Back to Journals » International Journal of Nanomedicine » Volume 9 » Issue 1
Preparation and characterization of self-assembled nanoparticles based on low-molecular-weight heparin and stearylamine conjugates for controlled delivery of docetaxel
Authors Kim D, Termsarasab U, Cho H, Yoon I, Lee J, Moon HT, Kim D ![]()
Received 16 September 2014
Accepted for publication 25 October 2014
Published 8 December 2014 Volume 2014:9(1) Pages 5711—5727
DOI https://doi.org/10.2147/IJN.S74353
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Thomas Webster
Dong-Hwan Kim,1 Ubonvan Termsarasab,1 Hyun-Jong Cho,2 In-Soo Yoon,3 Jae-Young Lee,1 Hyun Tae Moon,1 Dae-Duk Kim1
1College of Pharmacy and Research Institute of Pharmaceutical Sciences, Seoul National University, Seoul, Republic of Korea; 2College of Pharmacy, Kangwon National University, Chuncheon, Republic of Korea; 3College of Pharmacy and Natural Medicine Research Institute, Mokpo National University, Jeonnam, Republic of Korea
Abstract: Low-molecular-weight heparin (LMWH)–stearylamine (SA) conjugates (LHSA)-based self-assembled nanoparticles were prepared for intravenous delivery of docetaxel (DCT). 1-Ethyl-3-(3-dimethylaminopropyl) carbodiimide and N-hydroxysuccinimide were used as coupling agents for synthesis of LHSA conjugates. The physicochemical properties, in vitro antitumor efficacy, in vitro cellular uptake efficiency, in vivo antitumor efficacy, and in vivo pharmacokinetics of LHSA nanoparticles were investigated. The LHSA nanoparticles exhibited a spherical shape with a mean diameter of 140–180 nm and a negative surface charge. According to in vitro release and in vivo pharmacokinetic test results, the docetaxel-loaded LHSA5 (LMWH:SA =1:5) nanoparticles exhibited sustained drug release profiles. The blank LHSA nanoparticles demonstrated only an insignificant cytotoxicity in MCF-7 and MDAMB 231 human breast cancer cells; additionally, higher cellular uptake of coumarin 6 (C6) in MCF-7 and MDAMB 231 cells was observed in the LHSA5 nanoparticles group than that in the C6 solution group. The in vivo tumor growth inhibition efficacy of docetaxel-loaded LHSA5 nanoparticles was also significantly higher than the Taxotere®-treated group in the MDAMB 231 tumor-xenografted mouse model. These results indicated that the LHSA5-based nanoparticles could be a promising anticancer drug delivery system.
Keywords: amphiphilic polymer, docetaxel, drug delivery, low-molecular-weight heparin, self-assembled nanoparticle
Introduction
Over the past decade, nanoparticulate drug delivery systems containing anticancer agents have been investigated extensively due to their specific accumulation behavior at the tumor site.1,2 Nanoparticulate drugs can be distributed to tumor vasculatures by the enhanced permeability and retention (EPR) effect, taking advantage of the leaky vascular nature of tumor tissues that provides for passive tumor targeting.3,4 Additionally, these nanoparticulate drugs offer the advantages of prolonged systemic circulation by avoiding phagocytosis, improved efficacy, and reduced toxicity. Numerous biocompatible and biodegradable materials, such as poly(lactic acid),5,6 poly(glycolic acid),7 polycaprolactone,8,9 polysaccharides,10 proteins,11 and polypeptides,12 have been used for the preparation of polymeric nanoparticles. Among them, polysaccharides have been used extensively to prepare nanoparticles for drug delivery.2 Polysaccharides are natural biomaterials; thus, they are generally safe, nontoxic, biocompatible, and biodegradable. They have various derivable groups that can be modified with other chemicals. Among the drug delivery systems, self-assembled nanoparticles have been studied for the encapsulation and sustained release of poorly soluble drugs.13–15 Amphiphilic polymers can be used to form nanoparticles with a hydrophobic core and a hydrophilic shell. This structure enables the encapsulation and delivery of poorly soluble drugs, extending the circulation time of nanoparticles in the bloodstream.
Docetaxel (DCT), an antimitotic chemotherapy medication and a semisynthetic analog of paclitaxel, is a particularly important anticancer agent that has been used for the treatment of many cancers, exhibiting cytotoxic activity in breast, ovarian, prostate, and non-small-cell lung cancer cells.16–18 As with all chemotherapies, however, adverse effects are common; side effects such as neutropenia, peripheral neuropathy, musculoskeletal toxicity, and hypersensitivity reactions have been verified. In its commercial formulation (Taxotere®, Sanofi-Aventix, Bridgewater, NJ, USA), nonionic surfactant Tween 80 (polysorbate 80) and 13% ethanol in saline have been used to dissolve DCT, due to its poor solubility in water;19 however, this formulation is associated with unpredictable hypersensitivity reactions and other side effects.20 Thus, many new systems – including micelles, emulsions, and conjugates with other polymers – have been developed to deliver DCT to tumor sites with minimal adverse reactions.21–23
Heparin is a biocompatible, biodegradable, water-soluble natural polysaccharide coupled with various biological activities, including anticoagulation, anti-inflammatory, and antiangiogenesis effects.24,25 In addition to its well-known anticoagulant activity, the anticancer activity of heparin has been evaluated.26,27 Low-molecular-weight heparin (LMWH) exerts its anticancer activity by affecting the proliferation, adhesion, angiogenesis, migration, and invasion of cancer cells. Although the exact mechanism of LMWH interference with cancer biology still remains unclear, it may involve the inhibition of both coagulation and noncoagulation-dependent pathways of tumor progression.28 Furthermore, in a study of venous thromboembolism therapy, a significant reduction in the mortality rate was noted in the LMWH treatment group among cancer patients.26 Thus, these idiosyncratic properties facilitate the development of heparin-based delivery systems.29,30 However, unfractionated heparin (UFH) causes adverse effects, such as bleeding, which have limited its application. LMWH has a reduced anticoagulant activity and, thus far, appears to have a greater anticancer effect than UFH;31 thus, LMWH is preferable to UFH for the initial treatment of venous thromboembolism prevalent in cancer patients.32
In this study, we synthesized an amphiphilic polymer composed of LMWH and stearylamine (octadecylamine, SA) for the preparation of self-assembled nanoparticles for anticancer drug delivery. This negatively charged nanoparticle was prepared using LMWH-SA (LHSA) conjugates, with LMWH as the hydrophilic segment and SA as its hydrophobic counterpart. SA (18-carbon saturated fatty amine) is used for the synthesis of uniform nanoparticles.33 The physicochemical properties of LHSA-based nanoparticles containing DCT were characterized by determining size, zeta potential, and DCT-loading efficacy. DCT release from nanoparticles and the cytotoxicity of blank nanoparticles were also investigated. Moreover, the optimized formulation was evaluated via in vitro cellular uptake study using confocal laser scanning microscopy (CLSM) and fluorescence-activated cell sorter (FACS) measurements, and in vivo antitumor efficacy and pharmacokinetic studies.
Materials and methods
Materials
LMWH (101 IU mg−1), with an average molecular weight of ~4,500 Da, was purchased from Nanjing King-Friend Biochemical Pharmaceutical Co. Ltd (Nanjing, People’s Republic of China). 1-Ethyl-3-(3-dimethylaminopropyl) carbodiimide (EDC), N-hydroxysuccinimide (NHS), SA, coumarin 6 (C6), and pyrene were obtained from Sigma-Aldrich Co. (St Louis, MO, USA). DCT was purchased from Taihua Co. (Xi’an, People’s Republic of China). Formamide (FA), tetrahydrofuran (THF), and acetonitrile (HPLC [high-performance liquid chromatography] grade) were supplied by Fisher Scientific Korea Ltd. (Seoul, Republic of Korea). Dulbecco’s Modified Eagle’s Medium (DMEM), RPMI 1640 (developed by Roswell Park Memorial Institute), heat-inactivated fetal bovine serum, streptomycin, and penicillin were obtained from Gibco Life Technologies (Karlsruhe, Germany). Water was purified by distillation, deionization, and reverse osmosis (Milli-Q Plus). All other chemicals were of analytical grade, which were purchased from commercial sources.
Synthesis and characterization of LHSA conjugates
Amphiphilic LHSA conjugates were synthesized from LMWH and SA. LMWH (1 g) was dissolved in FA (200 mL) by gentle heating. To synthesize the LHSA conjugates, LMWH solutions were mixed with different amounts of EDC and NHS (3 mol/mol of SA, each) at room temperature. The solutions were then added with different amounts of SA (1, 3, and 5 mol/mol of LMWH) dissolved in THF (132 mL), followed by stirring at room temperature for 24 hours. The mixtures were then precipitated in excess cold acetone (at 5× the mixture volume) under vigorous agitation, filtered by a glass filter (G4), and stirred for 30 minutes. To remove excess EDC, NHS, and SA, the residues on the glass filter were carefully washed three times with cold acetone, followed by filtering with 50 mL of distilled deionized water (DDW). The filtrate was dialyzed against DDW for 48 hours using a dialysis membrane with molecular weight cutoff of 1 kDa (Spectrum Laboratories, Laguna Hills, CA, USA) and lyophilized. The LHSA conjugate was dissolved in D2O and analyzed by proton nuclear magnetic resonance (1H-NMR) spectroscopy (500 MHz).
