. These particular types of cells help to limit the passage of the active molecules from the vascular to the extracellular area of the brain.4–6 The present challenge is to develop a drug delivery approach that will allow the passage of active moieties through the BBB in a safe and efficient manner.
Out of numerous approaches that have been explored to increase the drug delivery to the brain, intranasal delivery system and lipid-mediated transport are among the frontrunners.7,8 The nasal route of administration is gaining popularity among pharmaceutical scientists as a potential route for achieving faster absorption and optimal bioavailability of a drug. The nasal cavity has an advantage of easy accessibility, larger surface area, porous endothelial membrane, increased blood flow, and avoidance of the first-pass metabolism.9 Although nasal delivery is routinely used for sinusitis as well as nasal allergy, infection, and congestion, the use of this route for systemic administration of a drug is limited due to local untoward effects including irritation or injury to nasal mucosa from drugs or their excipients.10 However, the nasal route is one of the focused delivery options for the treatment of CNS ailments due to a direct connection between brain and nose compartment through the olfactory route as well as peripheral circulation.11 This course of drug transport has gained much attention due to its ability to circumvent the BBB.
During the last decade, numerous efforts have been made to improve drug delivery using liposomes, polymeric solid lipid nanoparticles, and nanocolloidal systems.12 Liposomal drug formulations (emerged from self-forming enclosed lipid bilayers upon hydration) have shown significant improvement in drug delivery and therapeutic efficacy of many potent drugs, such as antimicrobial, antineoplastic, genetic materials, chelating agents, steroids, and vaccines.13 The efficient delivery of liposomal formulation results from a significant change in pharmacokinetics and pharmacodynamics of lipid-entrapped drugs.14 Besides optimal delivery of pharmaceuticals to the target organs, liposomal formulations have been shown to reduce the toxicity.15 To date, only a few attempts have been made to study the pharmacokinetics of liposomal drugs following intranasal route.16–19 In the present study, an attempt has been made to develop and evaluate the liposomal formulation of DNP (a prototype of an anti-Alzheimer’s drug, Figure 1) and to compare its brain and plasma pharmacokinetics following intranasal administration in rats.
 | Figure 1 Chemical structure of donepezil (CAS: 120014-06-4). |
Materials and methods
Materials
Donepezil used in the study was procured from Fluka, St Louis, MO, USA. Carboxymethyl cellulose, 1,2-distearyl-sn-glycero-3-phosphocholine (DSPC), cholesterol (CHE), polyethylene glycol (PEG), chloroform, acetonitrile, sodium dihydrogen phosphate, methanol, Sephadex G-25, sodium dodecyl sulfate, dichloromethane, and other chemicals were procured from Sigma-Aldrich (St Louis, MO, USA).
Formulation of DNP liposomes
The liposomes were prepared by traditional thin layer hydration method.14 DSPC, CHE, and PEG were dissolved in chloroform in a molar ratio of 1:2:0.5, and this solvent system was transferred to the round bottom 100 mL volumetric flask. The round bottom flask was carefully engrossed in a water bath connected to a rotary vacuum evaporator. The flask was rotated under vacuum until all the solvent got evaporated leaving behind a thin film of lipid. The overnight vacuum was maintained to confirm the complete removal of residual solvents. DNP was separately dissolved in phosphate buffer (pH 7.4) and added to the volumetric flask containing dried film of lipids. The flask was vortexed for 10 minutes and then frozen (−80°C) and thawed (30°C) three times. The prepared liposomes were stored at 4°C for further analysis.
Characterization of liposomal formulation
Morphology
The surface morphology of the liposomes was studied using scanning electron microscopy (Zeiss EVO LS10, Cambridge, UK).20 Samples were fixed on stubs using both side adhesive carbon tape (SPI Supplies, West Chester, PA, USA) and coated under vacuum with gold in a Q150R sputter coater unit obtained from Quorum Technologies Ltd. (East Sussex, UK) in an argon atmosphere at 20 mA for 120 seconds. After mounting, the samples were viewed and photographed. Particles size analyzer was used to determine the size of the liposomal entity (Mastersizer 2000, Malvern Instruments, Malvern, UK). Laser diffraction technique was applied to determine the liposomal size distribution by immersing liposomes in oil medium at a temperature of 25°C±1°C and scattering light at 90°.
Determination of encapsulation efficiency
The encapsulation efficiency (EE) of the liposomal formulation was determined by mini column centrifugation method.16 Cotton plugged syringes (1 mL) packed with Sephadex G-25 gel (1% m/v, soaked in 0.9% v/v saline for 4 h) were placed in centrifugal tubes and centrifuged at 6,700× g up to 15 minutes so that the bed gets dried completely. Liposomal formulation (500 μL) was then added to this dry bed and the entire assembly further centrifuged at 600× g for 15 minutes. The whole experiment was repeated six times to ensure the complete removal of the unencapsulated free drug. The amount of encapsulated drug was determined by liquid chromatography mass spectrometry (LCMS) analysis.
The following formula calculated the percentage of drug encapsulation:
|
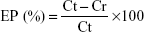
where EP is encapsulation percentage, Ct is concentration of total drug, and Cr is concentration of free drug.
