Back to Journals » OncoTargets and Therapy » Volume 7
Preparation and functional studies of hydroxyethyl chitosan nanoparticles loaded with anti-human death receptor 5 single-chain antibody
Authors Yang J, Huang X, Luo F ![]() , Cheng X, Cheng L, Liu B, Chen L, Hu R, Shi C, Zhuang G, Yin P
, Cheng X, Cheng L, Liu B, Chen L, Hu R, Shi C, Zhuang G, Yin P
Received 30 December 2013
Accepted for publication 11 March 2014
Published 21 May 2014 Volume 2014:7 Pages 779—787
DOI https://doi.org/10.2147/OTT.S59872
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Jingjing Yang,1,3,* Xiaoping Huang,1,3,* Fanghong Luo,1 Xiaofeng Cheng,3 Lianna Cheng,3 Bin Liu,4 Lihong Chen,2 Ruyi Hu,1,3 Chunyan Shi,1,3 Guohong Zhuang,1,3 Ping Yin2
1Anti-Cancer Research Center, Medical College, Xiamen University, Fujian, People's Republic of China, 2The Department of Pathology, Zhongshan Hospital, Xiamen University, Xiamen, People's Republic of China, 3Organ transplantation institution, Xiamen University, Xiamen, People's Republic of China, 4Jilin Vocational College of Industry and Technology, Jilin, People's Republic of China
*These authors contributed equally to this work
Objective: To prepare hydroxyethyl chitosan nanoparticles loaded with anti-human death receptor 5 single-chain antibody, and study their characteristics, functions, and mechanisms of action.
Materials and methods: The anti-human death receptor 5 single-chain antibody was constructed and expressed. Protein-loaded hydroxyethyl chitosan nanoparticles were prepared, and their size, morphology, particle-size distribution and surface zeta potential were measured by scanning electron microscopy and laser particle-size analysis. Mouse H22 hepatocellular carcinoma cells were cultured, and growth inhibition was examined using the CellTiter-Blue cell-viability assay. Flow cytometry and Hoechst 33342 were employed to measure cell apoptosis. Kunming mice with H22 tumor models were treated with protein-loaded hydroxyethyl chitosan nanoparticles, and their body weight and tumor size were measured, while hematoxylin and eosin staining was used to detect antitumor effects in vivo and side effects from tumors.
Results: The protein-loaded hydroxyethyl chitosan nanoparticles had good stability; the zeta potential was -24.2±0.205, and the dispersion index was 0.203. The inhibition of the protein-loaded hydroxyethyl chitosan nanoparticles on H22 growth was both time- and dose-dependent. Increased expressions of active caspase 8, active caspase 3, and BAX were detected following treatment. The average weight gain, tumor weight, and mean tumor volume of the protein and protein-loaded hydroxyethyl chitosan nanoparticle groups were significantly different (P<0.05) compared with the phosphate-buffered saline group.
Conclusion: The protein-loaded hydroxyethyl chitosan nanoparticles effectively suppressed tumor growth, indicating that nanotechnology has the potential for broad application in cancer therapy.
Keywords: anticancer effect, DR5, GCS-aDR5ScFv, H22
Introduction
Hepatocellular carcinomas (HCCs) are malignant tumors of the liver that include both primary and metastatic cancers. The annual global incidence of HCC patients is about 100 million, representing approximately a fifth of malignant tumors, and the incidence and mortality worldwide are the same as in 2012.1,2 The treatment of HCC is still dominated by surgery, radiation, and drug therapy. Therefore, new therapies that can specifically kill tumor cells, avoiding or reducing injury to normal adjacent tissues,3 such as molecule-targeting drugs, provide new options for HCC treatment.
Death receptor (DR)-5 (or tumor necrosis factor [TNF]-related apoptosis-inducing ligand [TRAIL] receptor 2), a member of the TNF-receptor superfamily, has been emerging as a favorable target for the design of anticancer reagents in recent years.4 Our studies have suggested that when the anti-human DR5 (aDR5) single-chain fragment variable (scFv) antibody binds to DR5, the cytoplasmic death domain of DR5 undergoes oligomerization, which results in the propagation of apoptotic signaling cascades through the formation of death-inducing signaling complexes.5–8 This selectively induces cell death in a wide variety of tumor cells, both in vitro and in vivo.7–10 One study11 found that both DR4 and DR5 are expressed in HCC and other cancers, where aDR5 scFv could be applied for treatment. Despite the advantages of using scFv molecules for immunotherapy, especially for chronic diseases, treatment efficacy is often compromised by the rapid blood clearance of infused scFv antibodies and the difficulty in maintaining high local concentrations of active molecules at the target site.12 Furthermore, antibodies are easily degraded in vitro, making their preparation difficult. Therefore, there is a need to discover a fine carrier that protects and enhances the drug effects of antibodies.
