Back to Journals » International Journal of Nanomedicine » Volume 14
Preparation and characterization of nimodipine-loaded nanostructured lipid systems for enhanced solubility and bioavailability
Authors Teng Z, Yu M, Ding Y, Zhang H, Shen Y, Jiang M, Liu P, Opoku-Damoah Y, Webster TJ ![]() , Zhou J
, Zhou J
Received 8 September 2018
Accepted for publication 29 October 2018
Published 21 December 2018 Volume 2019:14 Pages 119—133
DOI https://doi.org/10.2147/IJN.S186899
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Lei Yang
Zaijin Teng,1,* Miao Yu,1,* Yang Ding,1,* Huaqing Zhang,1 Yan Shen,1 Menglao Jiang,1 Peixin Liu,1 Yaw Opoku-Damoah,2 Thomas J Webster,3 Jianping Zhou1
1Department of Pharmaceutics, State Key Laboratory of Natural Medicines, China Pharmaceutical University, Nanjing 210009, China; 2Australian Institute for Bioengineering and Nanotechnology, The University of Queensland, St Lucia, Brisbane, QLD 4072, Australia; 3Department of Chemical Engineering, Northeastern University, Boston, MA 02115, USA
*These authors contributed equally to this work
Purpose: Nimodipine (NMP) is a clinical dihydropyridine calcium antagonist. However, the clinical application of NMP is limited by poor water solubility and low oral bioavailability. To overcome these drawbacks, this study designed optimal NMP-incorporated nanostructured lipid carriers (NLCs).
Methods: High-pressure homogenization was successfully applied to prepare NMP-NLC, and the nanoparticle morphology was observed by a transmission electron microscope. The existence form of NMP in NMP-NLC was investigated by powder X-ray diffraction, differential scanning calorimetry, and Fourier transform infrared spectroscopy, respectively. The in vitro release study was performed by the dialysis method, and in vivo studies including in situ intestinal perfusion and pharmacokinetics were investigated in rats with NMP detected by high-performance liquid chromatography.
Results: The obtained NMP-NLC shared a spherical shape of ~70 nm with a smooth surface and high encapsulation efficiency of 86.8%±2.1%. Spectroscopy indicated that the drug was in an amorphous state. The NMP-NLC exhibited a sustained release and diverse release profiles under different release medium, which mimicked the physiological environment. Moreover, an in situ intestinal perfusion experiment revealed that NMP-NLC could be mainly absorbed by the small intestine. Remarkable improvements in Cmax and AUC0–∞ from NMP-NLC were obtained from pharmacokinetic experiments, and the relative bioavailability of NMP-loaded nanostructured lipid systems was 160.96% relative to NMP suspensions.
Conclusion: Collectively, the NLCs significantly enhanced the oral bioavailability of NMP and might provide a promising nanoplatform for hydrophobic drug delivery.
Keywords: nimodipine, nanostructured lipid carriers, increased drug solubility, bioavailability improvement
Introduction
Nimodipine (NMP), a new generation of dihydropyridine calcium channel blocker, was originally developed by Bayer for cerebral dysfunction treatment, which was attributed to its high selectivity for brain tissue receptors and high blood–brain barrier permeability. It has been proposed as the first choice to prevent subarachnoid hemorrhage, as well as a decent opponent to treat Alzheimer’s disease, stroke, and hypertension.1 Conspicuously, since the drug was approved in West Germany in 1985 and the US in 1987, it has invariably been one of the top ten products in Bayer sales for the past 30 years and has an created economic value as much as $120 million. Currently, NMP has been widely applied in a variety of marketed products, such as injections, tablets, capsules, and oral liquids with the development of pharmaceutics, which promoted its clinical application. However, in terms of pharmacological properties, NMP belongs to Class II of the Biopharmaceutical Classification System with a half-life in plasma of 7–8 hours.2 In clinical applications, NMP has been limited due to a low dissolving capacity and vast first pass effect on low bioavailability.3 Thus, even for the optimal oral formulation of NMP tablets, Nimotop® marketed by Bayer, the patients were compromised to take 60 mg (about two tablets) every 4 hours for 21 consecutive days. Such high doses triggered a series of noncompliance and inconvenience and even limited its market prospect.4 Although intravenous injection could make up this defect, various safety problems were typically not negligible.5 Therefore, designing applicable formulations for the enhancement of bioactivity of NMP becomes an urgent need.