In vitro release of DNP from liposomal formulation
The in vitro release of DNP from the liposomal preparation was determined by dialysis sac technique using cellophane membrane (10 kDa). Simulated nasal fluid (SNF) was chosen as a medium for release studies.21 Dialysis sac, containing liposomal formulation was immersed in 100 mL of SNF (comprising 7.45 mg/mL NaCl, 1.29 mg/mL KCl, and 0.32 mg/mL CaCl2·2H2O) maintained at a pH of 6.2–6.8 and a temperature of 37°C, which was continuously stirred. The samples were drawn (1 mL) at predefined time periods of 5, 10, 15, 25, 35, 45, 60, 90, 120, 150, 180, 240, 300, and 360 minutes. The release of DNP in the samples was analyzed in triplicate by LCMS.
Stability studies
Physical stability studies of the liposomes were carried out to evaluate the leakage of the drug from these vesicles and their aggregation. The prepared liposomes were stored in covered transparent vials at 4°C±1°C and 25°C±1°C for 3 months. Samples from the liposomal formulation were withdrawn periodically. Over the study period, the stability of the liposomes was evaluated by the measurement of particle size, EE, physical appearance, aggregation, and sedimentation.
Pharmacokinetic studies
Pharmacokinetic studies were performed on male Wistar rats weighing 200±20 g. The protocol of the study was approved by the Institutional Ethics Committee of Prince Sultan Military Medical City for animal care. The rats were maintained on a 12 hour light and dark cycle at a temperature of 25°C and relative humidity of 45%. The animals were given food and water ad libitum and were closely monitored for any kind of behavioral changes during the experiment. Before the start of the pharmacokinetic study, the animals fasted overnight. As per the protocol, the rats were divided into three groups. The animals in group I and group II were administered 1 mg/kg body weight DNP by oral and nasal route respectively, whereas animals of group III received DNP liposomes (equivalent to 1 mg/kg body weight of DNP) intranasally. Six rats each were sacrificed at 0.5, 1, 1.5, 3, 4, 6, and 8 hours and blood and brain samples were collected for DNP analysis. Blood was centrifuged to separate plasma, and the brain tissues were washed thrice with saline, wiped with the soft fabric, weighed and stored at −80°C until analysis. The brain samples were homogenized in phosphate buffer for extraction and analyzed through LCMS for DNP level.
Pharmacokinetic parameters
Based on the brain and plasma level of DNP at various time intervals, following parameters were calculated by using the least squares program of SummitPK (Montrose, CO, USA): 22 area under concentration–time curve from time zero to time t (AUC0–t), area under concentration–time curve from time zero to infinity (AUC0–∞), terminal elimination half-life (T1/2), in agreement with terminal slope of a semilogarithmic concentration–time curve, maximum plasma drug level (Cmax), and rate of drug removal from the body (Kel).
Bioanalytical methodology
Liquid chromatography conditions
Chromatographic partition was achieved by using a Thermo Scientific, Aquasil C18 column (100×2.1 mm, 5 μm particle size) attached to Thermo Dionex Ultimate 3000 UHPLC with a quaternary pump, autosampler, solvent manager, and an MS-MS detector (Thermo Scientific LCQ Fleet, Ion Trap mass spectrometer, Serial# LCF 10356, San Jose, CA, USA). Ammonium acetate buffer (1 mM, pH 4.4) and methanol (20:80, v/v) were used as the mobile phase in nongradient elution mode at a flow rate of 300 μL/min. The injected sample volume was 10 μL. The column temperature was maintained at 40°C±2°C, and temperature of autosampler was maintained at 4°C±2°C. The chromatographic run time was 3 minutes.
Mass spectrometry conditions
The accurate mass was calculated using the Xcalibur software installed for the instrument. Analytes of interest were ionized in positive using electrospray ionization technique. The flow rate of sheath gas, auxiliary gas, sweep gas, capillary temperature, and collision energy were optimized for each analyte separately. Spray voltage was kept at 5 kV. Analyte (DNP) and internal standard (fluoxetine) were quantified on the basis of retention time and presence of parent ion, which was 380.25 for DNP and 310.24 for fluoxetine in selected ion monitoring mode.
Sample extraction protocol
The solid phase extraction method was developed for isolation of DNP from plasma. The plasma samples were thawed and vortexed to make it homogeneous. To 400 μL of each plasma sample, 100 μL of fluoxetine working solution was added and mixed in polypropylene tubes; 100 μL of ammonium acetate buffer was added into these polypropylene tubes and vortexed again for 30 seconds to achieve complete mixing. These samples were transferred to solid phase extraction cartridges (Agilent Bond Elut Plexa, Agilent Technologies, Santa Clara, CA, USA), which had been preconditioned using methanol and water (liquid chromatography grade). After centrifugation, the analytes were eluted from the cartridges with 30% methanol in water. Eluents were dried under a stream of nitrogen at 20 psi pressure and 40°C temperature. The dried residues were reconstituted with 300 μL of the mobile phase, and 10 μL was injected into the LCMS system for analysis. On the other hand, the brain samples were extracted using 4 mL of ethyl acetate and n-hexane (30:70). After homogenization, the samples were centrifuged, and the supernatant was transferred to another glass tube that was dried (20 psi, 40°C) under a stream of dry nitrogen and processed as per plasma samples.
Histopathological studies
After biodistribution studies, the rats from the last observation point (8 h) were used to investigate the histopathological changes in various tissues following administration of liposomal formulation by the nasal passage. Animals of test and control groups were anesthetized, and their heart, lung, kidney, spleen, liver, brain (midsection), and olfactory bulb were dissected and washed with physiological saline. The organs were cleaned and pressed with filter pads and stored in 10% formalin solution. The slides were prepared using the standard process and stained with hematoxylin and eosin for histopathological examinations. All the slides were examined by using a light microscope at a magnification of ×100.23
Statistical analysis
The statistical analysis was performed using the SPSS software (version 14.0, SPSS Inc., Chicago, IL, USA). Variables between two groups were compared by Student’s t-test. The analysis of variance followed by post hoc test was performed when more than two groups were compared. For all the tests, the differences of P<0.05 were considered as statistically significant.
