Back to Journals » International Journal of Nanomedicine » Volume 9 » Issue 1
Preparation and evaluation of cyclosporin A-containing proliposomes: a comparison of the supercritical antisolvent process with the conventional film method
Authors Karn PR, Jin S, Lee BJ, Sun BK, Kim M, Sung J, Hwang S ![]()
Received 30 June 2014
Accepted for publication 8 August 2014
Published 3 November 2014 Volume 2014:9(1) Pages 5079—5091
DOI https://doi.org/10.2147/IJN.S70340
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Thomas Webster
Pankaj Ranjan Karn,1 Su-Eon Jin,2 Benjamin Joon Lee,1,2 Bo Kyung Sun,1,2 Min-Soo Kim,3 Jong-Hyuk Sung,1,2 Sung-Joo Hwang1,2
1Yonsei Institute of Pharmaceutical Sciences, 2College of Pharmacy, Yonsei University, Yeonsu-gu, Incheon, Republic of Korea; 3College of Pharmacy, Pusan National University, Geumjeong-gu, Busan, Republic of Korea
Objectives: The objectives of this study were to prepare cyclosporin A (CsA)-containing proliposomes using the supercritical antisolvent (SAS) process and the conventional thin film method for the comparative study of proliposomal formulations and to evaluate the physicochemical properties of these proliposomes.
Methods: CsA-containing proliposomes were prepared by the SAS process and the conventional film method, composed of natural and synthetic phospholipids. We investigated particle size, polydispersity index, and zeta potential of CsA-containing proliposomes. In addition, both production yield and entrapment efficiency of CsA in different proliposomes were analyzed. Physicochemical properties of CsA-containing proliposomes were also evaluated, using differential scanning calorimetry and X-ray diffraction. The morphology and size of CsA-containing proliposomes were confirmed, using scanning electron microscopy. We checked the in vitro release of CsA from CsA-containing proliposomes prepared by different preparation methods, comparing them with Restasis® as a positive control and the stability of SAS-mediated proliposomes was also studied.
Results: CsA-containing proliposomes formed by the SAS process had a relatively smaller particle size, with a narrow size distribution and spherical particles compared with those of conventionally prepared proliposomes. The yield and entrapment efficiency of CsA in all proliposomes varied from 85% to 92% and from 86% to 89%, respectively. Differential scanning calorimetry and X-ray diffraction studies revealed that the anhydrous lactose powder used in this formulation retained its crystalline form and that CsA was present in an amorphous form. Proliposome powders were rapidly converted to liposomes on contact with water. The in vitro release study of proliposomal formulations demonstrated a similar pattern to Restasis®. The SAS-mediated CsA-containing proliposomes were stable on storage, with no significant changes in particle size, polydispersity index, and entrapment efficiency.
Conclusion: These results show promising features of CsA-containing proliposomal formulations, using the SAS process for the large-scale industrial application.
Keywords: proliposomes, cyclosporin A, supercritical antisolvent process, thin film method, Restasis®
Introduction
Cyclosporin A (CsA) is a powerful immunosuppressive drug mainly used to prevent graft rejection after organ transplantation, as well as for the treatment of several autoimmune and parasitic diseases.1–3 As an immunosuppressant, CsA at a low concentration has been proved useful for patients with various inflammatory ocular surface disorders (eg, dry eye syndrome and uveitis).4,5 For the enhanced delivery of CsA to an eventual site of action with minimum toxicity, several formulations of CsA are widely used. A topical preparation of Restasis® (0.05% CsA; Allergan, Irvine, CA, USA) is the major drug reported for dry eye in humans that is available in the form of oil-in-water emulsion eye drops.6 However, poor ocular tolerance, low bioavailability, and instability are major drawbacks associated with Restasis®.1,6 Other marketed drugs of CsA include Sandimmune® (Novartis International AG, Basel, Switzerland), an oil-based formulation,7,8 and Neoral® (Novartis International AG, Basel, Switzerland), a microemulsion-based preconcentrate formulation and both have issues because of their oil and alcohol contents.2,7,9 Both of these formulations have high intra- and interpatient variability.10 Considerable efforts have been made to improve the availability and the tolerance of topically applied CsA, but so far, none of the delivery systems have been fully satisfactory.4 Therefore, development of stable formulations of CsA continues to be of great importance.