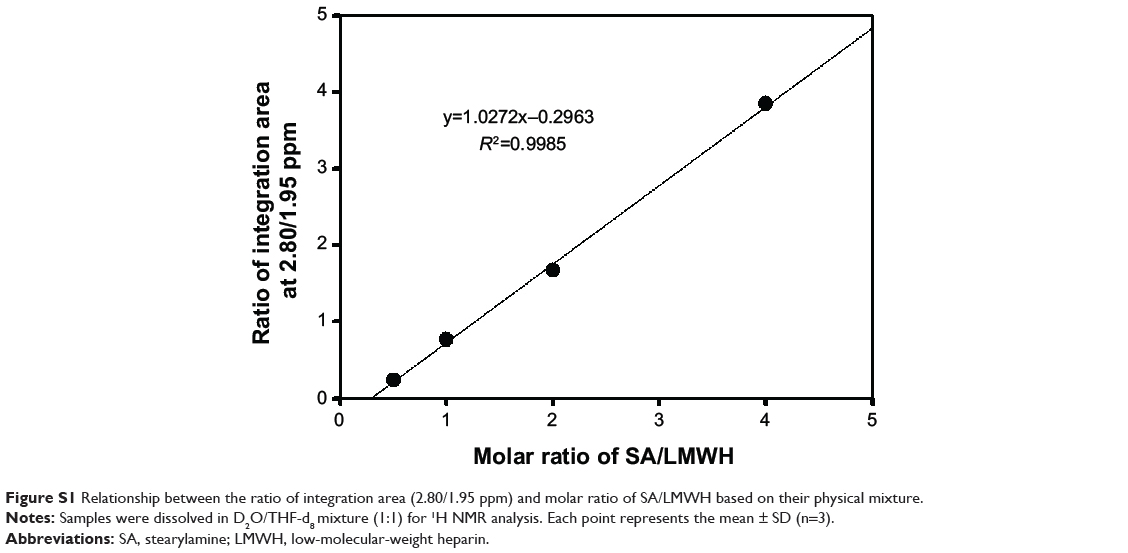
Aliquots of conjugates were dissolved in D2O (10 mg mL−1), and 1H-NMR spectra were obtained by using a Varian FT 500-MHz NMR spectrometer (Varian Inc., Palo Alto, CA, USA). The values of the proton peaks from the 1H-NMR spectra were integrated to determine the molar substitution of LHSA. Linear regression was prepared from the physical mixture of LMWH and SA at various molar ratios (LMWH:SA =0.5:1, 1:1, 1:2, and 1:4) dissolved in the mixture of D2O/THF-d8 (1:1). The ratio of the integration area (2.80/1.95 ppm) of each physical mixture was calculated.
The anticoagulant activity of LMWH and the LHSA conjugates was determined using the Coatest anti-FXa chromogenic assay (Chromogenix, Milan, Italy). The critical micellar concentration (CMC) of LHSA was investigated using pyrene as a fluorescence probe, as described previously.34 A pyrene solution in THF (2×10−6 M) was prepared, and the THF was evaporated under gentle nitrogen gas (N2) flow for 1 hour at 60°C. An LHSA solution in DDW (1 mL), in the concentration range of 10−5 to 1 mg mL−1, was added to each tube to achieve a final pyrene concentration of 6×10−7 M. The intensity ratio (I1/I3) in the excitation spectra was calculated and plotted. The excitation wavelength was fixed at 334 nm after scanning from 350 to 550 nm, and the slit openings for excitation and emission were set at 10 and 3 nm, respectively. Fluorescence measurements were performed using a fluorescent spectrometer FP-6500 (JASCO Co., Tokyo, Japan).
Preparation and characterization of LHSA-based nanoparticles
A simple modification of our previous methods was used for the self-assembly of LHSA and drug encapsulation.34 LHSA (5 mg) and 1 mg of DCT were completely solubilized in 1 mL of FA; the solvent was evaporated under gentle N2 flow for 5 hours at 80°C. LHSA and DCT-coated tubes were resuspended with 1 mL of DDW by vortexing for 3 minutes, followed by filtering through a 0.22 μm pore-size syringe filter (Minisart RC 15, Sartorius Stedium Biotech GmbH, Goettingen, Germany).
The mean diameter, polydispersity, and zeta potential of the nanoparticles were determined using a light scattering spectrophotometer (ELS-Z, Otsuka Electronics, Osaka, Japan) at a polymer concentration of 5 mg mL−1. The morphology of the LHSA nanoparticles was assessed by transmission electron microscopy (TEM) (JEM 1010, JEOL, Tokyo, Japan). The nanoparticle suspensions were stained with 2% (w/v) phosphotungstic acid, placed on a copper grid coated with carbon film, air-dried for 10 minutes, and photographed.
The nanoparticle suspensions were distilled with FA to disrupt the self-assembled structure. The drug encapsulation efficiency of drug-loaded nanoparticles was measured using HPLC.34 The HPLC instrument consisted of a dual pump (Waters 515, Waters Corporation, Milford, MA, USA), an autosampler (Waters 717 plus, Waters Corporation), an ultraviolet detector (Waters 2487, Waters Corporation), and a reverse-phase C-18 column (250×4.4 mm2, 5 μm, Shiseido, Japan). The mobile phase was a mixture of acetonitrile and DDW (55:45, v/v). The eluent was monitored at 230 nm and a flow rate of 1.0 mL min−1. The DCT concentrations were determined using 20 μL of injection volume at room temperature. The lower limit of quantitation was 500 ng mL−1.
In vitro DCT release studies
The LHSA5 nanoparticles (the volume equivalent of 100 μg of DCT) were loaded in semipermeable Mini-Gebaflex-tubes with a molecular weight cutoff of 6–7 kDa (Gene Bio-Application Ltd., Kfar Hanagid, Israel) and were immersed in the medium (10 mL). The dissolution medium was phosphate-buffered saline (PBS, pH: 7.4) containing 0.5% (w/v) Tween 80 at 37°C, and was rotated at 50 rpm. Aliquots (0.2 mL) of medium were collected at predetermined times (1, 2, 3, 4, 6, 9, 12, 24, 48, 72, 96, and 144 hours), and fresh media (37°C) of equal volume was added to maintain sink condition. The amounts of DCT released into the medium were analyzed by HPLC.
In vitro cytotoxicity of conjugate
MCF-7 and MDAMB 231 cells were purchased from the Korean cell line bank (Seoul, Republic of Korea) and were cultured in RPMI-1640 medium with 1% (v/v) penicillin (100 U mL−1), streptomycin (0.1 mg mL−1), and 10% (v/v) fetal bovine serum. They were incubated in a humidified 5% CO2 atmosphere at 37°C.
The cytotoxicity of LHSA5 conjugate polymer was evaluated in MCF-7 and MDAMB 231 cells by using MTS-based assays. MCF-7 and MDAMB 231 cells (1×104 cells) were seeded in 96-well plates. Various concentrations of LHSA5 polymer in the culture medium were incubated for 24 hours at 37°C, after which the cells were treated with an MTS-based CellTiter 96 AQueous One Solution Cell Proliferation Assay reagent (Promega Corp., Fitchburg, WI, USA) for 4 hours at 37°C following the manufacturer’s protocol. The absorbance of the medium was read at 490 nm using the Emax Precision Microplate Reader (Molecular Devices LLC, Sunnyvale, CA, USA).
In vitro antitumor efficacy studies
In vitro antitumor efficacy was evaluated using MTS-based assays. MCF-7 and MDAMB 231 cells (1×104 cells) were seeded in 96-well plates for 24 hours. After washing the cells twice with PBS, they were incubated with various concentrations (0.1, 1.0, and 5.0 μM) of DCT solution (Taxotere, Sanofi-Aventis) or DCT-loaded LHSA5 nanoparticles for 24 hours at 37°C. Then, the cells were treated with an MTS-based CellTiter 96 AQueous One Solution Cell Proliferation Assay reagent (Promega) for 4 hours at 37°C following the manufacturer’s instructions. The absorbance at 490 nm was read using an Emax Precision Microplate Reader (Molecular Devices LLC), and is expressed as percentage of viable cells compared with the control group (ie, without DCT).
Cellular uptake studies
The cellular uptake efficiency of the LHSA5 nanoparticles was investigated by CLSM with C6-loaded nanoparticles.34 C6 was also loaded in nanoparticles using a similar DCT loading method. Either MCF-7 or MDAMB 231 cells were seeded on culture plates (BD Falcon, Bedford, MA, USA) at a density of 1×105 per well (1.7 cm2 surface area) and incubated for 24 hours at 37°C. C6 (2 μg) was added alone or entrapped in the LHSA5 nanoparticles and incubated for 2 hours at 37°C. Then, the cells were washed with PBS (pH: 7.4) and were fixed with formaldehyde solution (4%, v/v) for 10 minutes. After completely drying the liquid content, a VECTASHIELD mounting medium with 4′,6-diamidino-2-phenylindole (H-1200 Vector Laboratories, Inc., Burlingame, CA, USA) was added to prevent fading. Finally, the cells on the slides were observed using CLSM (LSM 510, Carl-Zeiss, Thornwood, NY, USA).
The cellular uptake efficiency of the LHSA5 nanoparticles was also determined by flow cytometry. MCF-7 and MDAMB 231 cells were seeded on 24-well plates at a density of 5×105 per well and incubated overnight at 37°C. After removing the culture medium, the cells were incubated with free C6 or C6-loaded LHSA5 nanoparticles for 2 hours. The cells were then completely washed with PBS (pH: 7.4). After they were detached and centrifuged, the supernatant was removed. The cell pellets were suspended again with PBS containing 2% (v/v) fetal bovine serum. The uptake efficiency was analyzed by a BD FACSCalibur flow cytometry system equipped with the Cell Quest Pro software (Becton Dickinson Bioscience, San Jose, CA, USA).
In vivo antitumor efficacy
Tumor-xenografted mouse model was prepared using female BALB/c nude mice (5 weeks old; Charles River, Washington, MA) for in vivo anticancer efficacy evaluation. The mice were kept in a light-controlled room at 22°C±2°C temperature and 55%±5% relative humidity (Animal Center for Pharmaceutical Research, College of Pharmacy, Seoul National University, Seoul, Republic of Korea). The experimental protocols were approved by the Animal Care and Use Committee of the College of Pharmacy, Seoul National University. Tumor-xenografted mice were prepared by subcutaneous injection of MDAMD 231 cells (3×106 cells per mouse) in the right lateral flank. After measuring the tumor size with Vernier calipers, the tumor volume (mm3) was calculated by the following formula: volume =0.5× longest diameter × shortest diameter2. After 14 days, when the MDAMB 231 tumor became palpable, tumor volume and body weight measurements were commenced. The mice were randomly divided into four groups: the control (no treatment), blank LHSA5 nanoparticles, DCT solution (Taxotere), and DCT-loaded LHSA5 nanoparticles groups. DCT solution or DCT-loaded LHSA5 nanoparticles were intravenously injected at a dose of 30 mg kg−1 as DOC on days 0, 7, and 14. Tumor volume and body weight were measured for 18 days.
In vivo pharmacokinetic study in rats
Male Sprague Dawley rats (280–300 g body weight) were used for pharmacokinetic studies. Under light anesthesia (30 mg kg−1 Zoletil®, intramuscular injection, Virbac Korea Co. Ltd, Seoul, Republic of Korea), the femoral artery and vein of each rat were cannulated with a polyethylene tube (PE-50; Clay Adams, Parsippany, NJ, USA). DCT solution (Taxotere) or DCT-loaded LHSA5 nanoparticles (8 mg kg−1 as DCT) were administered to rats via the femoral vein within 1 minute. Blood samples (~300 μL) were collected via the femoral artery at 0 (control), 1, 5, 15, 30, 60, 120, 180, 240, 360, and 720 minutes after intravenous administration of Taxotere and DCT-loaded LHSA5 nanoparticles. After centrifugation of the blood samples, aliquots of plasma (100 μL) were stored at -70°C (Model DF8517; Ilshin Laboratory Co., Seoul, Republic of Korea) until liquid chromatography–mass spectrometry/mass spectrometry (LC–MS/MS) analysis of DCT. Immediately after each blood sampling, equal volume of 0.9% sodium chloride injectable solution containing 20 U mL−1 of heparin was injected.