Results
Bioanalytical method validation
The developed bioanalytical method was sensitive enough to quantify DNP in plasma and brain. There was no interference from the endogenous matrix at the retention time of DNP and internal standard, which suggests that the method was selective for the analytes. The calibration range was established between 1 and 500 ng/mL. Mean extraction recovery of DNP from plasma and brain tissue homogenates was 85.80%±2.2% and 81.20%±3.4%, respectively. The range of intra- and interday precision was found to be 4.22%–6.7% for plasma and 5.86%–7.3% for brain, respectively. The intra- and interday accuracy of the developed method was found to be 7.0% and 6.0%, respectively.
Characterization
In this study, lipid layer hydration technique was used to prepare the liposomes. The morphology of the prepared liposomes was evaluated by scanning electron microscopy. The liposomes were found to have a smooth surface, spherical shape, and existed mainly as a single unilamellar vesicle (Figure 2). However, some of the liposomes existed as multilamellar vesicles. Liposomes were nonaggregated with a particle size (>90%) of 102±3.3 nm. The mean % EE of the developed liposomes for DNP was found to be 84.91%±3.31%. The polydispersity index (PI), which is a measure of liposomal size distribution, was found to be 0.28±0.03. Low PI of the developed formulation showed that the formulation had a narrow size distribution. The zeta potential of the formulation was −28.31±0.85 mV.
 | Figure 2 SEM micrographs of the prepared liposomes of DNP. |
In vitro release of DNP from liposomal preparation
The cumulative release of DNP in SNF was studied by dialysis sac method. The release profile of liposomal DNP is given in Figure 3. The amount of drug release in SNF was measured at various time intervals up to 8 hours. The cumulative drug release data showed the drug release up to 8 hours was found to be 75.50%±3.03%. There was a burst release of the DNP during the first 150 minutes, after that the release rate was significantly reduced. The plateau was observed after 2.5 hours, which may be attributed to equilibration of drug concentration between the recipient and donor compartments.
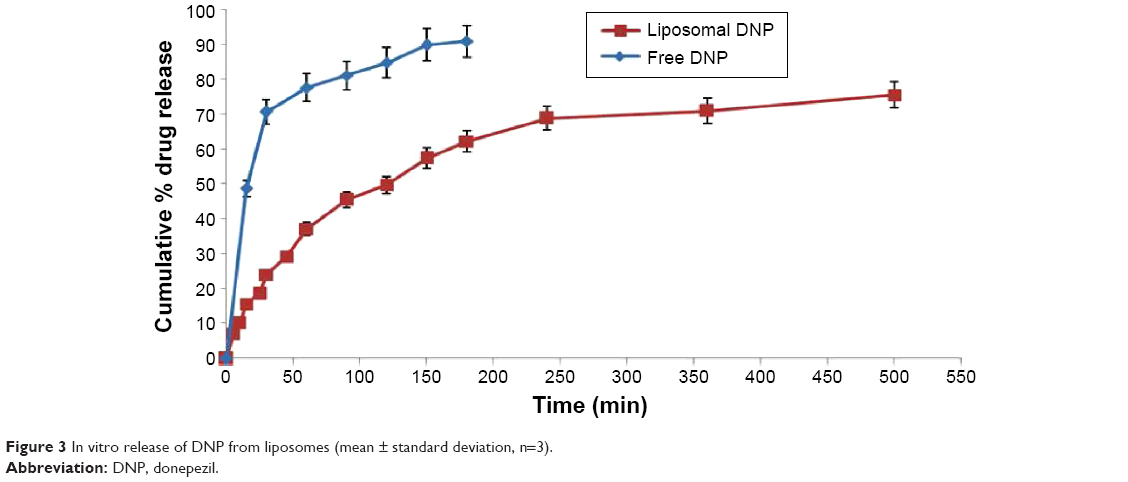 | Figure 3 In vitro release of DNP from liposomes (mean ± standard deviation, n=3). |
Stability studies
The data for the stability study of the prepared liposomes are shown in Table 1. There was no change in the EE and vesicle size of the liposomes stored at 4°C. However, an insignificant (P>0.5) change in the vesicle size and EE was observed in the liposomes stored at 25°C. Liposomes stored at refrigeration temperature (4°C) were able to maintain a higher percentage of the drug compared with room temperatures. There was no sedimentation, aggregation, or change of color in the prepared formulation over a period of 3 months at both temperature conditions.