The concept of proliposomes was first introduced by Payne et al in 1986; they described proliposomes as a dry, free-flowing, granular product that immediately forms a liposomal dispersion on contact with water or a biological fluid in the body.11,12 Since the discovery of liposomes in 1965, they have been widely investigated for encapsulating varieties of bioactive materials, but relatively few liposomal drug products have made it to the marketplace.13 The best examples of liposomal drug products are Doxil® (PEGylated doxorubicin; Janssen Pharmaceuticals, Inc., Titusville, NJ, USA) and AmBisome® (Amphotericin B; Astellas Pharma US Inc, Northbrook, IL, USA). AmBisome® is available in a lyophilized powdered form that can be reconstituted with saline water before use as a sterile injection.13 The main advantage of proliposomes over liposomes is that proliposomes avoid a liquid state that does not cause physicochemical instability to the vesicle. The aggregation, fusion of particles and hydrolysis, and oxidation of phospholipids are more prone to occur in a liquid state.14,15 Proliposomes are prepared in a dry and free-flowing form using several methods, such as freeze drying, spray drying, etc. They need complicated additional steps for proliposomal formulation, together with a cryoprotectant and high process temperature, which limits their wide application.2,13
Recently, the supercritical fluid of carbon dioxide (SCF-CO2) technology has emerged as one of the major techniques for the preparation of liposomes. This method has a notable advantage in that it controls the physicochemical properties of liposomes, including their size, shape, and yield altered by the solvent properties of the SCF.13,16 The supercritical antisolvent (SAS) process is one of the SCF-CO2 methods widely used for liposomal preparation. However, the preparation and evaluation of proliposomes using SCF-CO2 methods has not been fully explored yet, and there are only a few studies that deal with SCF-mediated proliposomes.13,15,17 Xia et al used the SAS process for preparing vitamin D3-loaded proliposomes and also studied lutein-encapsulated proliposomes using the same technique.15 To the best of our knowledge, the comparison of the physicochemical properties of conventional and SCF or SAS-mediated proliposomes is a novel approach and needs to be investigated.
In our previous studies with CsA2 and amphotericin B,18 the advantages of liposomes prepared using the SCF-CO2 method over the modified conventional Bangham method have already been investigated. To strengthen and broaden the application of the newly developed SCF-CO2 method, dry powder proliposomes containing CsA were examined in the present study. For this reason, we have selected different formulations of proliposomes containing natural and synthetic phospholipids. Proliposomes were also prepared by the conventional film method to compare the physicochemical and morphological properties of both conventional and SCF-mediated proliposomes. Therefore, the overall aim of this study was to prepare and evaluate dry powder proliposomes containing CsA, using a simple SAS process for scaled-up production.
Materials and methods
Materials
CsA was purchased from Concord Drugs Limited (Hyderabad, India). Cholesterol and hydrogenated soy phosphatidylcholine were obtained from Sigma-Aldrich (St Louis, MO, USA). Anhydrous lactose was purchased from Quest International (Itaska, IL, USA). Lipoid EPCS (phosphatidylcholine from egg lecithin) was generously gifted from Lipoid GmbH (Ludwigshafen, Germany). Distearoylphosphatidylcholine (DSPC) and distearoylphosphatidylglycerol, sodium salt (DSPG, Na) were purchased from Corden Pharma (Switzerland LLC). CO2 with a purity of 99.99% was supplied from Hanmi Gas Co., Ltd. (Seoul, Republic of Korea). Ethyl alcohol (purity 99.9%), methyl alcohol (purity 99.5%), chloroform (purity 99.5%), and high-performance liquid chromatography (HPLC)-grade acetonitrile were purchased from Samchun Pure Chemical Pvt Ltd. (Gyeonggi-do, Republic of Korea). Purified water of Milli-Q quality (Milli-Q Reference System, EMD Millipore®, Billerica, MA, USA) was used throughout the study.
Preparation of proliposomes
For the preparation of proliposomes, two kinds of compositions were selected in which formulation 1 consisted of naturally derived phospholipid from egg lecithin (Lipoid EPCS, GmbH), whereas formulation 2 consisted of a combination of natural (soy) and synthetic saturated phospholipids (DSPC and DSPG, Na), as shown in Table 1. The obtained proliposomes with formulations 1 and 2 by the SAS process were referred to as SCF-EPCS and SCF-DSPG proliposomes, respectively. In addition, the obtained proliposomes of formulations 1 and 2 by the conventional film method were referred to as Film-EPCS and Film-DSPG proliposomes, respectively.
 | Table 1 Composition of different proliposomes used in the study |
Preparation of proliposomes by the SAS process
Proliposomes containing CsA were prepared using the SCF-CO2 method2 with a slight modification: they lacked the final hydration step. The experimental apparatus, as shown in Figure 1, was made up of the following components: CO2 syringe pump; circular and cooling lines for maintaining the CO2 pump head and CO2, which flowed out of a storage tank (−7°C); and a reaction vessel (72 cm3) containing a magnetic stirrer, pressure indicator, and temperature indicator.
 | Figure 1 Schematic representation of the experimental apparatus for proliposome preparation by the SAS process. |
For both preparations, phospholipids, cholesterol, and CsA were dissolved in organic solvents followed by sonication (Ultrasonic Cleaner UC-20; Jeio Tech Co., Ltd., Seoul, Republic of Korea) until a clear and homogeneous solution was obtained. The CsA–lipid solution and lactose were then sealed in the reaction vessel. The supercritical CO2 was pumped to the vessel by a syringe pump (model 260D; ISCO Co, Louisville, KY, USA). The conditions of temperature and pressure of the reaction vessel were 45°C and 10 MPa for SCF-EPCS and 60°C and 10 MPa for SCF-DSPG. After approximately 30 minutes of stirring at equilibrium, additional supercritical CO2 continued to flow into the vessel for about 30 minutes to wash out any remaining solvents. The vessel was then slowly depressurized to atmospheric pressure, and CsA-phospholipid mixture coated the surface of lactose particles, forming a thin film. SCF-mediated proliposomes were then collected and stored at 4°C.