Nanotechnology has been widely applied in the cross-disciplinary field of biology and medicine since the 1990s. Notably, nanoparticle drug delivery systems, including liposomes, nano-emulsions, and polymeric nanoparticles, have gradually become emerging hotspots. Lipid nanoparticles, including solid lipid carriers and mixed lipid carriers at the nanostructure,6 have been identified as appropriate carriers for the delivery of drugs or other active ingredients. Solid lipid nanoparticles (SLN), the first generation of lipid carriers, comprise biocompatible solid lipid matrices. Similarly, inspired from SLN, nanostructured lipid carriers (NLC) comprise liquid and solid lipids.7 More importantly, it has been explored that the mixed lipid system could guarantee outstanding biological applicability, controlled drug release, and the potential of large industrial scale production when compared to SLN.8 Moreover, the miscellaneous lipid components in NLC could form an inner-defective structure and amorphous state to entrap more drugs and avoid the leakage of drugs during storage.9,10 In addition to the high efficiency of drug loading, NLCs possess numerous advantages on various administration methods by means of oral, pulmonary, dermal, and ocular, which have expanded applications for the nanoparticle delivery system. For oral routes, NLCs exhibit superior controlled drug release capacity over other lipid carriers in the gastrointestinal tract and different release mechanisms by adjusting the liquid lipid content in solid form. As for the promotion of low water-soluble drug absorption, it can maintain sufficient solubility in the intestinal absorption site on account of the small particle size and lipid solubilization.11 When interacting with bile salts to form micelles, the lipid components in NLCs degrade to mono- and diglycerides triggered by digestive enzymes to improve drug solubility in the static aqueous layer of the intestinal villi surface.8 Moreover, NLCs could enhance the inner drug absorption through the lymphatic circulation or the Peyer’s patches to achieve an optional uptake, thus avoiding the elimination of the liver.12 These clues suggest that NLC is a promising oral carrier for oral administration.
Generally, it is necessary to achieve a new purpose for traditional medicine by exploiting a more suitable drug formulation to enhance its therapeutic capacity.13 Thus, in this study, an NMP-loaded NLC (NMP-NLC) was designed to assess the potential of NLC for oral bioavailability enhancement. Here, the particle size, zeta potential, entrapment efficiency (EE%), morphology, physicochemical properties, and in vitro properties were assessed to describe the developed NMP-NLC. Moreover, biopharmaceutical properties of the optimized NMP-NLC were evaluated by an in situ intestinal perfusion study and pharmacokinetic study in rats to verify the feasibility of NMP-NLC on the absorption and bioavailability of NMP, respectively.
Materials and methods
Materials
NMP was purchased from Xinhua Pharmaceutical Co., Ltd. (Shandong, China). Precirol ATO 5 and glyceryl monostearate (GMS) were purchased from Gattefosse (Saint-Priest, France), medium chain triglycerides (MCTs), isopropyl myristate (IPM), ethyl oleate (EO), oleic acid (OA), and propylene glycol dicaprate (Labrafac PG) were purchased from Sinopharm Chemical Reagent Co., Ltd. (Shanghai, China). Poloxamer 188 and Tween 80 were purchased from Thai Chemical Co., Ltd. (Shanghai, China). Vitamin E polyethylene glycol succinate (TPGS), polyoxyethylene esters of 12-hydroxystearic acid (Solutol HS-15), polyoxyethylene castor oil (ELP), and hydrogenated polyoxyethylene castor oil (RH 40) were kindly supplied by BASF Co., Ltd. (Ludwigshafen, Germany). All the reagent chemicals complied with analytical grade standards.
Formulation screening
The method of formulation screening was based on previous research which was introduced to explore the partitioning behavior between different solid lipids and NMP via drug solubility investigation.13 Among various solid lipids, ATO5 and GMS were selected due to the higher solubility of NMP.14 Then, a liquid lipid screening was carried out by exploring the saturated solubility of NMP in various types of lipids. Briefly, the drug was dissolved in a liquid lipid (1 mL) and incubated in a shaking bath (HZC-250; Bing Lab Equipment Co. Ltd, Beijing, China) at 200 rpm at 37°C±0.5°C for 48 hours.15 The mixture was centrifuged (KL05R; Kaida Scientific Instrument Co., Ltd., Changsha, China) at 3,000 rpm for 30 minutes and the supernatant was filtered using a 0.22-micron membrane filter. The concentration of NMP in the supernatant was detected by high-performance liquid chromatography (HPLC) (Agilent 1260; Agilent Technologies, Santa Clara, CA, USA), which was equipped with a C18 column (250 mm × 4.6 mm, 5 μm).16 The mobile phase was water and methanol (30:70, v/v), the flow rate was 1 mL/min, and NMP was detected at 356 nm. In addition, different batches of NMP-NLC were prepared with a mass ratio of solid lipid and liquid lipid at 2:1, 7:3, 3:1, 4:1, and 5:1.17 The most appropriate ratio was identified by the particle size and the EE%.