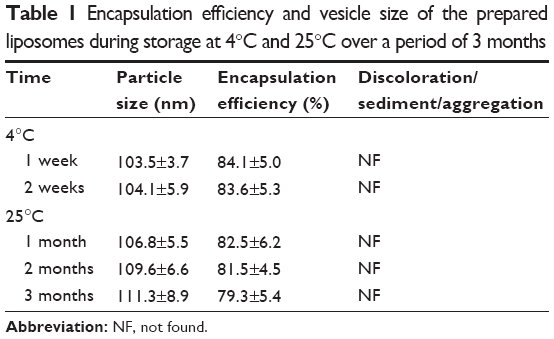 | Table 1 Encapsulation efficiency and vesicle size of the prepared liposomes during storage at 4°C and 25°C over a period of 3 months |
Comparative pharmacokinetics of DNP in plasma
The comparative mean plasma levels and time profile of free form and liposomal formulation of DNP administered by various routes are given in Figure 4. The absorption rate of both forms of the drug was significantly higher following nasal administration as compared with oral administration. Following oral administration of free drug, the maximum level in plasma was observed after 1 hour, whereas after intranasal administration, the peak level in plasma was reached in just 30 minutes. Moreover, the intranasally administered liposomal formulation was found to achieve a significantly higher level of DNP in plasma against oral or intranasal free drug administration. A significant increase in Cmax was observed when the free drug was given intranasally as compared with free drug administered orally (P<0.05). The AUC0–t and AUC0–∞ for intranasally administered DNP were also higher as compared with oral administration. Intranasal administration of liposomes showed a twofold higher AUC as compared with AUC following intranasal administration of free DNP. However, no significant difference was observed in the elimination rate constant and half-life (P>0.05) in these groups (Table 2).
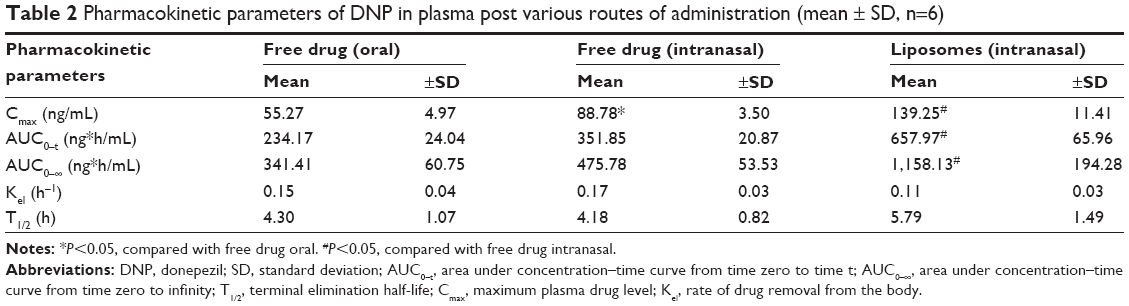 | Table 2 Pharmacokinetic parameters of DNP in plasma post various routes of administration (mean ± SD, n=6) |
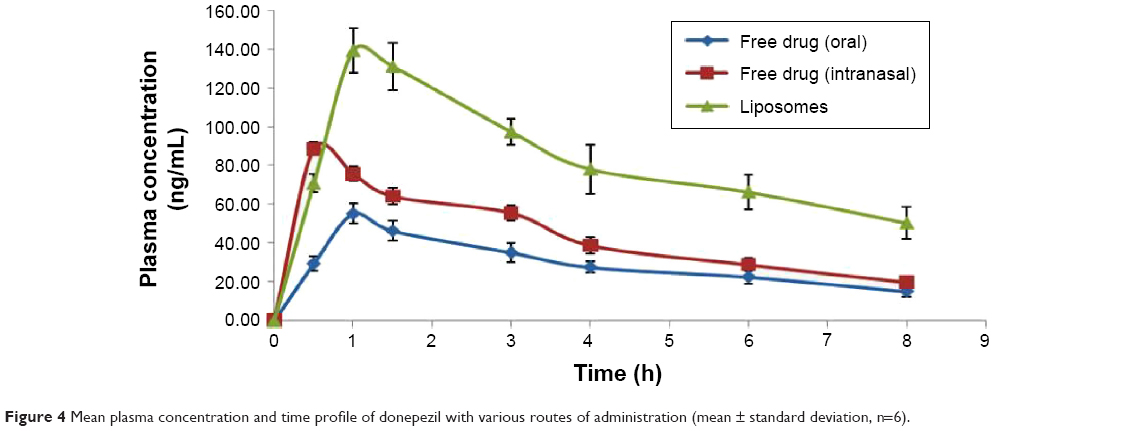 | Figure 4 Mean plasma concentration and time profile of donepezil with various routes of administration (mean ± standard deviation, n=6). |
Comparative pharmacokinetics of DNP in brain
The concentration of DNP in the brain following oral and nasal administration showed the same pattern as observed in the case of plasma, suggesting a better bioavailability of the drug following nasal delivery (Figure 5). The Cmax after intranasal liposomes administration was achieved at 1.5 hours and this concentration was maintained up to ~3 hours, which showed controlled and long-lasting release from the liposomal formulation. Comparative bioavailabilities of DNP in brain post free drug oral and free drug intranasal administration is given in Table 3. Oral administered free drug attained maximum brain level in 1.5 hours, whereas intranasal administration of free drug attained a Cmax at 1.0 hour. A significant difference in Cmax was observed when free DNP given orally was compared with free DNP intranasally (P<0.05). The AUC0–t and AUC0–∞ for intranasally administered free DNP followed the same pattern as in plasma. No significant difference was noticed in the elimination rate constant and half-life (P>0.05). It is evident from the data that intranasal administration of liposomal formulation achieves significantly higher (P<0.05) bioavailability in the brain when compared with free drug intranasal administration. The liposomal formulation also demonstrated a longer half-life (6.90±1.14 h) as compared with the intranasally administered free drug (5.55±1.04 h).