Preparation of proliposomes by the conventional film method
The thin film hydration method19,20 was adopted as the conventional method used to prepare proliposomes for comparison with proliposomes containing CsA that were prepared by a novel SAS process. Phospholipids, cholesterol, and CsA were dissolved in organic solvents, followed by sonication (Ultrasonic Cleaner UC-20; Jeio Tech Co., Ltd., Seoul, Republic of Korea) until a clear and homogeneous solution was obtained. Anhydrous lactose was then transferred to a round-bottomed flask into which the drug–lipid solution was slowly poured. The flask was then connected to a rotary evaporator (N-1110V-W; EYELA, Shanghai, People’s Republic of China) and water bath (SB-1200; EYELA), with the temperature maintained at 45°C for Film-EPCS and 60°C for Film-DSPG proliposomes with mixing. The organic solvent was then removed by reduced pressure and temperature to obtain a film on the wall of the vessel. Proliposomes were then collected and stored at 4°C.
Characterization of proliposomes
Determination of particle size, polydispersity index, and zeta potential of CsA-proliposomes
To determine the particle size, polydispersity index (PDI), and zeta potential of proliposomes, 10 mg proliposomes were dispersed in 10 mL Milli-Q water, and the mixture was agitated manually until it formed a clear dispersion. The average diameter, PDI, and zeta potential of liposomes were measured by dynamic light scattering (DLS) with a particle size analyzer (ELS-Z; Otsuka Electronics Co., Ltd., Osaka, Japan) at room temperature. The PDI was also determined to check the level of homogeneity of particle sizes. In general, a PDI value less than 0.1 represented monodispersion, whereas values greater than 0.1 represented polydispersion of liposomal vesicles.
Yield and entrapment efficiency of CsA-containing proliposomes
For the determination of CsA content in proliposomes, 10 mg proliposomes were dispersed in 5 mL Milli-Q water, and 1 mL aliquot was ruptured by methanol and the volume made up to 5.0 mL. Each sample was analyzed in triplicate by HPLC assay (Agilent 1200 Series System; Agilent Technologies, Santa Clara, CA, USA). The mobile phase was a mixture of acetonitrile and water (90%:10%, v/v) pumped at a flow rate of 1.0 mL/minute, and CsA was separated using a C18 column (4.6 mm ×150 mm, 3 μm; SUPELCO Analytical, Bellafonte, CA, USA). The sample injection volume was 10 μL. CsA detection was performed using an ultraviolet detector (Agilent Technologies) at a wavelength of 210 nm. The linearity range of the calibration curve was within 0.50–100 μg/mL, with a correlation coefficient of 0.9999. The percentage yield of CsA was calculated from Equation 1.
| (1) |

For entrapment efficiency (EE), 10 mg proliposomes were dispersed in 5 mL Milli-Q water, and 1 mL aliquot (dispersed proliposomes) was then placed in polycarbonate centrifuge tubes (Beckman Instruments, CA, USA) and centrifuged (Optima™ MAX-XP Ultracentrifuge; Beckman Instruments) at 50,000× g for 40 minutes at 4°C. Free drug concentration in the aqueous solution was determined by the HPLC assay (Agilent Technologies), and the EE of CsA was calculated using Equation 2.
| (2) |

where Wfree is the analyzed weight of free drug in the supernatant, and Wtotal is the analyzed weight of the drug in the dispersions.
Differential scanning calorimetry of proliposomes
The molecular state of the drugs in proliposomes was evaluated by differential scanning calorimetry (DSC; S-650, Scinco Co., Ltd., Seoul, Republic of Korea) of pure drug (CsA), cholesterol, lactose, and CsA-containing proliposome powders. An average sample weight of 2.5±1.0 mg was heated in a hermetically sealed aluminum pan over a temperature range of 20°C–250°C under constant nitrogen gas flow of 50 mL/minute at a heating rate of 10°C/minute. The instrument was calibrated with indium (calibration standard; purity >99.9%) for melting point and heat of fusion.
X-ray diffraction study of proliposomes
The crystalline characteristics of CsA and different proliposomes were analyzed to study the distribution of CsA in proliposomes by X-ray diffractometer (HR-XRD, SmartLAB, Rigako Corporation, Tokyo, Japan), with a rotating Cu anode. The Cu K-alpha radiation was generated at 20 mA and 40 kV and monochromatized by a Cu K-beta filter. Diffraction patterns recorded the X-ray intensity as a function of 2θ angle covering from 2.0° to 50.0°. The scanning rate was 6°/minute.