The concentration of DCT in the plasma samples was determined by LC-MS/MS. The LC-MS/MS system consisted of an Agilent 1260 series HPLC system (equipped with a G1312B binary pump, a G1367E high-performance autosampler, a G1322A vacuum degasser, a G1330B FC/ALS thermostat, and a G1316C TCC, Agilent Technologies, Santa Clara, CA, USA), an Agilent 6430 triple quadrupole mass spectrometer (Agilent Technologies), and the MassHunter Workstation software (version B.03.01, Agilent Technologies).
Plasma samples (100 μL) were deproteinized with acetonitrile (300 μL) and methanol (100 μL) containing 1 μL mL−1 paclitaxel (as an internal standard). They were vortexed and centrifuged at 16,000× g for 10 minutes. The supernatant (400 μL) was transferred to a clean tube and completely evaporated under a gentle stream of N2 gas at room temperature. Then, the residue was reconstituted with 60 μL of the mobile phase. After vortexing and centrifugation again, a 10 μL aliquot of supernatant was injected into a poroshell 120 EC-C16 column (2.7 μm, 4.6×50 mm, Agilent Technologies) under isocratic elution of the mobile phase consisting of ammonium formate and acetonitrile (30:70, v/v) at a flow rate of 0.2 mL min−1. The column temperature was maintained at 30°C. Data were acquired with electrospray ionization in the positive mode. Reaction monitoring was used to monitor the transitions from molecular ion to dominant product ion at charge-to-mass ratio of (m/z) 808 to 527 for DCT and m/z 876 to 591 for paclitaxel, respectively. The lower limit of quantitation value of DCT in rat plasma was 10 ng mL−1.
The following pharmacokinetic parameters were calculated using WinNonlin® (Pharsight, Mountain View, CA, USA): total area under the plasma concentration–time curve from time zero to the end time point (AUC), terminal half-life (t1/2), time-averaged total body clearance (CL), the apparent volume of the distribution under steady-state conditions (Vss), and the mean residence time.
Statistical analysis
Statistical analyses were performed using analysis of variance. P-values <0.05 indicated statistical significance. All experiments were performed at least three times, and the data were presented as the mean ± standard deviation.
Results
Synthesis and characterization of LHSA conjugates
Heparin and its derivatives have been widely used as drug delivery systems.35–37 In this study, SA (an aminated fatty acid) was conjugated to LMWH to produce an amphiphilic polymer, which is to form the self-assembled nanoparticles in aqueous environment (Figure 1A). As shown in Figure 1B, the amine group of SA was covalently coupled to the carboxylic group of LMWH in the presence of EDC and NHS, thus producing amphiphilic LHSA conjugates. In this coupling reaction, several LHSA conjugates were synthesized by adjusting the feed ratio of SA (mol) to LMWH (g) (LHSA1–LMWH:SA =1:1, LHSA3–LMWH:SA =1:3, and LHSA5–LMWH:SA =1:5). The synthesized LHSA conjugates were confirmed by 1H-NMR analysis, as shown in Figure 2 (Figure 2A, LMWH; Figure 2B, LHSA1; Figure 2C, LHSA3; Figure 2D, LHSA5; Figure 2E, SA). The 1H-NMR spectra of the LHSA conjugate exhibited proton signals for both LMWH and SA; a broad proton peak was observed for LMWH, and the presence of SA in the LHSA conjugate was confirmed by its chemical shift at 0.8–1.2 ppm. An alkyl chain (–CH2–) and a terminal methyl group (–CH3) of SA corresponded to the proton peaks at 1.10 and 0.90 ppm, respectively. These results indicated that the degree of substitution of LHSA increased with an increase in the feed ratio of SA. The physical mixtures of LMWH and SA with various molar ratios, prepared for calculation of the molar substitution ratio of LHSA, were analyzed by 1H-NMR. The linear regression line was plotted by the ratio of the integration area between the SA peak (2.80 ppm) and the LMWH peak (1.95 ppm). The degree of substitution of LHSA5 was calculated to be 34.0%, which indicates that approximately 1.7 of SA is conjugated to each LMWH (Figure S1).
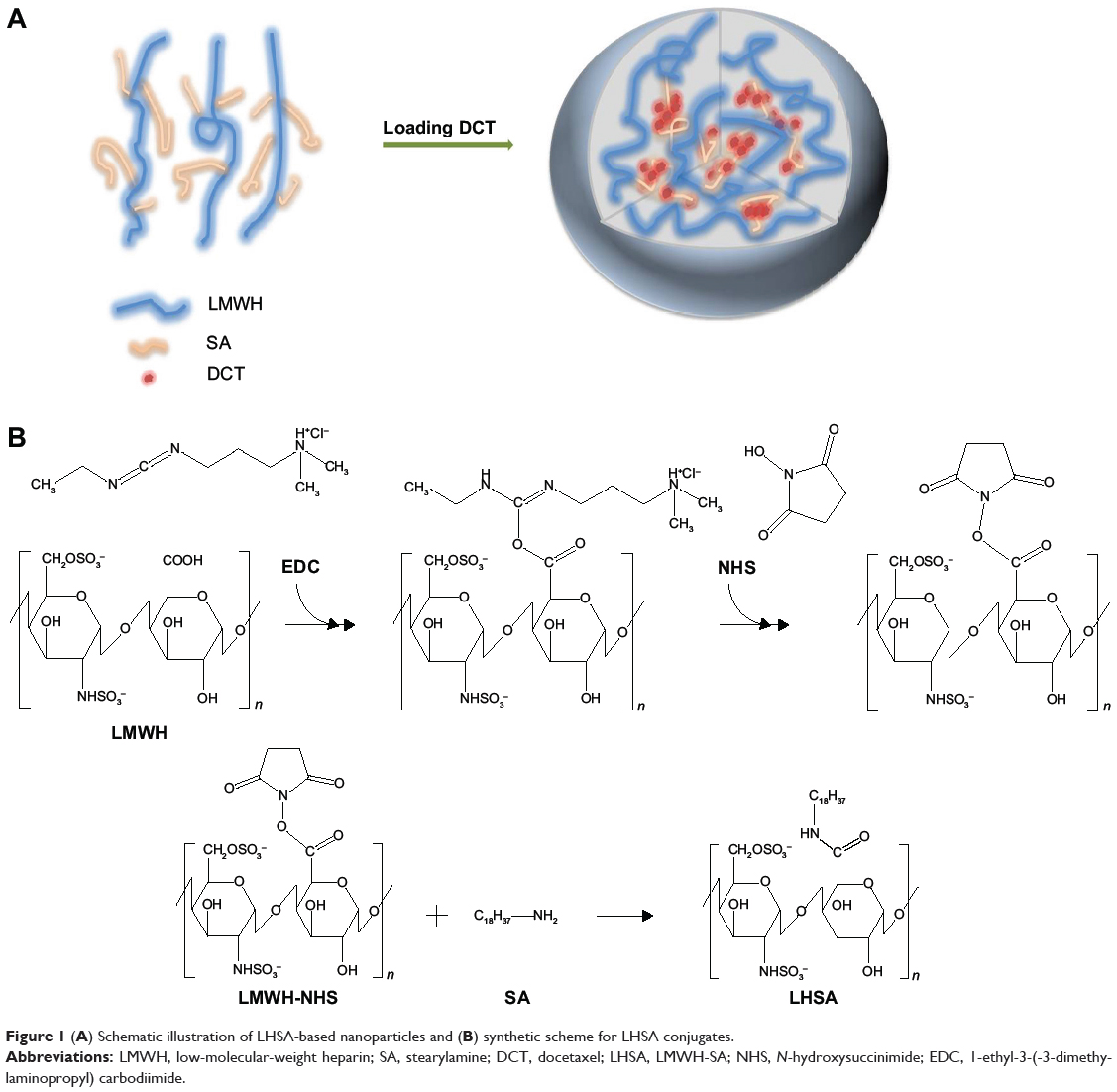
|
Figure 1 (A) Schematic illustration of LHSA-based nanoparticles and (B) synthetic scheme for LHSA conjugates. |
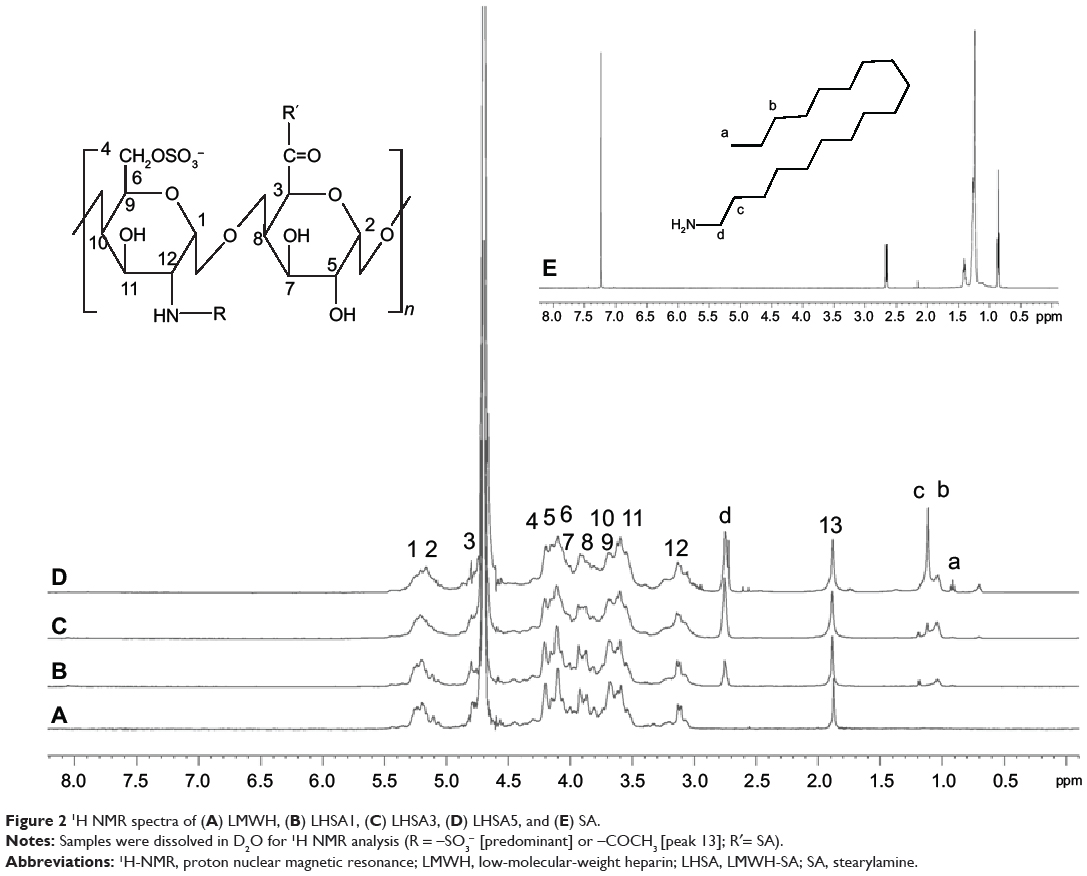
|
Figure 21H NMR spectra of (A) LMWH, (B) LHSA1, (C) LHSA3, (D) LHSA5, and (E) SA. |
The anticoagulant activities of LHSA1, LHSA3, and LHSA5 were 60.41%, 32.09%, and 31.92%, respectively, compared with that of free LMWH (Table 1). It is notable that the anticoagulant activity of LHSA5 did not significantly change with the encapsulation of DCT at 5:1 (LHSA5/DCT, w/w).