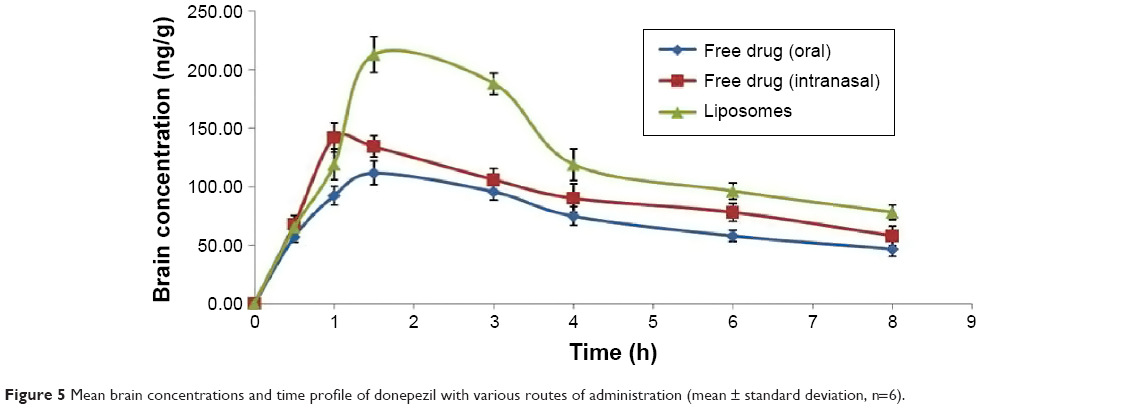 | Figure 5 Mean brain concentrations and time profile of donepezil with various routes of administration (mean ± standard deviation, n=6). |
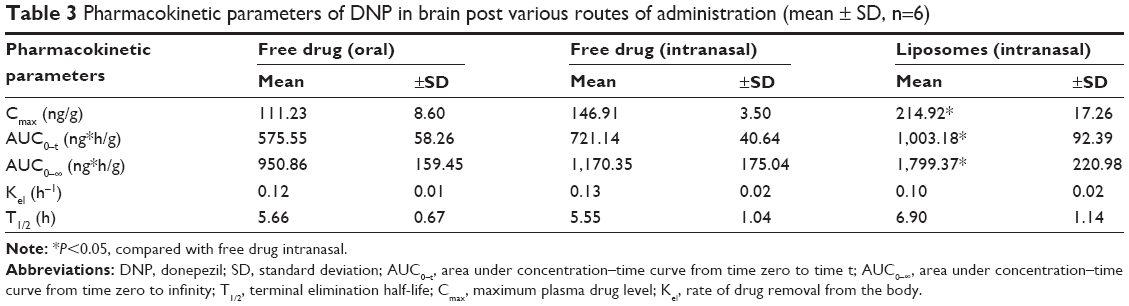 | Table 3 Pharmacokinetic parameters of DNP in brain post various routes of administration (mean ± SD, n=6) |
Histopathological studies
Microphotographs of rats were examined to detect any deleterious effect of liposomal formulation on liver, lung, heart, spleen, kidney, brain (midsection), and olfactory bulb (Figures 6 and 7). No microscopic or morphological changes were observed in the visceral tissues following nasal administration of the liposomal formulation of DNP in rats.
 | Figure 6 Histopathological examinations of heart, lung, kidney, spleen, and liver. |
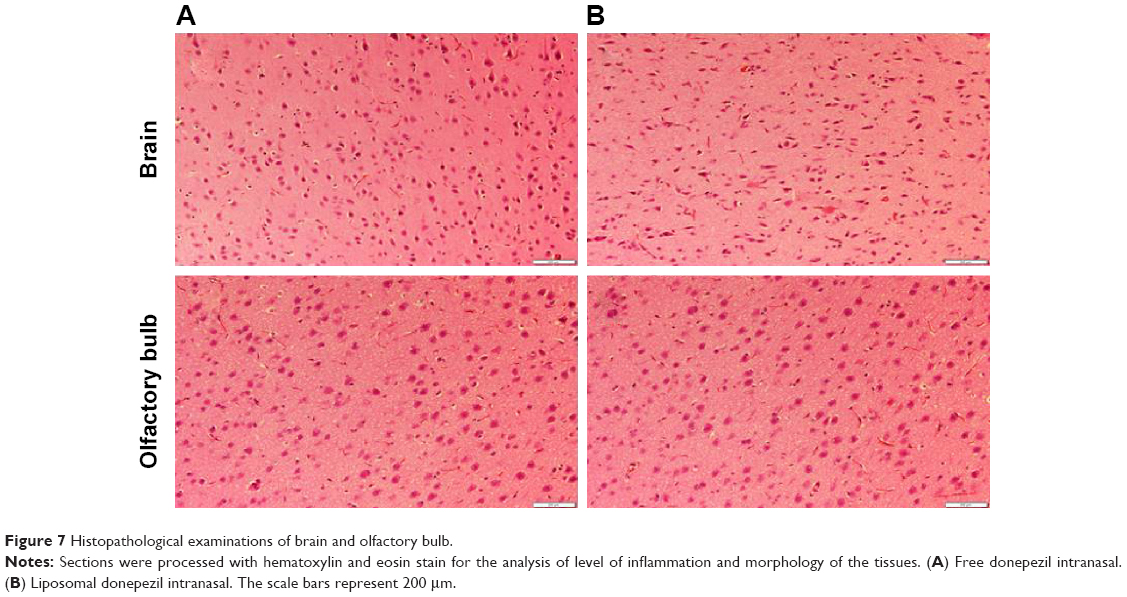 | Figure 7 Histopathological examinations of brain and olfactory bulb. |
Discussion
Treating CNS diseases is particularly challenging because the brain is designed to permit selective passage of molecules that are required for brain functions. In fact, the transport of pharmaceutical agents across BBB may be against its natural functions.24 As a result, many potent drugs are rendered ineffective due to lack of optimal drug delivery to the brain.25 Despite tremendous efforts, only limited success has been made to achieve desired bioavailability of drugs to the brain using the conventional route of administration. Intranasal cavity due to its peculiar anatomy, rich blood flow, and the large surface area has been shown to provide better bioavailability of several drugs as compared with other routes. On the other hand, the liposomal formulation has also been shown to improve drug delivery to the brain.13 However, only a few investigators have tried to study the brain bioavailability of drugs following nasal administration of liposomal formulations.16,26,27
The constituents used for liposomal formulation and their characteristics play an important role on the pharmacokinetics of drug and its bioavailability in the target organ. In this study, the liposomal formulation of DNP was prepared using DSPC, CHE along with PEG. This method of liposomal formulation has been successfully used for CNS drug delivery.16,28 Our studies on the characterization of liposomal formulation of DNP showed a uniform particle size (102±3.3 nm) and low PI (0.28±0.03), which is considered ideal for liposomal formulation.29 Furthermore, the smaller size of the vesicles has the potential to facilitate absorption of the drug.23 The liposomes demonstrated higher EE (84.91%±3.31%) for DNP, which may be attributed to the higher hydrophilicity of DNP and the composition of lipid used in the preparation of liposomal formulation. A higher EE of liposomal formulation has been associated with a better bioavailability of the drug.23,30,31 The formulation showed high zeta potential value (28.31±0.85 mV), which is an essential requirement of monodispersed stable system for efficient delivery of the drugs. Moreover, high zeta potential is associated with better physical stability and reduces the chances of coalescence.32
In vitro studies on free drug release from liposomal formulation showed an initial rapid release phase for 2 hours followed by a plateau (Figure 3). The rapid release phase could be related to the release of drug adsorbed to the surface of the liposomes. Whereas, in the second phase, the release rate slowed down, demonstrating typically sustained and prolonged drug release behavior. The results are in agreement with earlier reports suggesting the biphasic nature of liposomal dispersion.33 Drug release from liposomes mainly depends on lipid composition, the size of liposomes, and the extent of lipid packing.34 The in vitro drug release kinetics model from the DNP liposomal formulation fits well with the Weibull distribution equation (r=0.9861), which can enhance the absorption of DNP from nasal cavity (Table 4).12