Preparation of NMP-NLCs
As shown in Figure 1, the NMP-NLCs were prepared via a high-pressure homogenization method.18 Briefly, Labrafac PG (0.9 g) and Precirol ATO 5 (2.1 g) were mixed and melted at 80°C to form the oil phase. Then, NMP coarse powder (100 mg) was dissolved in ethanol (5 mL) and added into the oil phase mentioned above, followed by heating to evaporate the ethanol. The obtained mixture was added dropwise into hot water (30 mL, 80°C), which contained Tween 80 (1.4 g), followed by stirring at 15,000 rpm for 5 minutes by high-speed shear (FS-400D; Zhibolian Testing Instrument Sales Co., Ltd., Tianjin, China).19 The obtained pre-emulsion was transferred into a high-pressure homogenizer (AH-2010; ATS Engineering Inc, Suzhou Branch, Jiangsu, China) and homogenized at 100 MPa for six cycles.20 Then, the dispersion was curdled at room temperature to form nanoparticles and filtered through a 0.22-micron membrane, collected and stored at 4°C for further use.
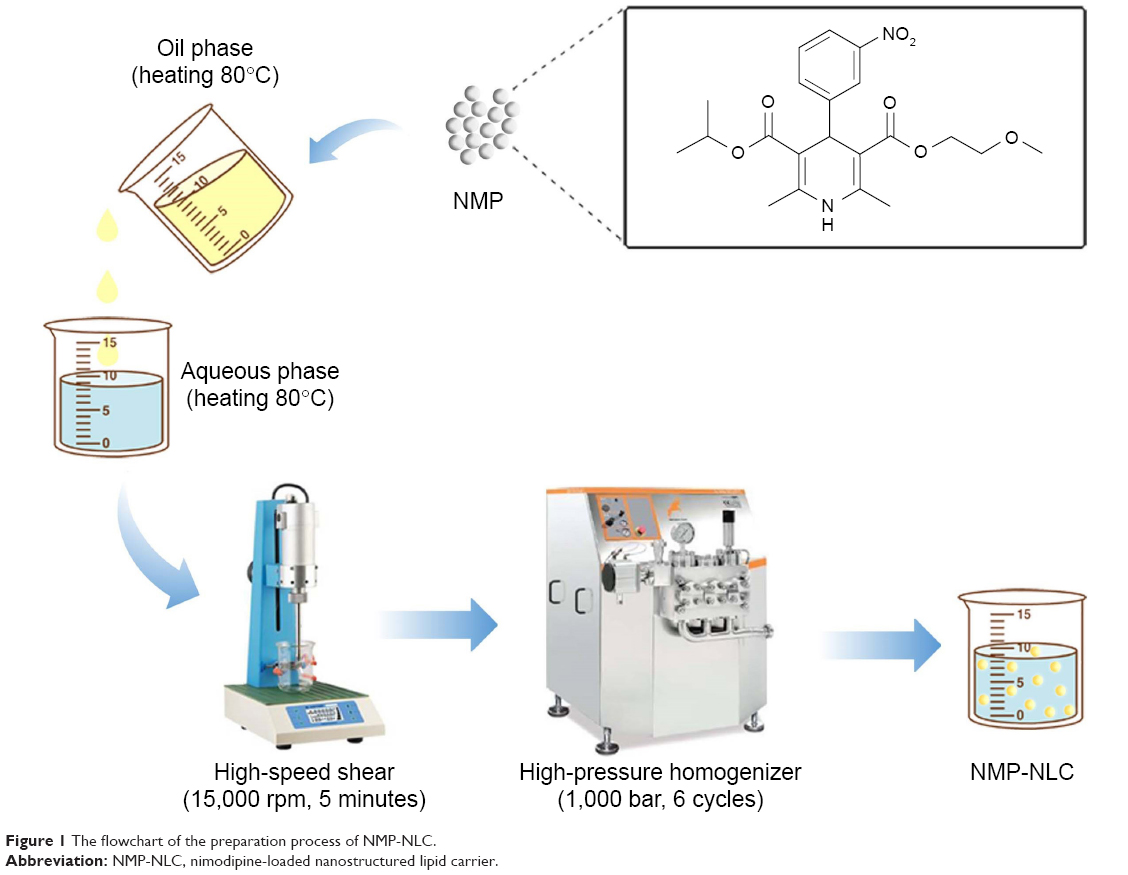 | Figure 1 The flowchart of the preparation process of NMP-NLC. |
Characterization of NMP-NLCs
Particle size and zeta potential
Particle size and zeta potential of the NMP-NLCs were detected by dynamic light scattering (DLS) using a Zetasizer Nano ZS (Malvern Instruments Ltd., Malvern, UK) at 25°C. The measurements were made in triplicate after a five times dilution with ultra-purified water.