Chitosan is a polycation gene carrier with non-toxic, no hemolysis, nonsensitized, biodegradable, biocompatible and a serious of favorable biological properties.13,14 Nanoparticles formed by rapid mixing of chitosan and deoxyribonucleic acid have significant potential as a gene-delivery vector. Modern nanotechnology, when used in effective drug-delivery systems, can deliver small molecules and biological macromolecular therapeutic drugs.15 Several carriers derived from chitosan have been developed as drug-delivery systems.16 Furthermore, both chitosan and its derivatives have become the focus of protein-nanoparticle research in recent years. Their specific structure and physicochemical properties give them good biological compatibility and biodegradability, low immunogenicity, and high biological activity, and make them good carriers for chemical drugs, antibodies, protein peptides, and genes. Nanoparticles are small and have a large surface area and high mucoadhesion properties, which are conducive to local administration17 and expansion to the contact area surrounding the intestinal wall, extended exposure time, and improved drug gastrointestinal stability. These properties improve oral drug-absorption bioavailability following administration of nanoparticle carriers.18 In this study, we prepared glycol chitosan (GCS)-aDR5 scFv and examined its characteristics, including the in vitro and in vivo antitumor effects and protein-inhibitory mechanisms, to study further the features and improve the effects of antibody drugs. This we hope will lay a solid foundation for future nanoparticle drug-targeting treatments for tumors.
Materials and methods
Characterization of aDR5 scFv
Expression and purification of human aDR5 scFv
The obtained recombination bacteria was selected and cultured at 37°C in 3 mL Luria–Bertani broth (LB medium) for 8–12 hours, added to 300 mL of fresh LB medium at 37°C/250 rpm. After 7 hours, when the cell density reached an optical density at 590 nm (OD590) of 0.6–0.8, isopropyl-β-d-thiogalactoside (IPTG) was added to 300 mL of LB medium to induce expression. The harvested Escherichia coli Rosetta-gami™ (EMD Millipore, Billerica, MA, USA) cells from shaker flasks were disintegrated with ultrasonication (300 W, 20 minutes), then precipitated by centrifugation (12,000 rpm, 20 minutes). The target protein was mainly present in inclusion bodies. The inclusion bodies were washed at 4°C for 7–8 hours in a washing fluid containing 2 mol/L urea. After washing, the inclusion bodies were dissolved in a solution containing 8 mol/L urea. The solution was centrifuged, and the supernatant was applied to a Ni-nitrilotriacetic acid affinity column (GE Healthcare Life Sciences, Piscataway, NJ, USA) to purify the protein. After loading and eluting, the targeted fractions were pooled and identified by sodium dodecyl sulfate (SDS) polyacrylamide gel electrophoresis (PAGE).
Immunofluorescence labeling with aDR5 scFv
SW480 cells were seeded on 22×22 mm glass coverslips and grown for 36 hours supplemented with 10% (volume/volume [v/v]) fetal bovine serum. Cells were washed with phosphate-buffered saline (PBS) and fixed for 10 minutes at −20°C in 50% (v/v) acetone/methanol and then air-dried. Slides were blocked by incubating for 2 hours in 5% (weight/volume [w/v]) bovine serum albumin in PBS and then incubated with fluorescein isothiocyanate (FITC)-labeled aDR5 scFv for 2 hours at room temperature. Stained cells were washed, then mounted in 50% (v/v) glycerol–PBS and examined by epifluorescence microscopy.
aDR5 scFv and aDR5 monoclonal antibody titers
Purified horseradish peroxidase-conjugated aDR5 scFv was diluted in a coating buffer (0.05 M carbonate-bicarbonate buffer, pH 9.6) to a final concentration of 4 μg/mL. Each well of 96-well microtiter plates was coated with 100 μL of the protein and left overnight at 4°C. Plates were washed to remove unbound peptides, and binding intensity of peptides was determined spectrophotometrically. The cutoff value was defined as the mean value plus three standard deviations of the mean OD.
Preparation and properties of GCS-aDR5 scFv
Preparation of GCS-aDR5 scFv
We put 4 mL 2 mg/mL GCS (82.1 kD) solution in a 25 mL three-neck flask, then added proper aDR5 scFv under magnetic stirring, and then applied glacial acetic acid to adjust the pH to 4.5. The particles formed spontaneously upon the addition of 20–40 drop/minute of a 1 g · L−1 TPP (mesotetraphenylporphyrin and 5,10,15,20-tetraphenyl-21H,23H-porphine, [C44H30N4]) solution until the solution presented opalescence, and then stirring was continued for 20 minutes. For the association of aDR5 scFv with GCS nanoparticles, aDR5 scFv was incorporated in the TPP solution.
Cell lines and mice
Hepatocellular H22 cells were obtained from the China Center for Type Culture Collection (Wuhan, People’s Republic of China). Cells were cultured in flasks with Dulbecco’s Modified Eagle’s Medium (GIBCO®DMEM, Thermo Fisher Scientific, Waltham, MA, USA) supplemented with 10% (v/v) fetal bovine serum and 1% penicillin–streptomycin at 37°C in a humidified atmosphere of 5% CO2. To facilitate attachment and growth, the culture medium was changed 4 hours later, after which culture-medium change was performed every 2–4 days. After immediate isolation, viable hepatocytes were at least 90%–95% of the total.
All the experiments were performed using 6- to 8-week-old female BALB/c mice and 5- to 7-week-old nude mice purchased from the Model Animal Research Center of Xiamen University Medical College. All the animals were housed under specific pathogen-free conditions with constant access to water and chow. Mice were allowed to acclimatize for at least 1 week before experiments. Briefly, all experimental procedures were carried out following approval of the Animal Care and Using Committee of Xiamen University.