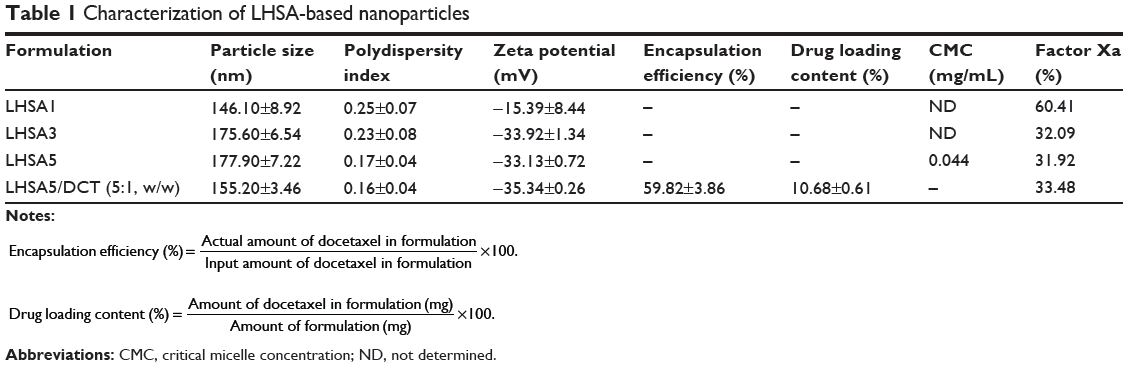
|
Table 1 Characterization of LHSA-based nanoparticles |

Preparation and characterization of LHSA-based self-assembled nanoparticles
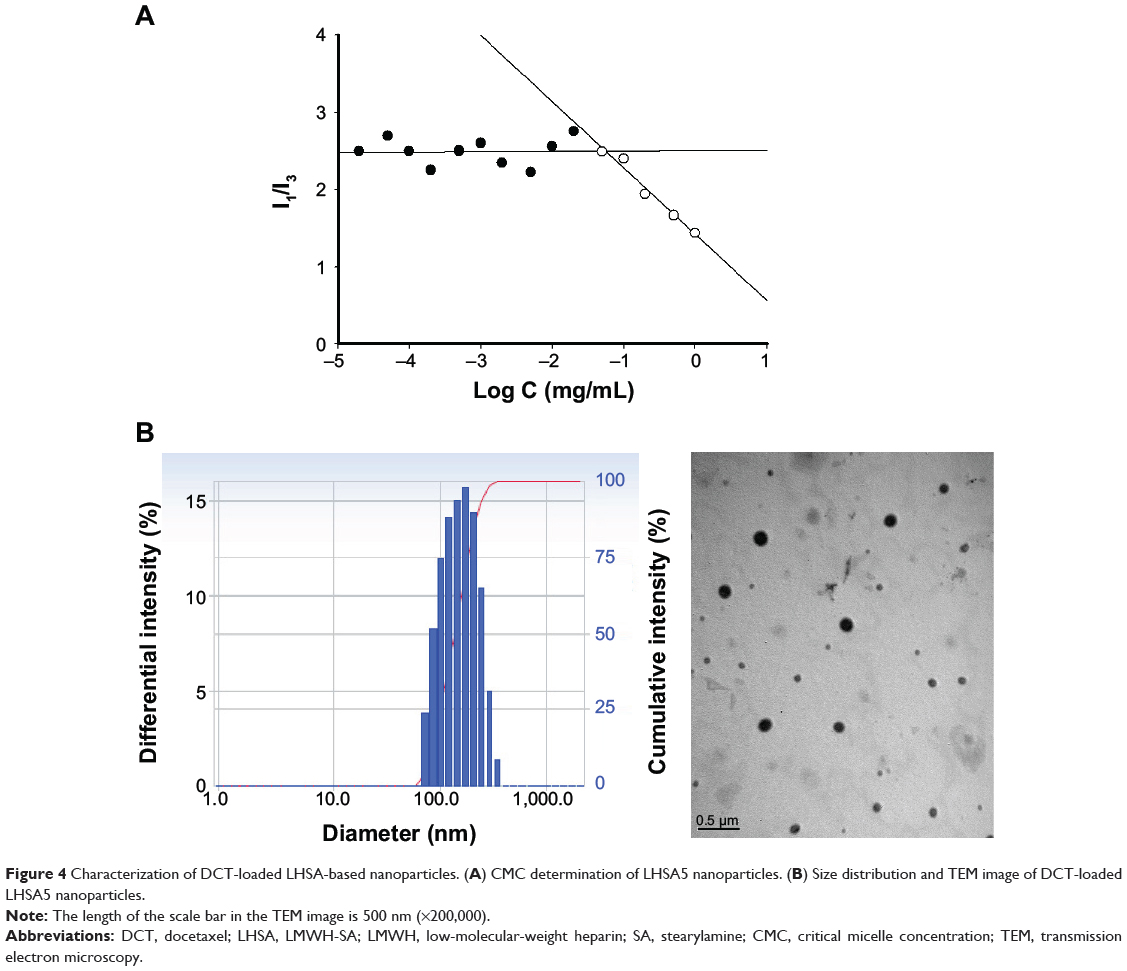
A solvent evaporation method was used to load DCT into the LHSA-based nanoparticles. Blank self-assembled LHSA nanoparticles were prepared without DCT loading by dissolving the LHSA conjugate in an aqueous solution. The mean diameter, zeta potential, and drug loading values are shown in Table 1. All samples successfully formed nanoparticles with a mean diameter of 140–180 nm, which was confirmed by the size distribution and TEM of LHSA nanoparticles (Figure 3A, LHSA1; Figure 3B, LHSA3; Figure 3C, LHSA5). The polydispersity index of blank and DCT-loaded nanoparticles were 0.171±0.04 and 0.161±0.04, respectively, indicating a narrow size distribution of the nanoparticles (Figure 3). It is interesting to note that the incorporation of DCT in the LHSA5 nanoparticles leads to a decrease of the particle size from 177.9 to 155.20 nm (Table 1). The loading of hydrophobic drugs in the inner core can enhance the stability of self-assembled nanoparticles by inducing morphology transition in order to decrease the total free energy, thereby resulting in the reduction of particle size.38,39 The nanoparticles showed negative zeta potential values due to the LMWH located in the shell. The CMC of LHSA nanoparticles was determined by measuring the fluorescence intensity in the presence of pyrene as a probe. After measuring the fluorescence intensity ratio (I1/I3) at various concentrations of the LHSA conjugate, the CMC was determined from the threshold concentration of self-assembled nanoparticles (Figure 4A). The CMC value of LHSA5 was 0.044 mg mL−1; however, the CMC values for LHSA1 and LHSA3 could not be determined (Table 1). This result suggests that the LHSA5 conjugate can form more stable nanoparticulate structures in an aqueous environment, compared with LHSA1 or LHSA3.
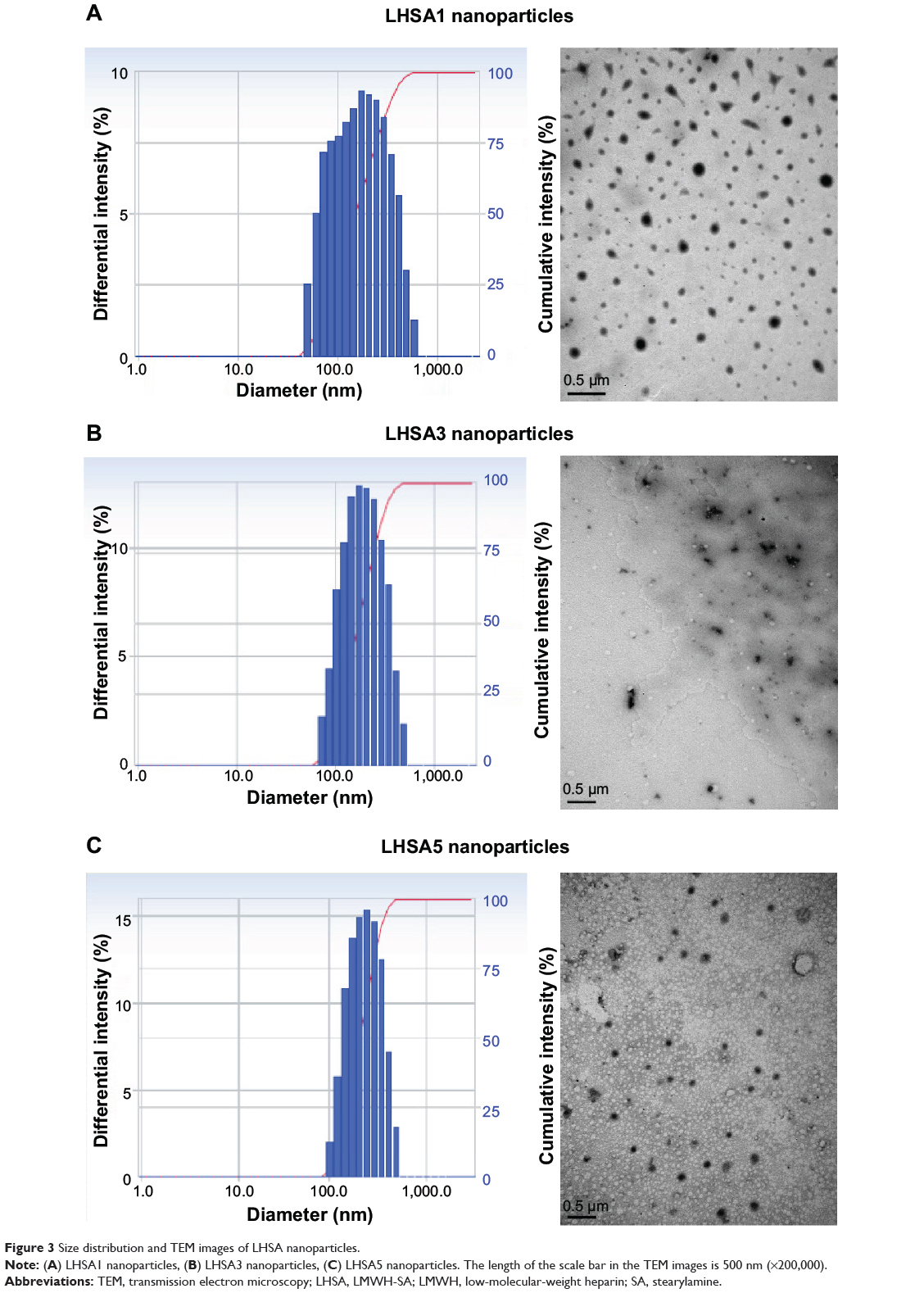
|
Figure 3 Size distribution and TEM images of LHSA nanoparticles. |
|
Figure 4 Characterization of DCT-loaded LHSA-based nanoparticles. (A) CMC determination of LHSA5 nanoparticles. (B) Size distribution and TEM image of DCT-loaded LHSA5 nanoparticles. |
The encapsulation efficiency and the drug loading content in the LHSA5/DCT nanoparticles was 59.82% and 10.68%, respectively (Table 1). TEM images showed that the DCT-loaded LHSA-based nanoparticles were also spherical in shape with narrow size distribution (Figure 4B).