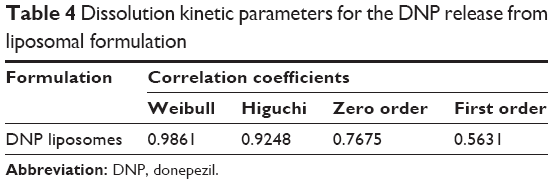 | Table 4 Dissolution kinetic parameters for the DNP release from liposomal formulation |
In vivo pharmacokinetic studies were undertaken to compare plasma and brain bioavailability of free DNP following oral and intranasal administration. The AUC of DNP following nasal administration of free DNP was significantly higher as compared with the AUC following oral route (P<0.05). Our findings are in agreement with several earlier reports suggesting better bioavailability of drugs in brain following intranasal administration as compared with oral or parenteral administration.35–37 The mechanism of drug permeation into the brain from the nasal cavity has been attributed to the rich vasculature of the nasal epithelium and olfactory pathway.24 Following intranasal administration, the transport of drug may involve endocytosis, vesicle carrier system, or neuronal transport.11 Although the olfactory pathway presents potential to bypass BBB, P-glycoproteins appear to be also functional in this area as they are present in both the olfactory epithelium and endothelial cells that surround the olfactory bulb.38 Moreover, transport of drugs from nasal cavity to CNS through trigeminal nerve has also been reported.39 Intranasal delivery of several drugs has been successfully used in brain tumors clinically and experimentally, as well as in animal models of Alzheimer’s disease.40–43
Pharmacokinetic studies on free and liposomal DNP following intranasal administration showed that the bioavailability of liposomal DNP in the brain was nearly two times higher as compared with free DNP (P<0.05). These results demonstrated rapid and greater transport of liposomal DNP into the brain. Our findings are in agreement with earlier reports showing significantly improved brain bioavailability of liposomal rivastigmine,16 indole-3-carbinol,26 and tacrine27 following intranasal administration. Jogani et al41 also reported a significant increase in brain tacrine level and fast reversal of memory following intranasal administration of emulsified drug in amnesic mice.
The lipid-based nanocarriers enhance the cellular uptake and transport of drugs across the capillary endothelium via receptor-mediated endocytosis and passage through BBB.44 Liposomal formulations have been shown to improve CNS drug delivery due to ability to encapsulate the drug molecule and protect them from fast excretion and metabolism.45 Potential advantages of liposomal drug delivery include targeted delivery, controlled release at the desired target, enhanced pharmacological activity, and reduced toxicity.46 Liposomal encapsulation also helps in delivering drugs to the brain without inflicting any damage to BBB. Our histopathological studies have clearly shown no sign of injury to the tissue structures following liposomal administration of DNP. These findings are in concurrence with an earlier report about the safety of the liposomal preparations.12
Overall, the results of the present study demonstrated that the intranasal route provided fast and enhanced bioavailability of DNP to the brain. The bioavailability of DNP further improved, when its liposomal formulation was administered by the nasal passage. The results of this study clearly suggest that intranasal route provides a safe, efficient, and convenient route of CNS drug delivery. Besides improving bioavailability, this strategy may reduce the side effects of drugs due to minimal systemic exposure. It may be concluded that nasal drug delivery of liposomal formulation provides an efficient alternate route to improving CNS delivery of DNP.