In vivo imaging
Hepatocellular H22 cells were collected at the logarithmic phase, and 6×107 cells were suspended in 0.1 mL serum-free medium, which was injected into the armpit of each nude mouse. The size of the transplanted tumors was observed twice every other day until they reached the target size. The drugs were injected into the mice through subcutaneous injection. One group was selected as the control with CY5.5 (C41H41K3N2O14S4), and the others were treated with CY5.5-labeled aDR5 scFv and GCS-aDR5 scFv. Fluorescence images were acquired before treatment, immediately after, and within 2 hours after treatment.
CellTiter-Blue method
Cell concentration was adjusted to 1×104, then seeded in a 96-well cell-culture plate. Different concentrations of GCS were added, and were calculated by five parallel holes each. Cells were cultured for 4 hours, assay plates removed from the 37°C incubator, and 20 μL/well CellTiter-Blue® (Promega, Fitchburg, WI, USA) added. They were then incubated using standard cell-culture conditions for 1–4 hours. The plate was shaken for 10 seconds, and fluorescence recorded at 570 nm. The experiment was repeated three times, and mean OD570 of each concentration of holes was taken each time. The proliferation-inhibition rate = (control well OD values - experimental well OD value)/control hole OD value ×100%.
Inhibition effect and cell-apoptosis detection
In vivo animal tumor model experiment
Tumors were generated in male Kunming mice by intramuscular injection of H22 cells (1×107 cells in 100 μL PBS) into the right hind leg of each mouse. All tumor-bearing mice were divided randomly into groups (eight mice/group). The treatments were initiated on day 3, when tumor volume reached about 40–50 mm3 (designated as day 0), and were performed once every 2 days for 2 weeks. The drugs were injected into mice subcutaneously (caudal vein). One group was selected as the control (sham-exposed), and the others were treated with PBS, GCS nanoparticles, aDR5 scFv, and GCS-aDR5 scFv. Mice were weighed daily after injection for clinical signs and the survival state.
Apoptosis assay by flow cytometry
To determine apoptosis, GCS-aDR5 scFv treated or untreated cells (25 μg/mL, 1 hour) were washed in PBS and resuspended in binding buffer at a concentration of 1×106 cells/L. After incubation, 195 μL of the solution was transferred to a 5 mL culture tube, with 5 μL annexin V-FITC (BD Biosciences, San Jose, CA, USA) added. The tube was then incubated for 30 minutes at room temperature in the dark. The cells were washed with binding buffer and resuspended in 190 μL binding buffer, with 10 μL propidium iodide added. Finally, the tube was gently vortexed and incubated for another 30 minutes in the dark. Cells were analyzed by FACSCalibur™ with CellQuest™ software (BD Biosciences).
Hoechst 33342 method
H22 cells were adjusted to 1×106/mL and resuspended. A coverslip was added to a six-well cell-culture plate first, cells were planted 2 mL/well in it, and the final cell concentration was 3×104/well. This was cultured at 37°C, 5% CO2, until cells covered 60% of the coverslip. GCS-aDR5ScFv, aDR5ScFv, and GCS (0.136 mg/mL, 2 mL) were respectively added as the experiment group; PBS was set as the control. The supernatant was cast after culturing in the plate for 4 hours. Cells were washed with PBS, and 1 mL Hoechst 33342/well was added. Another 20–30 minutes was necessary for the preparation of GCS-aDR5ScFv. The staining solution was cast and washed with PBS 2–3 times. Staining of apoptosis cells was detected by fluorescence microscopy.
Hematoxylin and eosin staining analysis
To distinguish the antitumor effects of PBS, GCS nanoparticles, aDR5ScFv and GCS-aDR5ScFv in vivo, hematoxylin and eosin staining was used. Tumor and liver tissues were harvested after mice had died (eight per group). The hematoxylin and eosin-stained tissues were examined for necrotic cells, tumor cells, or apoptotic cells. Hematoxylin and eosin-stained sections were viewed using a BHT microscope (Olympus, Tokyo, Japan).
Protein extraction and Western blot analysis
All cells were harvested at approximately 80% confluence. Protein concentration was determined using a bicinchoninic acid protein-assay kit (Pierce™; Thermo Fisher Scientific, Waltham, MA, USA). Protein (30 μg) samples were run on 12% SDS-PAGE, blotted onto a nylon membrane, probed with antibodies against β-actin, caspase 8, caspase 3, and BAX (Santa Cruz Biotechnology, Dallas, TX, USA), and incubated with a 1:2,000 dilution of horseradish peroxide-conjugated secondary antibodies (Promega) for 60 minutes at room temperature. Membranes were washed with 0.05% (v/v) Tween 20 in PBS (pH 7.6), and the bands were detected by using an enhanced chemiluminescence-detection system, with β-actin serving as the control. Scanning densitometry was performed using Adobe Photoshop (Apple, Cupertino, CA).
Analysis of toxicity
A mouse-weight curve was drawn by testing the weight of mice once a day from the first day of administration for the comparison of treatment group and control group mouse-weight changes.
Statistical analysis
Data are presented as means ± standard deviation per group. Statistical analysis was made for multiple comparisons using analysis of variance and Student’s t-test. P-values <0.05 were considered to be statistically significant.