In vitro DCT release
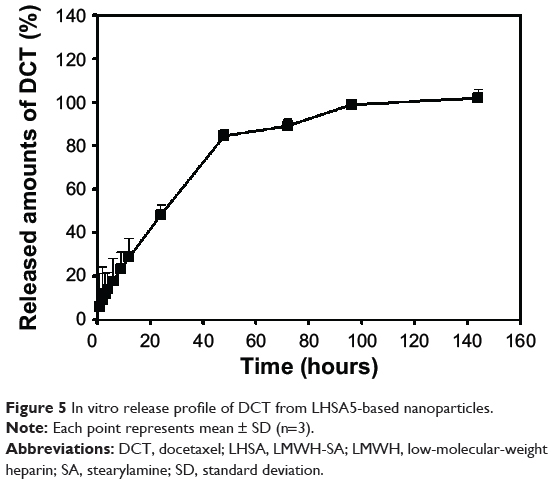
Figure 5 shows the in vitro DCT release profile. DCT was incorporated in the LHSA5 nanoparticles and the sink condition was adequately maintained in the release test. The in vitro release of DCT from LHSA5 nanoparticles was investigated for 6 days. After the initial release for 24 hours, the release rate of DCT slightly decreased with incubation time, and showed a sustained DCT release pattern. The release level of DCT within 24 hours was 48.24%; additional release continued up to 96 hours. A sustained DCT release pattern can lead to reduction in in vivo drug clearance, which results in maintenance of adequate drug concentrations for tumor growth inhibition.
|
Figure 5 In vitro release profile of DCT from LHSA5-based nanoparticles. |
In vitro cytotoxicity of LHSA conjugate
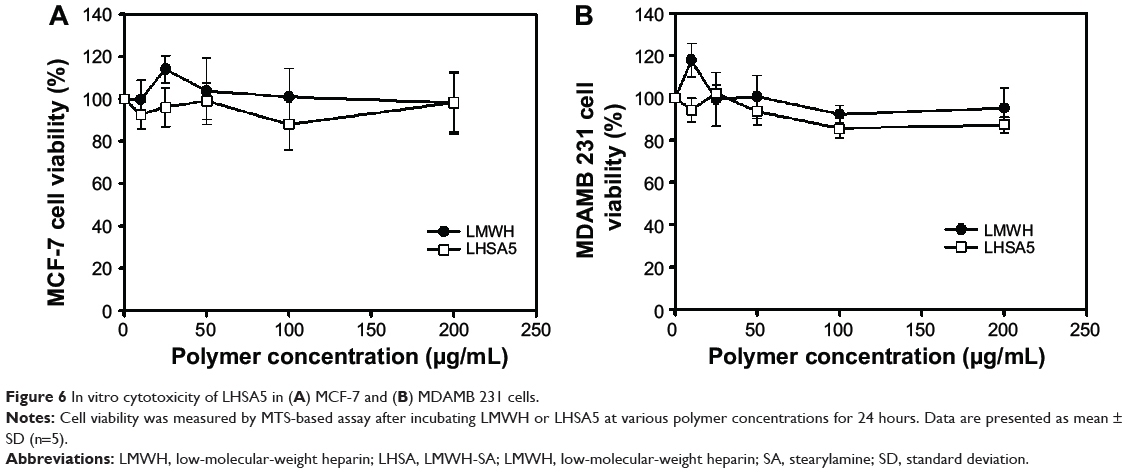
The cytotoxicity of blank LHSA5 conjugate was investigated in MCF-7 (Figure 6A) and MDAMB 231 (Figure 6B) breast cancer cell lines. Cell viability was measured after treating with various concentrations of LMWH and blank LHSA5 conjugate for 24 hours. At all blank LHSA5 conjugate polymer concentrations (0–200 μg mL−1), no significant viability differences between the LMWH and blank LHSA5 conjugate groups were observed, indicating that the blank LHSA5 conjugate polymer exerted no severe cytotoxicity on MCF-6 and MDAMB 231 cells.
|
Figure 6 In vitro cytotoxicity of LHSA5 in (A) MCF-7 and (B) MDAMB 231 cells. |
In vitro antitumor effect
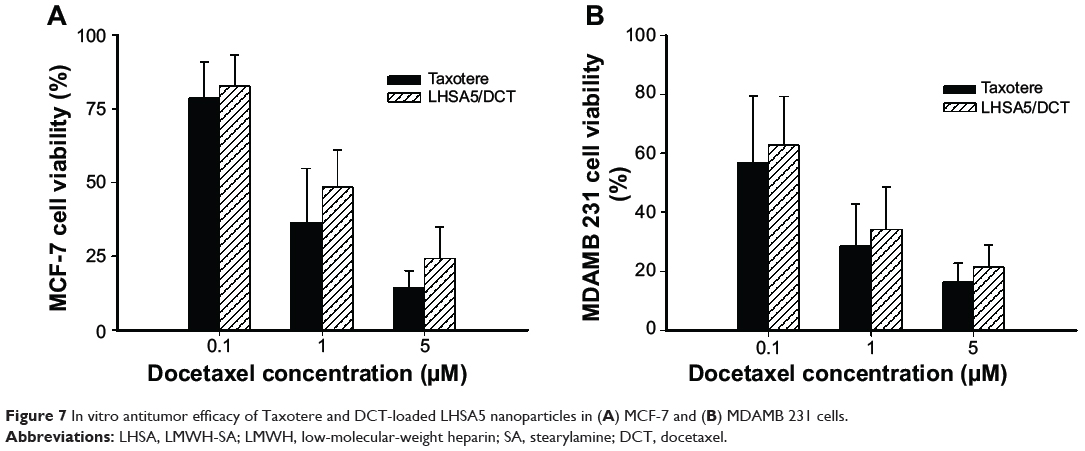
In vitro antitumor effects were evaluated using MCF-7 (Figure 7A) and MDAMB 231 (Figure 7B) cells. Free DCT (Taxotere) and DCT-loaded LHSA5 nanoparticles have antitumor efficacy against MCF-7 and MDAMB 231 cells. The DCT-loaded LHSA5 nanoparticle exhibited a similar or slightly lower efficacy than free DCT, possibly due to sustained release of free DCT from LHSA5 nanoparticles.
|
Figure 7 In vitro antitumor efficacy of Taxotere and DCT-loaded LHSA5 nanoparticles in (A) MCF-7 and (B) MDAMB 231 cells. |
In vitro cellular uptake study
MCF-7 and MDAMB 231 cells are human breast cancer cell lines, and were selected since DCT is mainly used for the treatment of breast cancer.40 To investigate the cellular uptake efficiency of LHSA5 nanoparticles, LHSA5 nanoparticles loaded with C6 were observed using CLSM, with 4′,6-diamidino-2-phenylindole staining of nuclei in MCF-7 and MDAMB 231 cells. C6 is a fluorescence probe that has been widely used in cellular uptake studies of nanoparticles.34,41 In Figure 8A, the stronger green fluorescent signals indicate higher intracellular accumulation of C6 from LHSA5 nanoparticles compared to that from the C6 solution.