Morphology of proliposomes
Scanning electron microscopy of proliposomes
Scanning electron microscopy (SEM) was used to examine the surface morphology of proliposomes and lactose anhydrous powder, using Field Emission-SEM (JSM-7100F, Jeol, Tokyo, Japan). The samples were spread on a double-sided adhesive carbon sheet and coated with platinum in an ion sputter coater.
Microscopy of CsA-containing proliposomes followed by hydration
The hydration process of proliposomes was studied using optical microscopy, as described by Song et al.21 Briefly, a small amount of SCF-EPCS proliposomes was placed onto a glass slide without a cover slip, a drop of water was added to the particles, and the images were immediately observed by optical microscope (Zeiss Obser D1, Carl Zeiss Microimaging GmbH, Germany).
In vitro release study of CsA from CsA-containing proliposomes
In vitro release of CsA from different proliposomes and a commercial product Restasis® (Allergan) was evaluated by a dynamic dialysis method.7,22 SCF-EPCS and Film-EPCS were used for this study, and Restasis® was used as control to compare the release profiles of CsA from formulations. Before the release test, proliposomes were dispersed in Milli-Q water, and total CsA content present in the samples was analyzed using HPLC assay. Two microliters of each sample was sealed in dialysis bags (MWCO 14,000 Da; dialysis tubing cellulose membrane, Sigma-Aldrich) by tightening the two ends of the tube by a thin thread. Dialysis tubes were soaked in Milli-Q water 24 hours before use. Then the dialysis bags containing a sample were immersed in a vial containing 10 mL simulated lacrimal fluid (SLF), which was maintained in a water bath at 32°C, based on the temperature of the eye surface, and stirred at a rotating speed of 50 rpm. The composition of SLF was 8.3 g NaCl, 0.084 g CaCl2·2H2O, and 1.4 g KCl in 1 L Milli-Q water.23 At time intervals of 0.5, 1, 2, 3, 4, 5, 6, and 24 hours, 1 mL of samples were withdrawn and immediately replaced with the equal volume of a fresh SLF solution. Withdrawn samples were then centrifuged and CsA contents were analyzed by the HPLC assay.
Stability of proliposomes
For a stability test of the proliposomes, SCF-EPCS proliposomes were stored at 4°C, and after regular intervals, 10 mg proliposomes were weighed and reconstituted with 10 mL Milli-Q water. The parameters of particle size, PDI, and EE were chosen to study the stability. The particle size of reconstituted proliposomes was measured by DLS (ELS-Z; Otsuka Electronic), and the EE (%) was analyzed by HPLC assay, using Equation 2.
Statistical analysis
Statistical analyses were performed using SPSS software, version 18.0, for Windows (IBM Corporation, Armonk, NY, USA). Student’s t-tests were used to compare the mean values between the two formulations, and one-way analysis of variance followed by post hoc tests was used to compare the means of more than two samples. The level of significance was set as P<0.05. All results were expressed as the mean ± standard deviation.
Results and discussion
Particle size, PDI, and zeta potential of proliposomes
Proliposomes containing CsA were successfully prepared using both the SAS and the conventional film methods. For the SAS process, it is necessary to find the optimum temperature and pressure for selective crystallization of the solute from its solution. In the previous study, an optimum pressure of 10 MPa and temperature of 45°C were reported.2 We have kept the same process pressure and temperatures for SCF-EPCS proliposomes, whereas process temperature was 60°C for SCF-DSPG, as DSPC and DSPG, Na have a phase transition temperature of 55°C. The yield of CsA from SCF-mediated proliposomes were mainly dependent on the rate of depressurization of a reaction vessel, and it was proved that the slower the depressurization, the higher the yield, with a relatively bigger size of proliposomes.24,25 It is well known that by altering the process parameters in the SCF method, the particle size, PDI, and production yield could be easily controlled.2,13
The particle size, PDI, and zeta potential results of all four proliposomes are shown in Table 2. The average diameter of proliposomes prepared using the SAS process varied from 700 nm to 1.2 μm, whereas the average particle size of conventional proliposomes ranged from 1.1 to 3.0 μm. The particle size measurements of conventional proliposomes showed less uniformity compared with SAS-mediated proliposomes, probably because of the agglomerates present in the preparations. None of the proliposomal particles were of monodispersed preparation, as observed by a PDI value higher than 0.1. However, the PDI values of SAS-mediated proliposomes were much smaller (P<0.05) than those of conventionally prepared proliposomes. Therefore, SAS-mediated proliposomes were supposed to be more homogeneously distributed. This could be further verified by observing Figure 2, which represents the volume distribution curves of SAS-mediated and conventional proliposomes. Distribution results indicate that conventional proliposomes consisted of more peaks compared with SAS-mediated proliposomes. Our results were in accordance with many published papers in which authors proved that SCF-CO2-mediated liposomes were of smaller size and more homogeneous compared with the conventionally prepared liposomes.25–27 All proliposomes were negatively charged, and the zeta potential values of DSPG proliposomes were much higher because of the presence of DSPC and DSPG, which have highly negative charges. Epstein-Barash et al28 reported the relatively higher zeta potential values of liposomes prepared using DSPG, DSPC, and cholesterol than those of CsA-containing proliposomes in the present study.