Results
Characterization of aDR5 scFv
The E. coli expression system was used to prepare aDR5 scFv in inclusion form. We identified that the appropriate inducing conditions were 0.6 mM IPTG at 37°C for 12 hours, following optimization using different induction times and IPTG concentrations. The purity of the target protein was >95% after isolation and purification. The mobility of the purified protein corresponded to a molecular weight of about 30 kDa by SDS-PAGE (Figure 1A), and the purified protein bound especially to eDR5. More eDR5 scFv complex was seen with increasing scFv concentration. As shown in Figure 1B, the 25 kD DR5 extracellular domain constructed by our lab (eDR5), was capable of binding to aDR5 scFv. eDR5 degeneration products (10 μg) were combined with 0.315 μg, 0.625 μg, 1.25 μg, 2.5 μg, 5 μg, and 10 μg aDR5 scFv for SDS-PAGE analysis by Coomassie staining to determine their binding capacity (Figure 1B). Figure 1B shows that all of the aDR5 scFvs combined with eDR5 to form complexes at a concentration of 0.315 μg aDR5 scFv, while the unbound eDR5 bands increased correspondingly with increased aDR5 scFv. Complex formation reached the maximum at 5 μg aDR5 scFv, indicating that eDR5 specifically binds to aDR5 scFv.
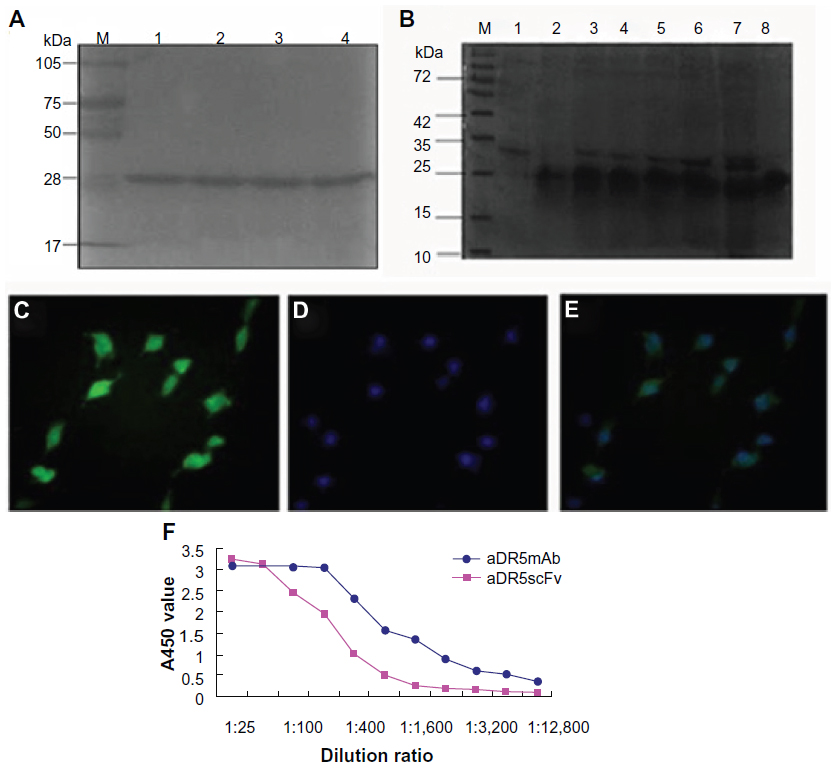 | Figure 1 (A–F) Purification and identification of recombinant aDR5ScFv protein. |
To assess the combination of recombinant aDR5 scFv and eDR5 further, SW480 cells were used as the model, as they express DR5 highly on the plasma membrane. For immunofluorescence analysis, aDR5 scFv was fluorescently labeled with FITC. Cells were treated with FITC-aDR5 scFv, and the observed green fluorescence was observed in almost all cells (Figure 1C), clearly revealing that FITC-aDR5 scFv could especially bind to DR5 on the cell membrane. Hoechst 33342 staining was used to stain the nuclei of all cells (Figure 1D). The combined image clearly shows that almost all cells were positive for FITC-aDR5 scFv (Figure 1E), which was consistent with the results assessed by SDS-PAGE. We then compared the affinity of an aDR5 scFv fragment with a commercially obtained anti-DR5 monoclonal antibody. Although the aDR5 scFv fragment had a lower affinity for its antigen, it displayed similar binding activity as the traditional anti-DR5 monoclonal antibody (Figure 1F).
Zeta-potential and distribution properties of GCS-aDR5 scFv
To improve the targeting properties of aDR5 scFv, we prepared GCS-aDR5 scFv nanoparticles. The average diameter of nanoparticles ranges from 10 to 1,000 nm, while the average particle size of GCS-aDR5 scFv was 203.7±0.186 nm, following protein adsorption on the surface. Furthermore, the GCS-aDR5 scFv was of good stability, polydispersity index (data not shown), and had a surface zeta potential of −24.2±0.205 mV. GCS and GCS-aDR5 scFv were dense spherical-shaped nanoparticles with uniform shape under scanning electron microscopy (Figure 2A and B). Compared with GCS, GCS-aDR5 scFv had a comparatively uniform morphology with minor parts of dispersion.