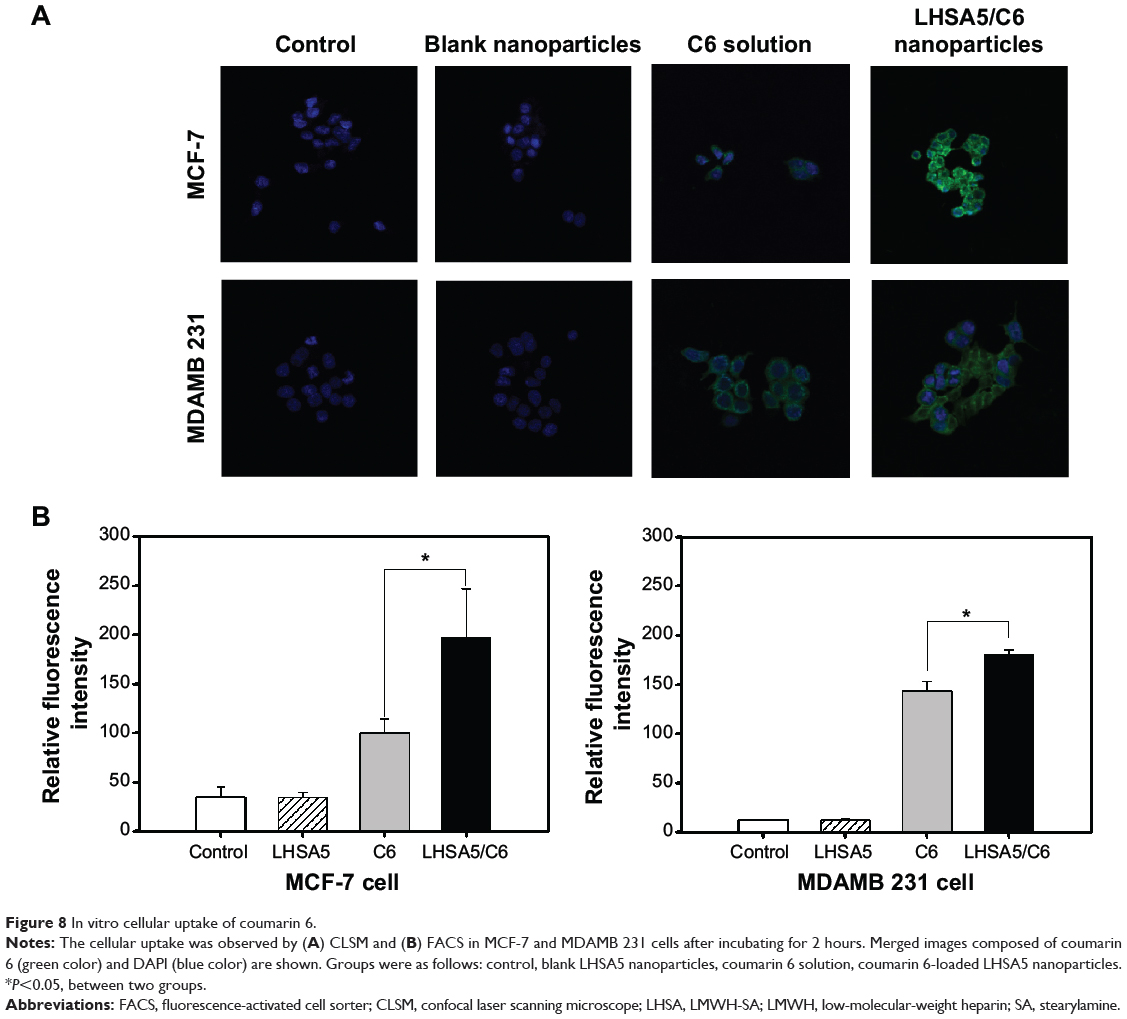
|
Figure 8 In vitro cellular uptake of coumarin 6. |
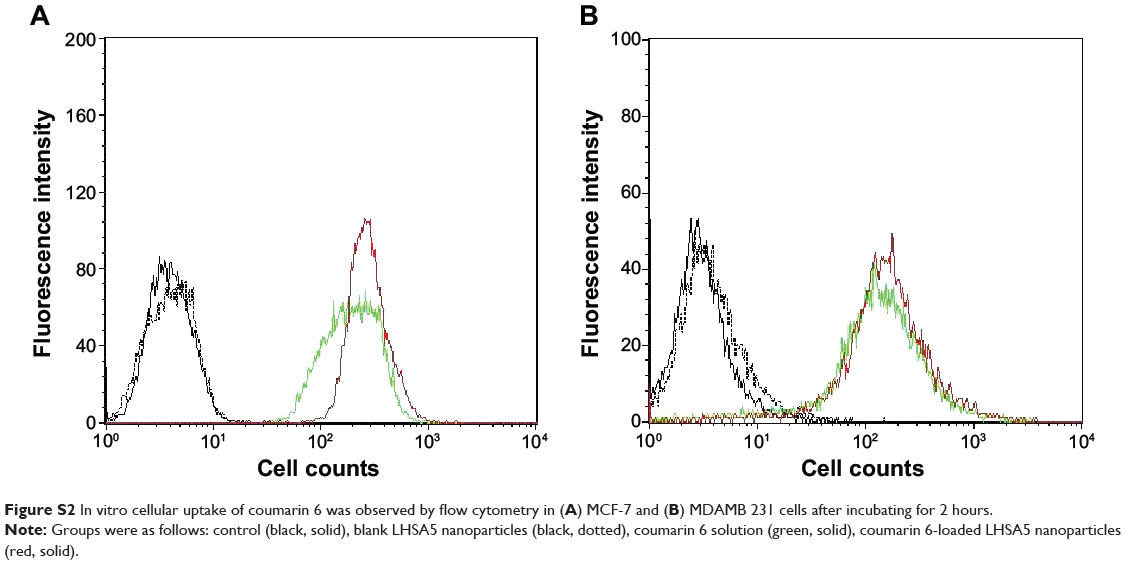
Cellular uptake efficiency was also quantitatively determined in MCF-7 (Figure S2A) and MDAMB 231 cells (Figure S2B) by a flow cytometry. The fluorescence intensity was assumed to be proportional to the amount of C6 in the cells; however, fluorescence was barely detected in the control (no treatment) and blank nanoparticle groups. Figure 8B shows that significantly higher amounts of C6 from LHSA5-based nanoparticles were taken up by the cells, compared with the C6 solution, which is consistent with the results of the CLSM study (Figure 8A). This result can be explained by the endocytosis of the nanoparticles.42,43 A previous study showed that fatty acid-modified polysaccharide-based nanoparticles form self-assembled nanoparticles with a multihydrophobic core.44 A hydrophobic minor core could facilitate the internalization of nanoparticles into cells via interaction with the plasma membrane. Although LHSA5-based nanoparticles are negatively charged due to their many sulfate and carboxyl groups, the nanoparticle was taken up in higher amounts, compared with the C6 solution.
In vivo antitumor efficacy
In vivo antitumor efficacy of the DCT-loaded LHSA5 nanoparticle formulation was evaluated in the MDAMB 231 tumor-bearing mouse model. Blank LHSA5 nanoparticles, DCT solution (Taxotere), and DCT-loaded LHSA5 nanoparticles were injected intravenously on days 0, 7, and 14. Then, tumor volume (mm3) and body weight (g) were monitored for 18 days.
The tumor volumes of the blank LHSA5 nanoparticle-treated, DCT solution (Taxotere)-treated, and DCT-loaded LHSA5 nanoparticle-treated groups on day 18 were 94.6%, 63.2%, and 47.1% that of the control group, respectively (Figure 9A). The most effective inhibition of tumor growth was observed in the group treated with DCT-loaded LHSA5 nanoparticles, while the blank LHSA5 nanoparticle treatment had little effect on tumor growth inhibition. Moreover, the tumor growth inhibition effect of the DCT-loaded LHSA5 nanoparticles was significantly greater than that of Taxotere (P<0.05).
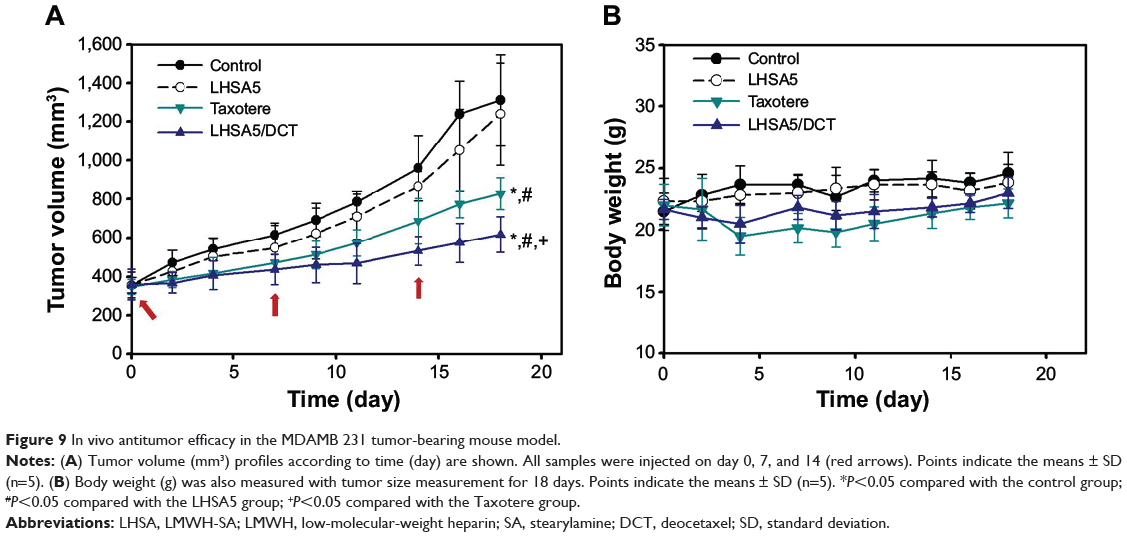
|
Figure 9 In vivo antitumor efficacy in the MDAMB 231 tumor-bearing mouse model. |
The safety of DCT-loaded LHSA nanoparticles was evaluated by measuring changes in body weight. For most groups, there was no significant difference in body weight (Figure 9B). However, the body weight of the Taxotere-treated group was significantly lower than that of the control group. The DCT-loaded LHSA5 nanoparticle-treated group showed no significant difference compared with the control group, suggesting that the DCT-loaded LHSA nanoparticle treatment was less toxic than Taxotere.
In vivo pharmacokinetic study
Figure 10 shows the plasma concentration–time profiles of DCT solution (Taxotere) and DCT-loaded LHSA5 nanoparticles, at a dose of 8 mg kg−1, in Sprague Dawley rats; relevant pharmacokinetic parameters are listed in Table 2. Our results showed that DCT was rapidly eliminated from the bloodstream in both the Taxotere and LHSA5 nanoparticle groups over the first 30 minutes after dosing. However, after dose normalization, the DCT in LHSA5 nanoparticles yielded a higher plasma concentration than did Taxotere. Noncompartmental analysis of the plasma concentrations showed a significant change in the pharmacokinetic parameters of DCT in LHSA5 nanoparticles. Compared with Taxotere, the LHSA5 nanoparticles provided a significantly higher AUC (2.48-fold), terminal t1/2 (3.02-fold), and mean residence time (3.64-fold). The LHSA5 nanoparticles also significantly decreased the clearance (CL, 2.54-fold) of DCT. These in vivo pharmacokinetic properties were thought to be related to its in vitro sustained release pattern. This result indicates that the LHSA5 nanoparticles prolonged the circulation of DCT in the bloodstream, thereby improving its therapeutic efficacy (Figure 9A).