 | Figure 2 Size distribution (volume) of lipoid EPCS (phosphatidylcholine from egg lecithin)-based proliposomes prepared using (A) the conventional film method and (B) the SAS process. |
 | Table 2 Particle size, PDI, and zeta potential of proliposomes |
Yield and EE of proliposomes
The percentage yield and EE of four different proliposomes are summarized in Table 3. The EEs of SCF-EPCS, Film-EPCS, SCF-DSPG, and Film-DSPG proliposomes were 87.77±1.56, 87.74±0.40, 88.94±1.29, and 86.78±0.89, respectively. Analysis of variance showed there were no significant differences in the yield (P>0.05) or EE (P>0.05) of all the four formulations; therefore, EE of a drug in a particular case was independent of the preparation method. The higher values of EE of CsA in proliposomes seemed to be obvious because the CsA is highly lipophilic compound (log P=2.91).29 The EE of CsA in proliposomes is mainly dependent on the amount of cholesterol present.30,31 Similar results with a high EE percentage of CsA in proliposomes have been reported in previous studies.2,7,20,32
 | Table 3 Yield and EE of proliposomes |
DSC analysis of proliposomes
DSC studies were performed to evaluate the physical state of the drug in the formulation and to analyze the thermal properties of the drug.33 DSC study of raw CsA, anhydrous lactose, cholesterol, and different proliposomes is shown in Figure 3. The major characteristic peaks of CsA were found at 65°C and 132°C (Figure 3A). The absence of the melting transitions from a CsA thermogram in the proliposomal formulations indicated the amorphous nature of CsA. However, as an indirect approach, interaction between the drug and anhydrous lactose powder could be explained by DSC thermograms. The thermogram of the raw anhydrous lactose showed that one endothermic peak entered at 235°C (Figure 3B), which is considered a phase transition (Tm). The characteristic peak of cholesterol was found at 150°C (Figure 3C) and was seen to be unchanged in all proliposomal formulations. Moreover, none of the proliposomes (Figure 3D–G) showed a significant decrease (P>0.05) in melting peaks of lactose powder, suggesting there was no chemical interaction of lactose in proliposomes and, therefore, lactose was retaining its physical state (ie, a crystalline form or a nonamorphous form). The absence of the characteristic DSC peaks corresponding to CsA confirmed that CsA was present in the amorphous or noncrystalline form within proliposomes. The findings of the DSC analyses were further supported by the X-ray diffraction (XRD) data.
 | Figure 3 DSC thermograms of (A) CsA, (B) anhydrous lactose, (C) cholesterol, (D) proliposomes prepared using lipoid EPCS (phosphatidylcholine from egg lecithin) by SAS process, (E) proliposomes prepared using lipoid EPCS by film method, (F) proliposomes prepared using HSPC, DSPC, and DSPG by the SAS process, and (G) proliposomes prepared using HSPC, DSPC, and DSPG by the film method. |
XRD study of proliposomes
Figure 4 represents the XRD pattern of CsA (Figure 4A) physical mixture (Lipoid EPCS, cholesterol, and CsA) prepared according to the preparation method of proliposomes without lactose (Figure 4B), anhydrous lactose (Figure 4C), and different proliposomes (Figure 4D and E). The X-ray diffractogram pattern of CsA (Figure 4A) showed that CsA has major peaks at 6.78°, 9.12°, 15.06°, and 16.80° that disappeared in all proliposomal formulations (Figure 4D and E). However, the typical peaks of CsA were still present in the physical mixture, as seen in Figure 4B. It was believed that CsA was distributed in the thin lipid film layer in the form of a molecule. Furthermore, the crystalline character of lactose and proliposome powders were also estimated, and a diffractogram pattern of lactose and proliposomes were the same and superimposable, which confirmed that the crystalline character of lactose was not changed in the process of the preparation. Similar results were also obtained by Xu et al,34 in which the crystalline character of sorbitol did not alter in the process of proliposome preparation. The amount of lactose in the proliposomal powders was 72%, whereas the amount of CsA was 4%. Therefore, lactose was not added in the physical mixture to detect CsA in the mixture. These results suggest CsA is dispersed homogeneously in the preparation or embedded completely in the proliposomes and that anhydrous lactose behaves neutrally in the formulations. Hence, CsA was present in an amorphous or noncrystalline form, whereas lactose was present in the crystalline form in the final formulation of all proliposomes.