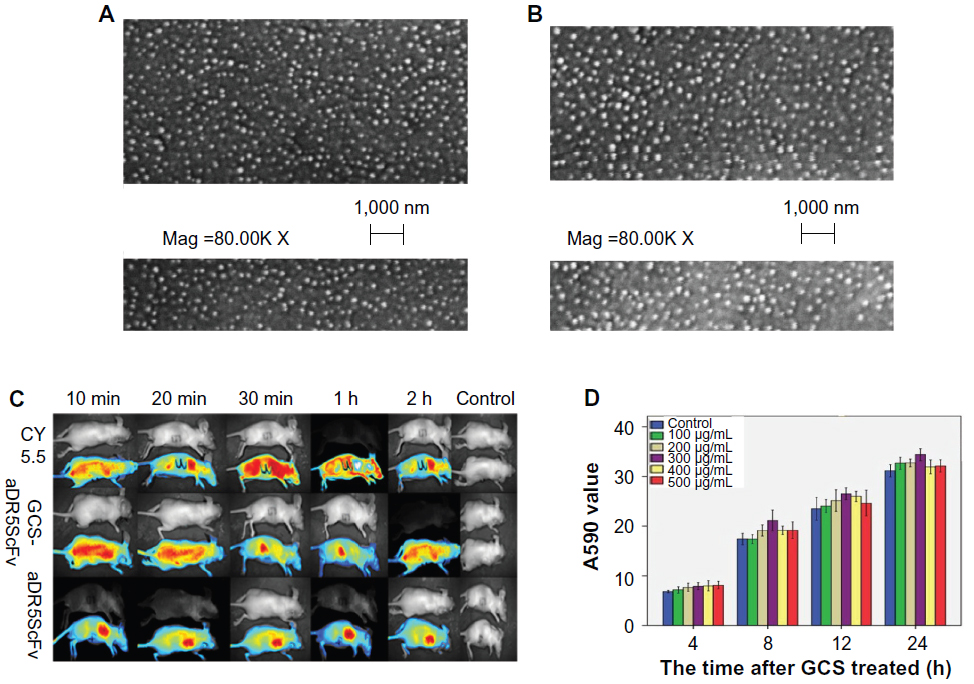 | Figure 2 (A–D) Distribution and morphological properties of glycol chitosan (GCS). |
The distribution properties of the prepared GCS-aDR5 scFv were also tested. To investigate the tumor-targeting ability of GCS-aDR5 scFv and aDR5 scFv in vivo, we intramuscularly injected H22 cells into the armpit of each mouse. The results showed that GCS-aDR5 scFv was widely distributed within the first 10 minutes, following which it gradually targeted the tumors. The control inhibited widespread fluorescence until 30 minutes and decreased afterwards. The administration of aDR5 scFv alone resulted in enrichment at the injection site (Figure 2C). The results showed that GCS-aDR5 scFv was targeted to the tumors much more efficiently than healthy organs, whereas the control nanoparticles without antibody had no such targeting ability.
Next, we examined the toxic effects of GCS-aDR5 scFv. H22 cells were treated with GCS-aDR5 scFv for 4, 8, 12, and 24 hours (Figure 2D). The CellTiter-Blue assay was used to examine cell viability, based on the mechanism that living cells convert resazurin into a fluorescent end product, whereas dead cells do not generate any fluorescent signal. The OD value of various concentrations of GCS alone were compared with a PBS control at different time points. The difference was not significant during the early time points, but with increasing time the concentrations required for the same effects differed (Figure 2C). Therefore, GCS had almost no toxic effects on H22 tumor cells.
Inhibitory effect analysis
GCS-aDR5 scFv and aDR5 scFv had lethal effects on H22 cells. The growth-inhibition rate increased with increasing dosage and treatment period with GCS-aDR5 scFv and aDR5 scFv (Figure 3A). Flow-cytometry results suggested that 0.136 mg/mL aDR5 scFv, GCS-aDR5 scFv, or GCS treatment of H22 cells for 12 hours caused 52.5%, 25.2%, and 3.3% apoptosis, respectively (Figure 3B). This indicates that GCS-aDR5 scFv and aDR5 scFv induced tumor cell apoptosis in vitro, with GCS having basically no toxic effects.20 Hoechst 33342 staining was used to detect H22 cell apoptosis after 0.136 mg/mL aDR5 scFv, GCS-aDR5 scFv, or GCS treatment for 24 hours (Figure 3C). The results showed that the nuclei of cells in the GCS-aDR5 scFv and aDR5 scFv groups exhibited significant fragmentation and apoptosis, whereas in the GCS group the cell shape was round, and nuclear pyknosis was observed with almost no apoptosis.