Conclusion
The liposomal formulation of DNP was found to be stable with high EE and sustained-release behavior. Intranasal administration of DNP liposomes significantly increased the brain bioavailability of the drug as compared with conventional dosage form and route of administration. The nasal administration of the liposomal formulation of CNS drug holds promise for bypassing the BBB and eliminates the risk of spillover effects of the drug to normal tissues. The advantages of intranasal delivery of liposomal formulation include rapid delivery to the CNS, avoidance of hepatic first-pass metabolism, elimination of the need for systemic delivery, and reducing unwanted systemic side effects. Moreover, this route provides painless and convenient self-administration for patients and may decrease the frequency of administration due to sustained-release behavior.
Acknowledgments
The authors are thankful to Dyanidhi Yesunayagam at Prince Sultan Military Medical City for providing us the rat plasma for the method development and validation for the study. The authors are also thankful to Manea Al Ahmari for the assistance in sample preparation and LCMS analysis.
Disclosure
The authors report no conflicts of interest in this work.
References
Whiteford HA, Ferrari AJ, Degenhardt L, Feigin V, Vos T. The global burden of mental, neurological and substance use disorders: an analysis from the global burden of disease study 2010. PLoS One. 2015;10:e0116820. | ||
Girardin F. Membrane transporter proteins: a challenge for CNS drug development. Dialogues Clin Neurosci. 2006;8(3):311–321. | ||
Matsumoto J, Takata F, Machida T, et al. Tumor necrosis factor-α-stimulated brain pericytes possess a unique cytokine and chemokine release profile and enhance microglial activation. Neurosci Lett. 2014;578:133–138. | ||
Upadhyay RK. Drug delivery systems, CNS protection, and the blood brain barrier. Biomed Res Int. 2014;2014:869269. | ||
Garcia-Garcia E, Andrieux K, Gil S, Couvreur P. Colloidal carriers and blood-brain barrier (BBB) translocation: a way to deliver drugs to the brain? Int J Pharm. 2005;298(2):274–292. | ||
Patel MM, Goyal BR, Bhadada SV, Bhatt JS, Amin AF. Getting into the brain. CNS Drugs. 2009;23(1):35–58. | ||
Chen H, Chen CC, Acosta C, Wu SY, Sun T, Konofagou EE. A new brain drug delivery strategy: focused ultrasound-enhanced intranasal drug delivery. PLoS One. 2014;9(10):e108880. | ||
Miyake MM, Bleier BS. The blood-brain barrier and nasal drug delivery to the central nervous system. Am J Rhinol Allergy. 2015;29(2):124–127. | ||
Sousa F, Castro P, Fonte P, Sarmento B. How to overcome the limitations of current insulin administration with new non-invasive delivery systems. Ther Deliv. 2015;6(1):83–94. | ||
Klossek JM. Efficacy and safety of mometasone furoate nasal spray in the treatment of sinusitis or acute rhinosinusitis. Rev Laryngol Otol Rhinol (Bord). 2007;128(3):187–192. | ||
Vyas TK, Shahiwala A, Marathe S, Misra A. Intranasal drug delivery for brain targeting. Curr Drug Deliv. 2005;2(2):165–175. | ||
Chen J, Jiang H, Wu Y, Li Y, Gao Y. A novel glycyrrhetinic acid-modified oxaliplatin liposome for liver-targeting and in vitro/vivo evaluation. Drug Des Devel Ther. 2015;9:2265–2275. | ||
Spuch C, Navarro C. Liposomes for targeted delivery of active agents against neurodegenerative diseases (Alzheimer’s disease and Parkinson’s disease). J Drug Deliv. 2011;2011:469679. | ||
Law SL, Huang KJ, Chou HY. Preparation of desmopressin-containing liposomes for intranasal delivery. J Control Release. 2001;70(3):375–382. | ||
Samad A, Sultana Y, Aqil M. Liposomal drug delivery systems: an update review. Curr Drug Deliv. 2007;4(4):297–305. | ||
Arumugam K, Subramanian G, Mallayasamy S, Averineni R, Reddy M, Udupa N. A study of rivastigmine liposomes for delivery into the brain through intranasal route. Acta Pharm. 2008;58(3):287–297. | ||
Mistry A, Stolnik S, Illum L. Nanoparticles for direct nose-to-brain delivery of drugs. Int J Pharm. 2009;379(1):146–157. | ||
Hamblin KA, Wong JP, Blanchard JD, Atkins HS. The potential of liposome-encapsulated ciprofloxacin as a tularemia therapy. Front Cell Infect Microbiol. 2014;4:79. | ||
Ali J, Ali M, Baboota S, et al. Potential of nanoparticulate drug delivery systems by intranasal administration. Curr Pharm Des. 2010;16(14):1644–1653. | ||
Zeng S, Ye M, Qiu J, et al. Preparation and characterization of genipin-cross-linked silk fibroin/chitosan sustained-release microspheres. Drug Des Devel Ther. 2015;9:2501–2514. | ||