 | Figure 4 XRD patterns of (A) CsA, (B) physical mixture (lipid+CsA+cholesterol), (C) lactose, (D) SCF and film-method-based proliposomes prepared using lipoid EPCS (phosphatidylcholine from egg lecithin), and (E) SCF and film-method-based proliposomes prepared using HSPC, DSPC, and DSPG. The characteristic peaks of CsA were found at 6.78° (a), 9.12° (b), 15.06° (c) and 16.80° (d). |
SEM images of proliposomes
The surface morphology of lactose and different proliposomal powders is illustrated in Figure 5. A SEM image of Film-EPCS proliposomes showed a few spherical particles present in the preparation. However, with the same formulation, the proliposome prepared using the SAS process showed much better morphology at different magnifications, and most of the particles present were spherical and relatively more homogeneously distributed (Figure 5A and B). Similarly, the morphology of SCF-DSPG proliposomes appeared to be spherical and more homogeneously distributed compared with the conventionally prepared proliposomes (Figure 5C and D). At higher magnification, the surface morphology of lactose was obviously different from the proliposomes (Figure 5E). The size of proliposomes obtained by the SEM was lower compared with that obtained by the DLS, as expected, and found in ranges from 600 to 900 nm. However, on the basis of the DLS results, the mean diameter of SCF-EPCS proliposomes was about 0.8–1.0 μm. This small difference in the measurement was obvious because the sample preparation of SEM requires drying that apparently causes shrinkage, so the mean diameter determined by SEM is significantly smaller than that determined by DLS and atomic force microscopy.35
 | Figure 5 SEM images of (A) Film-EPCS proliposomes (B) SCF-EPCS proliposomes (C) Film-DSPG proliposomes (D) SCF-DSPG proliposomes and (E) anhydrous lactose powder. |
The results of SEM confirmed that proliposomes prepared using a SAS process had better morphology in terms of more spherical particles, less aggregation, and more homogeneous distribution of the particles in the preparations.
Hydration of proliposomes
Figure 6 illustrates how the hydration of proliposomes quickly progressed within a minute on contact with the water. This rapid conversion of proliposomes to liposomes confirmed that on contact with the physiological fluid of the body, proliposomes could be converted into liposomes rapidly. As shown in Figure 6, the complete conversion of proliposome to multilamellar vesicles took place within 2 minutes of hydration. This hydration study does not have any direct correlation with the ophthalmic application, but this study strongly supports proliposomes being converted quickly once they came in contact with water so that reconstituted liposomal solution (formed on contact with sterile saline water) could be used as an ophthalmic solution. The multilamellar structure of liposomes was also confirmed from Figure 6. The reconstituted proliposomes were spherical and more uniformly distributed.
 | Figure 6 Series of time-lapse photomicrographs of 0 (A), 30 seconds (B), 1 minute (C), and 2 minutes (D), depicting the process of hydration of SCF-EPCS proliposomes at 100×. |
In vitro release
The in vitro release profile of Film-EPCS, SCF-EPCS proliposomes, and Restasis® (Allergan) is shown in Figure 7. There was no significant difference (P>0.05) in the overall release of CsA from Restasis® and proliposomes for 24 hours. All three formulations have a total release rate ranging between 40% and 45% after 24 hours. The release rates appeared to be slow because the solubility of CsA in SLF is extremely low. The results obtained with the release study were quite different from those of Guan et al7 and those of Aksungur et al.36 In the case of the study by Guan et al,7 they have maintained the sink condition, and thereby the release rate was very low. Aksungur et al used a different procedure to evaluate the release rate. Interestingly, proliposomes were shown to have a similar release profile as that of Restasis® formulation. The result obtained with in vitro release helped further to carry out the in vivo study.6
 | Figure 7 In vitro drug release profile of CsA from CsA-containing proliposomes and Restasis® in simulated lacrimal fluid (n=3). |
Stability of proliposomes
The stability of proliposomes was evaluated using SCF-EPCS proliposomes. The initial particle size and PDI of proliposomes were 1,012±170 nm and 0.34±0.03, respectively, which changed to 995±179 nm (P>0.05) and 0.38±0.01 (P>0.05), respectively, after 3 months of storage at 4°C (Figure 8). In addition, EE (%) of SCF-EPCS proliposomes was changed from 87.77±1.56 to 86.68±2.45 (P>0.05). None of the changes shown in this study was statistically significant. The stability of proliposomes prepared with the SAS process might be explained by the static repulsion of the carbonic acids incorporated into the bilayer membrane.37
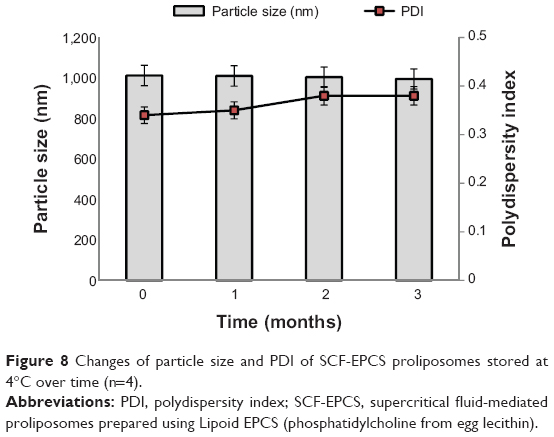 | Figure 8 Changes of particle size and PDI of SCF-EPCS proliposomes stored at 4°C over time (n=4). |
In general, liposomes have some stability issues with long-term storage. In this regard, proliposomes containing CsA are of particular interest as a new drug delivery system that may address the stability problems associated with liposomes. Many studies described improving the therapeutic potential of CsA using liposomal formulation; however, none of the studies has shown their full potential for clinical approval, mainly because of their small-scale preparation abilities and stability issues.