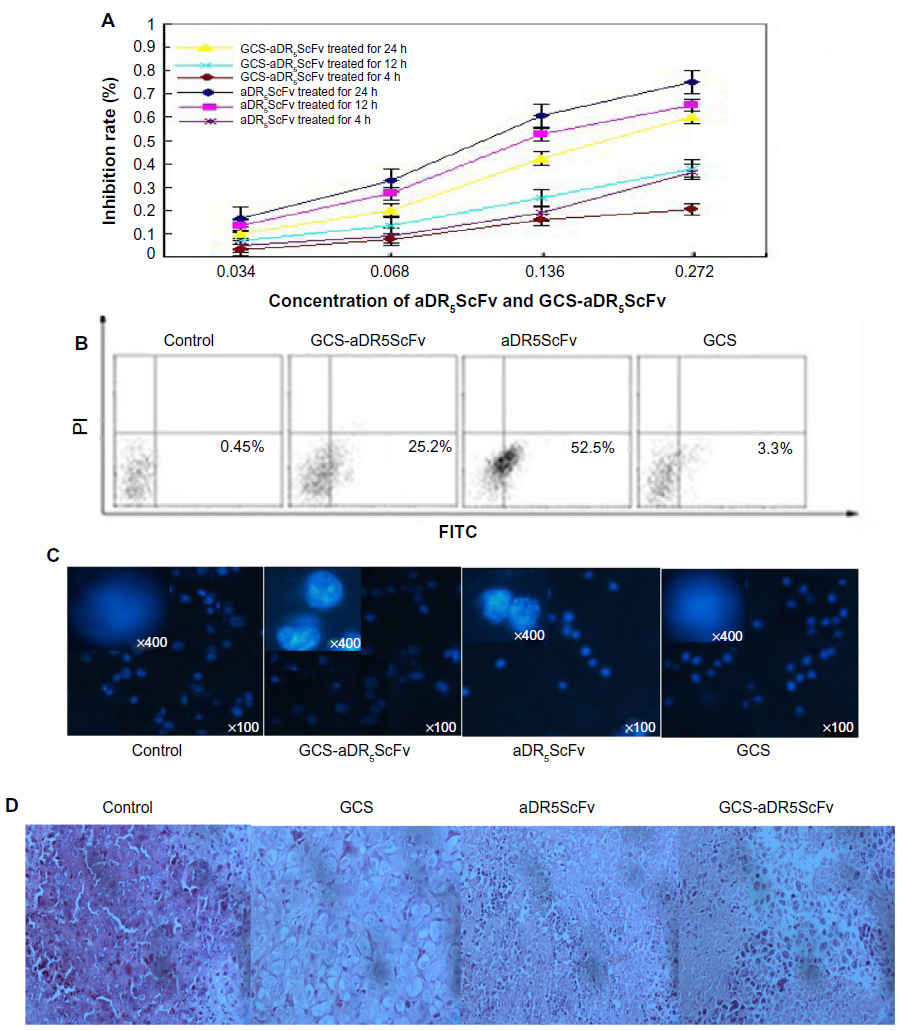 | Figure 3 (A–D) In vivo and in vivo studies on the inhibition effects of glycol chitosan (GCS) aDR5 single-chain fragment variable (scFv). |
Hematoxylin and eosin staining revealed a large volume of tumor cells in the PBS-group tumor sections, the size of which was significant and interspersed in the fibrous tissue. Part of the cytoplasm was transparent, and some myxoid changes could be seen. The tumor cells of the GCS-aDR5 scFv-treatment group were obviously undergoing death, with more visibly dead cells located in areas of lymphocytic infiltration and a multifocal necrotic zone around the tumor. The ADR5 scFv-treatment group tumor cells were damaged, with some areas of necrosis and distinct hyperplasia around tumors, and scattered dead cells and infiltrating lymphocytes visible in the tumor tissues. Compared with the PBS group, the GCS group had no typically round cells, with badly stained nuclei, irregular chromatin, or condensed cytoplasm (Figure 3D).
Mechanism of cell apoptosis
Detection of active caspase 8, active caspase 3, and BAX protein expressions
Using the PBS group as control, Western blotting was applied to detect the expression of active caspase 8, active caspase 3, and BAX after treatment with 0.136 mg/mL GCS-aDR5 scFv, aDR5 scFv, or GCS for 24 hours to determine their effect on hepatocellular H22 cells. In the GCS-aDR5 scFv and aDR5ScFv groups, active caspase 8, active caspase 3, and BAX expressions were upregulated compared with the PBS group; however, the GCS group did not change significantly. In the GCS-aDR5 scFv group, active caspase 8, active caspase 3, and BAX expressions were not obviously increased when compared with the aDR5 scFv group (Figure 4).
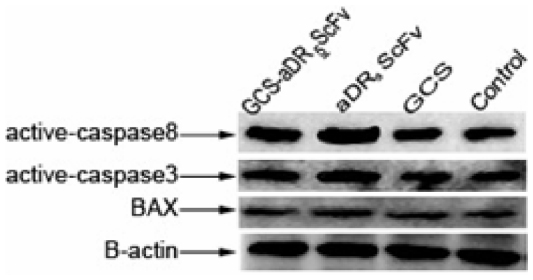 | Figure 4 The mechanism of cell apoptosis. |
Analysis of toxicity
Mouse-weight curves were obtained by measuring the weight of mice every day from the first day of administration for comparison of the treatment- and control-group weight changes. The body weight of mice did not change significantly until day 4 after administration. The average weight gain in mice of the GCS-aDR5 scFv group was 8.2 g, which was higher than that of the PBS group (2.5 g), and the difference was significant (P<0.05) between these groups from days 5 to 12 (Figure 5). The weight of mice was not significantly different between the GCS and PBS groups. Furthermore, the mice were in good condition and had active behavior throughout the study, suggesting that GCS-aDR5 scFv and aDR5 scFv administration had no obvious side effects.