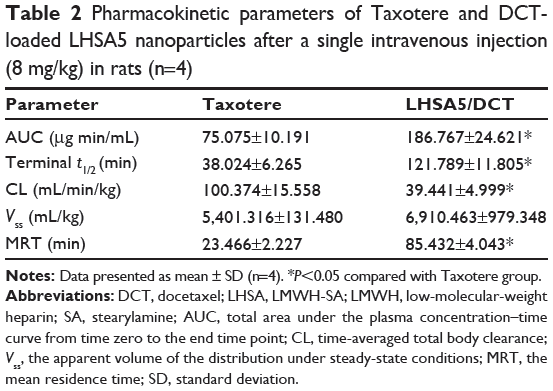
|
Table 2 Pharmacokinetic parameters of Taxotere and DCT-loaded LHSA5 nanoparticles after a single intravenous injection (8 mg/kg) in rats (n=4) |
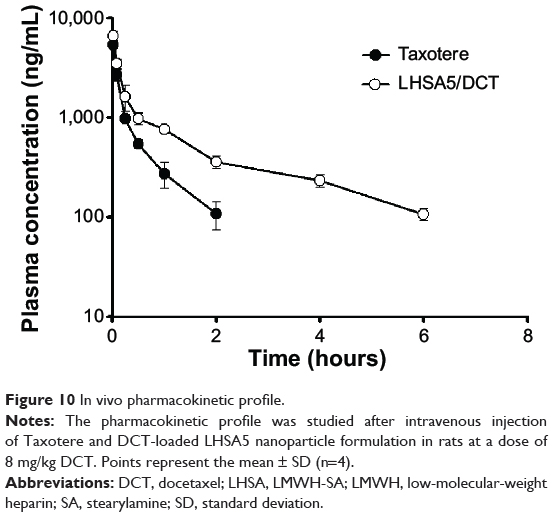
|
Figure 10 In vivo pharmacokinetic profile. |
Discussion
The synthetic scheme for the amphiphilic LHSA conjugate is shown in Figure 1. Amphiphilic LHSA conjugates with various LMWH to SA molar ratios were synthesized to investigate the feasibility of forming stable self-assembled nanoparticles. From 1H-NMR analysis of LHSA conjugates, the proton peak of SA (chemical shifts at 0.8–1.2 ppm) in LHSA1 and LHSA3 was lower than that for LHSA5 (Figure 2). An accurate substitution ratio (SA to LMWH) can be determined from the 1H-NMR spectrum of the physical mixtures of LMWH and SA. In our previous study,45 the correlation of the molar ratio of arachidic acid to chitosan oligosaccharide (CSO) was evaluated using 1H-NMR from a physical mixture of arachidic acid and CSO in DMSO-d6. However, LMWH has a strong hydrophilic property, while SA has a strong hydrophobic property; thus, an NMR cosolvent consisting of D2O and THF-d8 was used to dissolve LMWH and SA. The molar substitution ratios increased with increasing feed SA molar ratio. Note that these results are contrary to those of the anticoagulant activity of LHSA conjugates calculated using an anti-FXa chromogenic assay (Table 1); the anticoagulant activity of LHSA conjugates decreased as the amount of chemically coupled SA increased (Table 1), which is consistent with previously reported heparin derivative studies.46,47
All LHSA1, LHSA3, and LHSA5 conjugates prepared in this study formed self-assembled nanoparticles that were 140–180 nm in size. It is known that nanoparticles with a mean diameter <200 nm accumulate in tumors via the enhanced permeability and retention effect and exhibit reduced uptake by the reticuloendothelial system.3,48 Among them, the LHSA5 nanoparticles showed a narrower size distribution and a more stable spherical morphology than the LHSA1 and LHSA3 nanoparticles, as shown in Figure 3. However, the LHSA1 and LHSA3 conjugates did not possess the hydrophobicity necessary for the formation of stable nanoparticles due to the low substitution ratio of SA. Additionally, the CMC value of the LHSA5 conjugate was significantly lower (44 μg mL−1) than those of other low-molecular-weight surfactants or amphiphilic polymers,49,50 indicating that it can form stable nanoparticle structures, even in the presence of the reduced polymer concentrations after dilution with body fluids.
The LMWH of LHSA creates a hydrophilic outer shell of the nanoparticles in an aqueous environment. The SA of LHSA provides an internal hydrophobic core that can be used to encapsulate insoluble drugs.51 It was interesting to note that the zeta potential values of LHSA nanoparticles were negative, indicating that the LMWH was located on the surface of the shell. Therefore, LHSA could possess heparin activity after nanoparticle formation, yet the anticoagulant activity of LHSA was lower than that of the unmodified LMWH. Because LMWH reduces the risk of embolic events in patients with malignancy and acute venous thromboembolism,52 its use in cancer patients is recommended by numerous guidelines and is now regarded as a standard of care.53 Thus, the self-assembled LHSA5 nanoparticle could be attractive as an anticancer drug carrier, which has a synergic effect with poorly water-soluble anticancer drugs encapsulated in the core.
After characterization of the blank LHSA-based nanoparticles, we evaluated the DCT-loaded LHSA nanoparticles using the LHSA5 conjugate, due to its more stable nanoparticle configuration compared with the other structures. The encapsulation efficacy of the DCT-loaded LHSA5 nanoparticle was 59.82%, with TEM images revealing a spherical shape. The in vitro release of DCT from the LHSA5 nanoparticle continued for ~96 hours, as shown in Figure 5. From these results, we expected a reduction in the in vivo drug clearance, allowing a drug concentration adequate for tumor growth inhibition to be sustained. When intravenously administered in Sprague Dawley rats, the concentration profile of DCT in plasma was sustained for 6 hours, as compared with 2 hours for Taxotere administration (Figure 10).
The effects of the DCT-loaded LHSA5 nanoparticles on in vitro cytotoxicity and antitumor efficacy in MCF-7 and MDAMB 231 cells were investigated, as shown in Figures 6 and 7. The LMWH and blank LHSA5 nanoparticles exhibited no severe in vitro cytotoxicity. Free DCT and DCT-loaded LHSA5 nanoparticles were cytotoxic to MCF-7 and MDAMB 231 cells. DCT-loaded LHSA5 nanoparticles had a similar or slightly lower efficacy than free DCT, thus contributing to the sustained release of free DCT from LHSA5 nanoparticles. As shown in Figure 5, only 40% of the free DCT was released from LHSA5 nanoparticles in 24 hours in the in vitro release study.
C6 has been widely used in cellular uptake studies,34,41 and was used in this study as a fluorescence probe to investigate the cellular uptake efficiency of the LHSA nanoparticle. LHSA5 nanoparticles loaded with C6 in MCF-7 and MDAMB 231 cells were visualized by CLSM (Figure 8A). A significant difference was observed in the fluorescence intensities of the C6 solution and the C6-loaded LHSA nanoparticles. In addition, a significantly higher amount of C6 from the LHSA5 nanoparticle was taken up by the cells compared to that from the C6 solution, as shown in Figure 8B. This result could be attributable to endocytosis of the nanoparticles.42,43 Moreover, the fatty acid-modified polysaccharide-based nanoparticles are capable of forming self-assembled nanoparticles with a multihydrophobic core.44 The hydrophobic part of the nanoparticles could facilitate drug internalization into the cells via interactions with the cell membrane. Although the LHSA5-based nanoparticles were negatively charged due to their sulfate and carboxyl groups, the nanoparticles were taken up in significantly higher amounts compared with the C6 solution. In addition to drug uptake by nanoparticles, long-term cellular drug retention can play key roles in drug efficacy in microtubule-targeted anticancer drugs.54 Thus, LHSA5 nanoparticles could induce a synergy effect by enhancing drug uptake and retention time, which are shown in in vivo antitumor efficacy study (Figure 9).
The DCT-loaded LHSA5 nanoparticles exhibited significant inhibitory effects on MDAMB 231 tumor growth, compared with the other groups (Figure 9A). This can be explained in terms of the physicochemical properties of the drug-loaded nanoparticles that induce sustained drug release (Figure 10) and passive targeting to tumor tissue via an enhanced permeability and retention effect. Although the blank LHSA5 nanoparticles showed insignificant in vivo inhibition of MDAMB 231 tumor growth in this study, others have reported the antitumor effects of several heparin derivatives in tumor-bearing mice.36,55,56 This discrepancy can be attributed to the differences in tumor type, heparin injection frequency and capacity, administration route, and heparin-modifying molecules.
The concentration profile of DCT in plasma was sustained up to 6 hours (Figure 10). This result indicates that LHSA5 nanoparticles contributed to prolonged circulation of DCT in the bloodstream and is consistent with the improved therapeutic efficacy (Figure 9A). Compared with the DCT solution, the DCT-loaded LHSA5 nanoparticles provided a significantly higher AUC, t1/2, and mean residence time and a lower time-averaged total body clearance, as shown in Table 2. These in vivo pharmacokinetic parameters were related to its in vitro sustained DCT release pattern. Moreover, the increased retention time and decreased clearance of the DCT from plasma may contribute to enhanced in vivo antitumor efficacy in the tumor xenograft model, as shown in Figure 9A.
Conclusion
LHSA conjugates were successfully synthesized, and self-assembled nanoparticles based on LHSA were prepared. The LHSA-based nanoparticles have an LMWH moiety on the outer shell resulting in a negative surface charge. Among the synthesized conjugates, LHSA5 formed stable self-assembled nanoparticles in an aqueous environment, with a narrow size distribution. Because the LHSA5 nanoparticles showed 30% of the anticoagulant activity exhibited by free LMWH, it likely also maintains the other heparin activities. The DCT-loaded LHSA5 nanoparticle showed sustained drug-release profiles, in vitro and in vivo, compared with Taxotere. The nanoparticulate structure influenced cellular uptake; consequently, the DCT-loaded LHSA5 nanoparticle formulation improved the half-life of DCT and significantly inhibited MDAMB 231 tumor growth in xenograft mice. Thus, LHSA5-based self-assembled nanoparticles may be useful as an anticancer drug delivery system.
Acknowledgments
This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korean government (MSIP) (Number 2009-0083533) and the MarineBio Research Program (NRF-C1ABA001-2011-0018561).
Disclosure
The authors report no conflicts of interest in this work.