Conclusion
This study represents the first report on the physicochemical comparison of proliposomes prepared using a SAS process and the conventional thin film method. The two major barriers with a conventional method, namely, scale-up production and stability issues, could be overcome using a SAS process. We formulated SAS-mediated dry powder CsA-containing proliposomes, which had a relatively smaller particle size of distinct spherical vesicles with narrow distribution, and a similar EE of CsA compared with the conventional thin film method-based CsA-containing proliposomes. In addition, we proposed a suitable formulation for the development of a liposomal drug, as there were no significant differences in the physicochemical properties of SCF-mediated proliposomes. Both SCF-EPCS and SCF-DSPG proliposomes have shown similar potentials. CsA-containing SCF-EPCS proliposomal powders could be stored for the long term without any thermodynamic changes, and on reconstitution with sterile saline water, the resulting liquid liposomal dispersion could be used for the ophthalmic delivery to cure dry eye syndrome. It can have the potential to be used as an ophthalmic formulation in an experimentally induced dry eye model. Additional in vivo studies have been tried to confirm these advantages of proliposomal formulations compared with conventional formulations in a separate study.
Acknowledgments
This work (2014R1A2A2A01005059) was supported by the Mid-career Researcher Program through a National Research Foundation grant funded by the Ministry of Science, ICT and Future Planning, Republic of Korea. The authors cordially thank Ms Hee Sun Lee from the Advanced Characterization Centre, Yonsei Institute of Convergence Technology, Yonsei University, Republic of Korea, for her assistance in SEM and XRD studies.
Disclosure
The authors report no conflicts of interest in this work.
References
Gupta C, Chauhan A. Ophthalmic delivery of cyclosporine A by punctal plugs. J Control Release. 2011;150(1):70–76. | ||
Karn PR, Cho W, Park HJ, Park JS, Hwang SJ. Characterization and stability studies of a novel liposomal cyclosporin A prepared using the supercritical fluid method: comparison with the modified conventional Bangham method. Int J Nanomedicine. 2013;8:365–377. | ||
Malaekeh-Nikouei B, Jaafari MR, Tabassi SAS, Samiei A. The enhancement of immunosuppressive effects of cyclosporine A on human T-cells using fusogenic liposomes. Colloids Surf B Biointerfaces. 2008;67(2):238–244. | ||
Lallemand F, Felt-Baeyens O, Besseghir K, Behar-Cohen F, Gurny R. Cyclosporine A delivery to the eye: a pharmaceutical challenge. Eur J Pharm Biopharm. 2003;56(3):307–318. | ||
Utine CA, Stern M, Akpek EK. Clinical review: topical ophthalmic use of cyclosporin A. Ocul Immunol Inflamm. 2010;18(5):352–361. | ||
Karn PR, Kim HD, Kang H, Sun BK, Jin SE, Hwang SJ. Supercritical fluid-mediated liposomes containing cyclosporin A for the treatment of dry eye syndrome in a rabbit model: comparative study with the conventional cyclosporin A emulsion. Int J Nanomedicine. 2014;9(1): 3791–3800. | ||
Guan P, Lu Y, Qi J, et al. Enhanced oral bioavailability of cyclosporine A by liposomes containing a bile salt. Int J Nanomedicine. 2011;6: 965–974. | ||
Ranjita S. Nanosuspensions: a new approach for organ and cellular targeting in infectious diseases. J Pharm Invest. 2013;43(1):1–26. | ||
Klyashchitsky BA, Owen AJ. Drug delivery systems for cyclosporine: achievements and complications. J Drug Target. 1998;5(6):443–458. | ||
Beauchesne PR, Chung NS, Wasan KM. Cyclosporine A: a review of current oral and intravenous delivery systems. Drug Dev Ind Pharm. 2007;33(3):211–220. | ||
Payne NI, Timmins P, Ambrose CV, Ward MD, Ridgway F. Proliposomes: a novel solution to an old problem. J Pharm Sci. 1986;75(4): 325–329. | ||
Payne NI, Browning I, Hynes CA. Characterization of proliposomes. J Pharm Sci. 1986;75(4):330–333. | ||
Karn PR, Cho W, Hwang SJ. Liposomal drug products and recent advances in the synthesis of supercritical fluid-mediated liposomes. Nanomedicine (Lond). 2013;8(9):1529–1548. | ||
Hiremath PS, Soppimath KS, Betageri GV. Proliposomes of exemestane for improved oral delivery: formulation and in vitroevaluation using PAMPA, Caco-2 and rat intestine. Int J Pharm. 2009; 380(1–2):96–104. | ||
Xia F, Hu D, Jin H, Zhao Y, Liang J. Preparation of lutein proliposomes by supercritical anti-solvent technique. Food Hydrocoll. 2012;26(2): 456–463. | ||