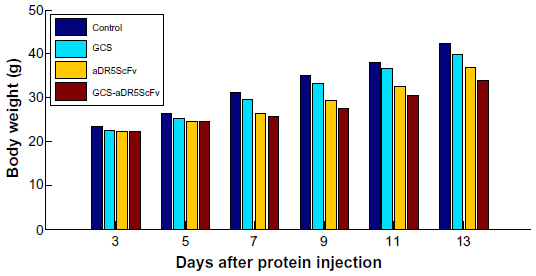 | Figure 5 Analysis of toxicity. Comparison of body weight in H22 tumor-bearing mice of each group. |
Discussion
Our laboratory successfully prepared and purified a highly pure and stable aDR5 scFv, which could specifically bind to eDR5. FITC-labeled aDR5 scFv binds to SW480 cells especially, which express membrane-associated DR5 highly, with green fluorescence seen in almost all cells. We also constructed and expressed blank GCS nanoparticles and GCS-aDR5 scFv by ion gelation. Scanning electron microscopy and laser particle-size analysis indicated that GCS and GCS-aDR5 scFv had good stability and similar morphology by measuring their surface zeta potential and polydispersity index.
CellTiter-Blue detection revealed that GCS had no toxic effects on H22 tumor cells, indicating that GCS is a favorable drug carrier with good biological compatibility, which confirms previous reports that GCS has no toxic effects on living tissues.20 GCS-aDR5 scFv and aDR5 scFv have lethal effects on H22 cells. The H22 growth-inhibition rate also increased with increasing dose and time of GCS-aDR5 scFv and aDR5 scFv treatments. Flow cytometry was used to determine the optimum time and concentration for inducing death. The results suggested that 0.136 mg/mL aDR5 scFv, GCS-aDR5 scFv, or GCS treatment of H22 cells for 12 hours caused 52.5%, 25.2%, and 3.3% apoptosis, respectively. Together with Hoechst 33342 staining, this indicates that GCS-aDR5 scFv and aDR5 scFv both induce apoptosis of tumor cells in vitro.
In vitro experiments also indicated that the efficiency of HCC apoptosis induced by GCS-aDR5 scFv was less than pure aDR5 scFv. Tumor blood is rich in blood vessels, and it has a special composition, characterized by larger vascular endothelial gaps of approximately 400–800 nm, and less lymphatic return compared with normal tissue.21 Nanodrugs can be targeted selectively to tumor tissue to bring higher drug concentrations, with these effects in the tumor tissue called the enhanced permeability and retention effect. Targeting nanodrugs further improves tumor targeting through surface-specific ligand-mediated endocytosis, which simultaneously reduces accumulation in normal tissue, thereby increasing tolerance toward chemotherapy, improving the clinically complete remission rate of patients, and reducing drug resistance in tumor cells.22–24 The inferiority of GCS-aDR5 scFv to aDR5 scFv at inducing a high rate of apoptosis in H22 cells is possibly due to the enhanced permeability and retention effect not existing in the in vitro environment.
In vivo antitumor experiments found that the mean weight and tumor volume in mice of the GCS-aDR5 scFv and aDR5 scFv groups were significantly different (P<0.05) compared with the PBS group. Hematoxylin and eosin staining revealed that the tumor cells of the GCS-aDR5 scFv-treatment group underwent increased death○. Compared with the PBS group, the GCS group had no typically round cells, badly stained nuclei, irregular chromatin, or condensed cytoplasm, indicating that GCS had no toxic effects on live tissue,25 which was in agreement with the literature.
Western blotting indicated that the mechanism of increased internal tumor cell apoptosis induced by GCS-aDR5 scFv and aDR5 scFv may result in the upregulation of active caspase 8, active caspase 3, and BAX protein expressions. Currently, TRAIL-induced apoptosis involves c-FLIP, XIAP, members of the BCL-2 family, DISC formation, and regulation of caspase 8 activity.26 TRAIL-induced apoptosis involves both the exogenous and endogenous apoptotic pathways. The increased expression of active caspase 8, active caspase 3, and BAX in the experiment may have been activated upon binding of aDR5 scFv with DR5 to induce apoptosis and activate active caspase 8, active caspase 3, and BAX.
The targeting of therapies and drugs specific to tumor sites has become a hotspot of research in recent years. Furthermore, cancer-research studies have increasingly investigated nanoparticles as a delivery carrier for treatment using tumor-specific drugs. This study showed that the ion-gelation preparation of GCS-aDR5 scFv was effective in the inhibition of tumor growth, with GCS-aDR5 scFv exhibiting very good stability. These results may help lay the foundation for the future use of protein nanoparticles for drug targeting in cancer-treatment research.
Acknowledgments
This work was partially supported by the Natural Science Foundation of China (81072472) and the Xiamen Municipal Science and the Technology Foundation of China (3502Z20124045).
Disclosure
The authors report no conflicts of interest in this work.