References
Kwon GS, Okano T. Polymeric micelles as new drug carriers. Adv Drug Deliv Rev. 1996;21(2):107–116. | ||
Liu Z, Jiao Y, Wang Y, et al. Polysaccharides-based nanoparticles as drug delivery systems. Adv Drug Deliv Rev. 2008;60(15):1650–1662. | ||
Maeda H, Wu J, Sawa T, et al. Tumor vascular permeability and the EPR effect in macromolecular therapeutics: a review. J Control Release. 2000;65(1):271–284. | ||
Maeda H. Tumor-selective delivery of macromolecular drugs via the EPR effect: background and future prospects. Bioconjugate Chem. 2010;21(5):797–802. | ||
Verrecchia T, Spenlehauer G, Bazile D, et al. Non-stealth (poly(lactic acid/albumin)) and stealth (poly(lactic acid-polyethylene glycol)) nanoparticles as injectable drug carriers. J Control Release. 1995;36(1):49–61. | ||
Shao X, Liu Q, Zhang C, et al. Concanavalin A-conjugated poly(ethylene glycol)-poly(lactic acid) nanoparticles for intranasal drug delivery to the cervical lymph nodes. J Microencapsul. 2013;30(8):780–786. | ||
Labhasetwar V, Song C, Humphrey W, et al. Arterial uptake of biodegradable nanoparticles: effect of surface modifications. J Pharm Sci. 1998;87(10):1229–1234. | ||
Molpeceres J, Guzman M, Aberturas MR, et al. Application of central composite designs to the preparation of polycaprolactone nanoparticles by solvent displacement. J Pharm Sci. 1996;85(2):206–213. | ||
Aishwarya S, Mahalakshmi S, Sehgal PK. Collagen-coated polycaprolactone microparticles as a controlled drug delivery system. J Microencapsul. 2008;25(5):298–306. | ||
Sharma S, Benson HA, Mukkur TK, et al. Preliminary studies on the development of IgA-loaded chitosan-dextran sulphate nanoparticles as a potential nasal delivery system for protein antigens. J Microencapsul. 2013;30(3):283–294. | ||
Jahanshahi M, Babaei Z. Protein nanoparticle: a unique system as drug delivery vehicles. Afr J Biotechnol. 2008;7(25):4926–4934. | ||
Wu Y, MacKay JA, McDaniel JR, et al. Fabrication of elastin-like polypeptide nanoparticles for drug delivery by electrospraying. Biomacromolecules. 2008;10(1):19–24. | ||
Kwon GS, Okano T. Soluble self-assembled block copolymers for drug delivery. Pharm Res. 1999;16(5):597–600. | ||
Saravanakumar G, Min KH, Min DS, et al. Hydrotropic oligomer-conjugated glycol chitosan as a carrier of paclitaxel: synthesis, characterization, and in vivo biodistribution. J Control Release. 2009;140(3):210–217. | ||
Sun P, Zhang Y, Shi L, et al. Thermosensitive nanoparticles self-assembled from PCL-b-PEO-b-PNIPAAm triblock copolymers and their potential for controlled drug release. Macromol Biosci. 2010;10(6):621–631. | ||
Lyseng-Williamson KA, Fenton C. Docetaxel. Drugs. 2005;65(17):2513–2531. | ||
Baker SD, Sparreboom A, Verweij J. Clinical pharmacokinetics of docetaxel. Clin Pharmacokinet. 2006;45(3):235–252. | ||
Michael A, Syrigos K, Pandha H. Prostate cancer chemotherapy in the era of targeted therapy. Prostate Cancer Prostatic Dis. 2008;12(1):13–16. | ||
Persohn E, Canta A, Schoepfer S, et al. Morphological and morphometric analysis of paclitaxel and docetaxel-induced peripheral neuropathy in rats. Eur J Cancer. 2005;41(10):1460–1466. | ||
Engels FK, Mathot RA, Verweij J. Alternative drug formulations of docetaxel: a review. Anticancer Drugs. 2007;18(2):95–103. | ||
Immordino ML, Brusa P, Arpicco S, et al. Preparation, characterization, cytotoxicity and pharmacokinetics of liposomes containing docetaxel. J Control Release. 2003;91(3):417–429. | ||
Yin Y-M, Cui F-D, Mu C-F, et al. Docetaxel microemulsion for enhanced oral bioavailability: preparation and in vitro and in vivo evaluation. J Control Release. 2009;140(2):86–94. | ||
Esmaeili F, Dinarvand R, Ghahremani MH, et al. Docetaxel–albumin conjugates: preparation, in vitro evaluation and biodistribution studies. J Pharm Sci. 2009;98(8):2718–2730. | ||
Buller H, Gent M, Gallus A, et al. Low-molecular weight heparin in the treatment of patients with venous thromboembolism: the Columbus Investigators. N Engl J Med. 1997;337:657–662. | ||
Young E. The anti-inflammatory effects of heparin and related compounds. Thromb Res. 2008;122(6):743–752. | ||
Niers TMH, Klerk CPW, DiNisio M, et al. Mechanisms of heparin induced anti-cancer activity in experimental cancer models. Crit Rev Oncol Hematol. 2007;61(3):195–207. | ||
Castelli R, Porro F, Tarsia P. The heparins and cancer: review of clinical trials and biological properties. Vasc Med. 2004;9(3):205–213. | ||
Marchetti M, Vignoli A, Russo L, et al. Endothelial capillary tube formation and cell proliferation induced by tumor cells are affected by low molecular weight heparins and unfractionated heparin. Thromb Res. 2008;121(5):637–645. | ||
Bae KH, Mok H, Park TG. Synthesis, characterization, and intracellular delivery of reducible heparin nanogels for apoptotic cell death. Biomaterials. 2008;29(23):3376–3383. | ||
Casu B, Guerrini M, Guglieri S, et al. Undersulfated and glycol-split heparins endowed with antiangiogenic activity. J Med Chem. 2004;47(4):838–848. | ||
Smorenburg SM, Van Noorden CJ. The complex effects of heparins on cancer progression and metastasis in experimental studies. Pharmacol Rev. 2001;53(1):93–106. | ||
Farge D, Debourdeau P, Beckers M, et al. International clinical practice guidelines for the treatment and prophylaxis of venous thromboembolism in patients with cancer. J Thromb Haemost. 2013;11(1):56–70. | ||
Min Y, Akbulut M, Kristiansen K, et al. The role of interparticle and external forces in nanoparticle assembly. Nat Mater. 2008;7(7):527–538. | ||
Cho H-J, Yoon HY, Koo H, et al. Self-assembled nanoparticles based on hyaluronic acid-ceramide (HA-CE) and Pluronic® for tumor-targeted delivery of docetaxel. Biomaterials. 2011;32(29):7181–7190. | ||
Park K, Lee GY, Park R-W, et al. Combination therapy of heparin–deoxycholic acid conjugate and doxorubicin against squamous cell carcinoma and B16F10 melanoma. Pharm Res. 2008;25(2):268–276. | ||
Park K, Lee GY, Kim Y-S, et al. Heparin–deoxycholic acid chemical conjugate as an anticancer drug carrier and its antitumor activity. J Control Release. 2006;114(3):300–306. | ||
Lee JS, Go DH, Bae JW, et al. Synthesis and characterization of heparin conjugated Tetronic®–PCL copolymer for protein drug delivery. Curr Appl Phys. 2007;7:e49–e52. | ||
Li S, Byrne B, Welsh J, et al. Self-assembled poly(butadiene)-b-poly (ethylene oxide)polymersomes as paclitaxel carriers. Biotechnol Prog. 2007;23(1):278–285. | ||
Lim Soo P, Eisenberg A. Preparation of block copolymer vesicles in solution. J Polym Sci B Polym Phys. 2004;42(6):923–938. | ||
Clarke SJ, Rivory LP. Clinical pharmacokinetics of docetaxel. Clin Pharmacokinet. 1999;36(2):99–114. | ||
Mu CF, Balakrishnan P, Cui FD, et al. The effects of mixed MPEG-PLA/Pluronic copolymer micelles on the bioavailability and multidrug resistance of docetaxel. Biomaterials. 2010;31(8):2371–2379. | ||
He C, Hu Y, Yin L, et al. Effects of particle size and surface charge on cellular uptake and biodistribution of polymeric nanoparticles. Biomaterials. 2010;31(13):3657–3666. | ||
Conner SD, Schmid SL. Regulated portals of entry into the cell. Nature. 2003;422(6927):37–44. | ||
You J, Hu F-Q, Du Y-Z, et al. High cytotoxicity and resistant-cell reversal of novel paclitaxel loaded micelles by enhancing the molecular-target delivery of the drug. Nanotechnology. 2007;18(49):495101. | ||
Termsarasab U, Cho H-J, Kim DH, et al. Chitosan oligosaccharide–arachidic acid-based nanoparticles for anti-cancer drug delivery. Int J Pharm. 2013;441(1–2):373–380. | ||
Park K, Kim K, Kwon IC, et al. Preparation and characterization of self-assembled nanoparticles of heparin-deoxycholic acid conjugates. Langmuir. 2004;20(26):11726–11731. | ||
Rosenberg RD. Heparin, antithrombin, and abnormal clotting. Annu Rev Med. 1978;29(1):367–378. | ||
Gaucher G, Dufresne M-H, Sant VP, et al. Block copolymer micelles: preparation, characterization and application in drug delivery. J Control Release. 2005;109(1):169–188. | ||
Lee JS, Go DH, Bae JW, et al. Heparin conjugated polymeric micelle for long-term delivery of basic fibroblast growth factor. J Control Release. 2007;117(2):204–209. | ||
Kratohvil JP, Hsu WP, Kwok DI. How large are the micelles of di-α-hydroxy bile salts at the critical micellization concentrations in aqueous electrolyte solutions? Results for sodium taurodeoxycholate and sodium deoxycholate. Langmuir. 1986;2(2):256–258. | ||
Rizkalla N, Range C, Lacasse F-X, et al. Effect of various formulation parameters on the properties of polymeric nanoparticles prepared by multiple emulsion method. J Microencapsul. 2006;23(1):39–57. | ||
Lee AY, Levine MN, Baker RI, et al. Low-molecular-weight heparin versus a coumarin for the prevention of recurrent venous thromboembolism in patients with cancer. N Engl J Med. 2003;349(2):146–153. | ||
Nishioka J, Goodin S. Low-molecular-weight heparin in cancer-associated thrombosis: treatment, secondary prevention, and survival. J Oncol Pharm Pract. 2007;13(2):85–97. | ||
Jordan MA, Kamath K. How do microtubule-targeted drugs work? An overview. Curr Cancer Drug Targets. 2007;7(8):730–742. | ||
Thorpe PE, Derbyshire EJ, Andrade SP, et al. Heparin-steroid conjugates: new angiogenesis inhibitors with antitumor activity in mice. Cancer Res. 1993;53(13):3000–3007. | ||
Lapierre F, Holme K, Lam L, et al. Chemical modifications of heparin that diminish its anticoagulant but preserve its heparanase-inhibitory, angiostatic, anti-tumor and anti-metastatic properties. Glycobiology. 1996;6(3):355–366. |
Supplementary materials
|
Figure S1 Relationship between the ratio of integration area (2.80/1.95 ppm) and molar ratio of SA/LMWH based on their physical mixture. |
|
Figure S2 In vitro cellular uptake of coumarin 6 was observed by flow cytometry in (A) MCF-7 and (B) MDAMB 231 cells after incubating for 2 hours. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.