Pasquali I, Bettini R, Giordano F. Supercritical fluid technologies: an innovative approach for manipulating the solid-state of pharmaceuticals. Adv Drug Deliv Rev. 2008;60(3):399–410. | ||
Xia F, Jin H, Zhao Y, Guo X. Supercritical antisolvent-based technology for preparation of vitamin D3 proliposome and its characteristics. Chin J Chem Eng. 2011;19(6):1039–1046. | ||
Hwang S-J, Park H-J, Cho W, et al, inventors; Foundation of University-Industry Research Collaboration of Chungnam National University, assignee. New methods and apparatus for preparing liposomes. Korean patent KR2011-00969622011. 2011 Aug 31. | ||
Bangham AD, Standish MM, Watkins JC. Diffusion of univalent ions across the lamellae of swollen phospholipids. J Mol Biol. 1965;13(1): 238–252. | ||
Shah NM, Parikh J, Namdeo A, Subramanian N, Bhowmick S. Preparation, characterization and in vivo studies of proliposomes containing Cyclosporine A. J Nanosci Nanotechnol. 2006;6(9–10):2967–2973. | ||
Song K-H, Chung S-J, Shim C-K. Preparation and evaluation of proliposomes containing salmon calcitonin. J Control Release. 2002; 84(1–2):27–37. | ||
Lai J, Lu Y, Yin Z, Hu F, Wu W. Pharmacokinetics and enhanced oral bioavailability in beagle dogs of cyclosporine A encapsulated in glyceryl monooleate/poloxamer 407 cubic nanoparticles. Int J Nanomedicine. 2010;5:13–23. | ||
Paulsson M, Edsman K. Controlled drug release from gels using surfactant aggregates: I. Effect of lipophilic interactions for a series of uncharged substances. J Pharm Sci. 2001;90(9):1216–1225. | ||
Otake K, Imura T, Sakai H, Abe M. Development of a new preparation method of liposomes using supercritical carbon dioxide. Langmuir. 2001;17(13):3898–3901. | ||
Otake K, Shimomura T, Goto T, et al. Preparation of liposomes using an improved supercritical reverse phase evaporation method. Langmuir. 2006;22(6):2543–2550. | ||
Kadimi US, Balasubramanian DR, Ganni UR, Balaraman M, Govindarajulu V. In vitro studies on liposomal amphotericin B obtained by supercritical carbon dioxide-mediated process. Nanomedicine. 2007; 3(4):273–280. | ||
Naik S, Patel D, Surti N, Misra A. Preparation of PEGylated liposomes of docetaxel using supercritical fluid technology. J Supercrit Fluids. 2010;54(1):110–119. | ||
Epstein-Barash H, Orbey G, Polat BE, et al. A microcomposite hydrogel for repeated on-demand ultrasound-triggered drug delivery. Biomaterials. 2010;31(19):5208–5217. | ||
el Tayar N, Mark AE, Vallat P, Brunne RM, Testa B, van Gunsteren WF. Solvent-dependent conformation and hydrogen-bonding capacity of cyclosporin A: evidence from partition coefficients and molecular dynamics simulations. J Med Chem. 1993;36(24):3757–3764. | ||
Al-Angary A, Bayomi M, Khidr S, Al-Meshal M, Al-Dardiri M. Characterization, stability and in vivo targeting of liposomal formulations containing cyclosporin. Int J Pharm. 1995;114(2):221–225. | ||
Al-Meshal MA, Khidr SH, Bayomi MA, Al-Angary AA. Oral administration of liposomes containing cyclosporin: a pharmacokinetic study. Int J Pharm. 1998;168(2):163–168. | ||
Guo J, Ping Q, Chen Y. Pharmacokinetic behavior of cyclosporin A in rabbits by oral administration of lecithin vesicle and Sandimmun Neoral. Int J Pharm. 2001;216(1–2):17–21. | ||
Demetzos C. Differential Scanning Calorimetry (DSC): a tool to study the thermal behavior of lipid bilayers and liposomal stability. J Liposome Res. 2008;18(3):159–173. | ||
Xu H, He L, Nie S, et al. Optimized preparation of vinpocetine proliposomes by a novel method and in vivo evaluation of its pharmacokinetics in New Zealand rabbits. J Control Release. 2009;140(1):61–68. | ||
Dubes A, Parrot-Lopez H, Abdelwahed W, et al. Scanning electron microscopy and atomic force microscopy imaging of solid lipid nanoparticles derived from amphiphilic cyclodextrins. Eur J Pharm Biopharm. 2003;55(3):279–282. | ||
Aksungur P, Demirbilek M, Denkbaş EB, Vandervoort J, Ludwig A, Unlü N. Development and characterization of Cyclosporine A loaded nanoparticles for ocular drug delivery: Cellular toxicity, uptake, and kinetic studies. J Control Release. 2011;151(3):286–294. | ||
Bothun GD, Knutson BL, Strobel HJ, Nokes SE. Liposome fluidization and melting point depression by pressurized CO2 determined by fluorescence anisotropy. Langmuir. 2005;21(2):530–536. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.