Zaki NM, Awad GA, Mortada ND, ElHady SSA. Enhanced bioavailability of metoclopramide HCl by intranasal administration of a mucoadhesive in situ gel with modulated rheological and mucociliary transport properties. Eur J Pharm Sci. 2007;32(4):296–307. | ||
Al-Asmari AK, Ullah Z, Al-Sabaan F, Tariq M, Al-Eid A, Al-Omani SF. Effect of vitamin D on bioavailability and lipid lowering efficacy of simvastatin. Eur J Drug Metab Pharmacokinet. 2015;40(1):87–94. | ||
Wang X, Guan Q, Chen W, Hu X, Li L. Novel nanoliposomal delivery system for polydatin: preparation, characterization, and in vivo evaluation. Drug Des Devel Ther. 2015;9:1805–1813. | ||
Illum L. Is nose-to-brain transport of drugs in man a reality? J Pharm Pharmacol. 2004;56(1):3–17. | ||
Misra A, Ganesh S, Shahiwala A, Shah SP. Drug delivery to the central nervous system: a review. J Pharm Pharm Sci. 2003;6(2):252–273. | ||
Song JM, Kirtane AR, Upadhyaya P, et al. Intranasal delivery of liposomal indole-3-carbinol improves its pulmonary bioavailability. Int J Pharm. 2014;477(1–2):96–101. | ||
Corace G, Angeloni C, Malaguti M, et al. Multifunctional liposomes for nasal delivery of the anti-Alzheimer drug tacrine hydrochloride. J Liposome Res. 2014;24(4):323–335. | ||
van den Berg MP, Verhoef JC, Romeijn SG, Merkus FW. Uptake of estradiol or progesterone into the CSF following intranasal and intravenous delivery in rats. Eur J Pharm Biopharm. 2004;58(1):131–135. | ||
Haeri A, Alinaghian B, Daeihamed M, Dadashzadeh S. Preparation and characterization of stable nanoliposomal formulation of fluoxetine as a potential adjuvant therapy for drug-resistant tumors. Iran J Pharm Res. 2014;13(Suppl):3–14. | ||
Bhujbal SV, de Vos P, Niclou SP. Drug and cell encapsulation: alternative delivery options for the treatment of malignant brain tumors. Adv Drug Deliv Rev. 2014;67–68:142–153. | ||
Ungaro F, d’Angelo I, Coletta C, et al. Dry powders based on PLGA nanoparticles for pulmonary delivery of antibiotics: modulation of encapsulation efficiency, release rate and lung deposition pattern by hydrophilic polymers. J Control Release. 2012;157(1):149–159. | ||
Heurtault B, Saulnier P, Pech B, Proust JE, Benoit JP. Physico-chemical stability of colloidal lipid particles. Biomaterials. 2003;24(23):4283–4300. | ||
Catalin Balaure P, Mihai Grumezescu A. Smart synthetic polymer nanocarriers for controlled and site-specific drug delivery. Curr Top Med Chem. 2015;15(15):1424–1490. | ||
Nagayasu A, Uchiyama K, Kiwada H. The size of liposomes: a factor which affects their targeting efficiency to tumors and therapeutic activity of liposomal antitumor drugs. Adv Drug Deliv Rev. 1999;40(1):75–87. | ||
Furubayashi T, Kamaguchi A, Kawaharada K, et al. Evaluation of the contribution of the nasal cavity and gastrointestinal tract to drug absorption following nasal application to rats. Biol Pharm Bull. 2007;30(3):608–611. | ||
Varshosaz J, Sadrai H, Heidari A. Nasal delivery of insulin using bioadhesive chitosan gels. Drug Delivery. 2006;13(1):31–38. | ||
Ikechukwu Ugwoke M, Kaufmann G, Verbeke N, Kinget R. Intranasal bioavailability of apomorphine from carboxymethylcellulose-based drug delivery systems. Int J Pharm. 2000;202(1–2):125–131. | ||
Graff CL, Pollack GM. Nasal drug administration: potential for targeted central nervous system delivery. J Pharm Sci. 2005;94(6):1187–1195. | ||
Dhuria SV, Hanson LR, Frey WH 2nd. Intranasal delivery to the central nervous system: mechanisms and experimental considerations. J Pharm Sci. 2010;99(4):1654–1673. | ||
Hashizume R, Ozawa T, Gryaznov SM, et al. New therapeutic approach for brain tumors: intranasal delivery of telomerase inhibitor GRN163. Neuro Oncol. 2008;10(2):112–120. | ||
Jogani VV, Shah PJ, Mishra P, Mishra AK, Misra AR. Intranasal mucoadhesive microemulsion of tacrine to improve brain targeting. Alzheimer Dis Assoc Disord. 2008;22(2):116–124. | ||
Jogani VV, Shah PJ, Mishra P, Mishra AK, Misra AR. Nose-to-brain delivery of tacrine. J Pharm Pharmacol. 2007;59(9):1199–1205. | ||
Doyle KP, Yang T, Lessov NS, et al. Nasal administration of osteopontin peptide mimetics confers neuroprotection in stroke. J Cereb Blood Flow Metab. 2008;28(6):1235–1248. | ||
Martin-Banderas L, Holgado MA, Venero JL, Alvarez-Fuentes J, Fernandez-Arevalo M. Nanostructures for drug delivery to the brain. Curr Med Chem. 2011;18(34):5303–5321. | ||
Jain KK. Nanobiotechnology-based strategies for crossing the blood-brain barrier. Nanomedicine (Lond). 2012;7(8):1225–1233. | ||
Tosi G, Costantino L, Ruozi B, Forni F, Vandelli MA. Polymeric nanoparticles for the drug delivery to the central nervous system. Expert Opin Drug Deliv. 2008;5(2):155–174. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.