References
Marrero JA. Hepatocellular carcinoma. Curr Opin Gastroenterol. 2006;22:248–253. | |
Motola-Kuba D, Zamora-Valdés D, Uribe M, Méndez-Sánchez N. Hepatocellular carcinoma. An overview. Ann Hepatol. 2006;5:16–24. | |
Vasir JK, Red MK, Labhasetwar VD. Nanosystems in drug targeting: opportunities and challenges. Curr Nanosci. 2005;1:47–64. | |
French LE, Tschopp J. Protein-based therapeutic approaches targeting death receptors. Cell Death Differ. 2003;10:117–123. | |
Sheridan JP, Marsters SA, Pitti RM, et al. Control of TRAIL-induced apoptosis by a family of signaling and decoy receptors. Science. 1997;277:818–821. | |
Chaudhary PM, Eby M, Jasmin A, Bookwalter A, Murray J, Hood L. Death receptor 5, a new member of the TNFR family, and DR4 induce FADD-dependent apoptosis and activate the NF-κB pathway. Immunity. 1997;7:821–830. | |
Kischkel FC, Lawrence DA, Chuntharapai A, Schow P, Kim KJ, Ashkenazi A. Apo2L/TRAIL-dependent recruitment of endogenous FADD and caspase-8 to death receptor 4 and 5. Immunity. 2000;12:611–620. | |
Suliman A, Lam A, Datta R, Srivastava RK. Intracellular mechanisms of TRAIL: apoptosis through mitochondrial-dependent and -independent pathways. Oncogene. 2001;20:2122–2133. | |
Hesry V, Piquet-Pellorce C, Travert M, et al. Sensitivity of prostate cells to TRAIL-induced apoptosis increases with tumor progression: DR5 and caspase 8 are key players. Prostate. 2006;66:987–995. | |
Walczak H, Miller RE, Ariail K, et al. Tumoricidal activity of tumor necrosis factor related apoptosis-inducing ligand in vivo. Nat Med. 1999;5:157–162. | |
Miyashita T, Kawakami A, Nakashima T, et al. Osteoprotegerin (OPG) acts as an endogenous decoy receptor in tumour necrosis factor-related apoptosis-inducing ligand (TRAIL)-mediated apoptosis of fibroblast-like synovial cells. Clin Exp Immunol. 2004;137:430–436. | |
Hudson PJ, Souriau C. Engineered antibodies. Nat Med. 2003;9:129–134. | |
Kim TH, Jiang HL, Nah JW, Cho MH, Akaike T, Cho CS. Receptor-mediated gene delivery using chemically modified chitosan. Biomed Mater. 2007;2:95–100. | |
Zhang X, Ercelen S, Duportail G, et al. Hydrophobically modified low molecular weight chitosans as efficient and nontoxic gene delivery vectors. J Gene Med. 2008;10:527–539. | |
Nam HY, Kwon SM, Chung H, et al. Cellular uptake mechanism and intracellular fate of hydrophobically modified glycol chitosan nanoparticles. J Control Release. 2009;135:259–267. | |
Park JH, Kwon SG, Nam JO, et al. Self-assembled nanoparticles based on glycol chitosan bearing 5β-cholanic acid for RGD peptide delivery. J Control Release. 2004;95:579–588. | |
Huang M, Wu W, Qian J, Wan DJ, Wei XL, Zhu JH. Body distribution and in situ evading of phagocytic up take by macrophages of long-circulating poly(ethylene glycol) cyanoacrylate-co-n-hexadecyl cyanoacrylate nanoparticles. Acta Pharmacol Sin. 2005;26:1512–1518. | |
Maeda H, Wu J, Sawa T, Matsumura Y, Hori K. Tumor vascular permeability and the EPR effect in macromolecular therapeutics: a review. J Control Release. 2000;65:271–278. | |
Kranjc S, Cemazar M, Grosel A, Sentjurc M, Sersa G. Radiosensitising effect of electrochemotherapy with bleomycin in LPB sarcoma cells and tumors in mice. BMC Cancer. 2005;5:115. | |
Saboktakin MR, Tabatabaie RM, Maharramov A, Ramazanov MA. Synthesis and characterization of pH-dependent glycol chitosan and dextran sulfate nanoparticles for effective brain cancer treatment. Int J Biol Macromol. 2011;49:747–751. | |
Allen TM, Cullis PR. Drug delivery system: entering the mainstream. Science. 2004;303:1818–1822. | |
Liu Y, Miyoshi H, Nakamura M. Nanomedicine for drug delivery and imaging: a promising avenue for cancer therapy and diagnosis using targeted functional nanoparticles. Int J Cancer. 2007;120:2527–2537. | |
Jain KK. Nanomedicine: application of nanobiotechnology in medical practice. Med Princ Pract. 2008;17:89–101. | |
Allen TM. Ligand-targeted therapeutics in anticancer therapy. Nat Rev Cancer. 2002;2:750–763. | |
Shi J, Liu Y, Zheng Y, et al. Therapeutic expression of an anti-death receptor 5 single-chain fixed-variable region s tumor growth in mice. Cancer Res. 2006;66:11946–11953. | |
Zhang L, Zhang X, Barrisford GW, Olumi AF. Lexatumumab (TRAIL-receptor 2 mAb) induces expression of DR5 and promotes apoptosis in primary and metastatic renal cell carcinoma in a mouse orthotopic model. Cancer Lett. 2007;251:146–157. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.