EE% and loading efficiency (LE%)
The EE% was detected by an ultra-centrifugation method.21 Briefly, 1 mL of a NMP-NLC suspension was diluted in methanol and demulsified by an ultrasound water bath. The dispersion was added into an ultrafiltration centrifuge tube and centrifuged at 4,000 rpm for 20 minutes. The ultrafiltrate containing free drug was filtered through a 0.22-micron membrane filter and detected by HPLC as mentioned above. Similarly, LE% was the ratio of the incorporated drug to lipid (w/w). The EE% and LE% were calculated by the following Eq. (1) and (2), respectively.
|
|
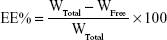
|
|
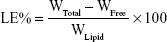
where, WTotal was the content of the total drug in NLC, WFree was the content of the unentrapped drug in ultrafiltrate, and WLipid was the content of the lipid in the system.
Transmission electron microscopy (TEM)
TEM analysis was employed to observe the morphology of the nanoparticles. Briefly, a diluted NMP-NLC solution was deposited on formvar-coated copper grids, followed by negative staining with 1% (w/v) phosphotungstic acid for 10 minutes. The obtained sample was observed by TEM (H-7650; Hitachi Ltd., Tokyo, Japan) at an acceleration voltage of 200 kV.
In vitro release study
The in vitro release study of NMP from NMP-NLC was detected as per the dialysis method.22 Dialysis bags (MWCO 12–14 kDa; Ruida Henghui Technology Development Co., Ltd., Beijing, China) were initially activated according to the manufacturer’s protocol. To investigate the release profile of NMP from NMP-NLC in response to different physiological pH, HCl (0.1 M, pH 1.2, 0.5% Tween 80) and PBS (0.01 M, pH 6.8, 0.5% Tween 80) were introduced as the release medium to stimulate succus gastricus and intestinal fluids, respectively. The obtained NMP-NLC (1 mL, 2.5 mg NMP) and the equivalent NMP suspension were added into the dialysis bags, followed by submerging into the released medium (100 mL, 37°C), and were incubated in a shaking bath at 100 rpm. At predetermined time intervals, including 0.5, 1, 2, 4, 8, 12, 24, and 48 hours, 1 mL of the release samples was withdrawn for HPLC analysis as mentioned above. The whole medium was refreshed with warmed released medium. Release profiles were obtained by calculating the percent of cumulative drug release against time and estimated according to Eq. (3):
|
|

Stability study
A preliminary study was conducted on the physical stability of NMP-NLCs. The freshly prepared NMP-NLC was stored in a closed glass vial at 4°C and 25°C, respectively. Particle size and polydispersity index were detected at 0, 1, and 2 weeks during the 2-week monitoring period.
Spectral analysis of NMP-NLC
Differential scanning calorimetry (DSC)
DSC analysis was analyzed by using a differential scanning calorimeter (DSC 8000/8500; PerkinElmer Inc., Waltham, MA, USA).23 The samples, including NMP coarse powder, physical mixtures of NMP and lipids with the same mass ratio as NMP-NLC, and lyophilized NMP-NLC powder, were accurately weighed for 3 mg and sealed in an aluminum pan, followed by heating at a rate of 10°C/min over a defined temperature ranging from 40°C to 200°C. Meanwhile, the whole process was protected under nitrogen protection. An empty aluminum pan was used as the reference. The indium and zinc standards were carried to calibrate the enthalpy scales and temperature, respectively.
Powder X-ray diffraction analysis (PXRD)
PXRD analysis was employed to identify the composition and structure based on the diffraction characteristics produced when X-rays pass through the lattice of the substance. The PXRD patterns were obtained using an powder X-ray diffractometer (XRD-6000; Shimadzu Group Co., Ltd., Kyoto, Japan).24 Different samples, including NMP coarse powder, physical mixtures, and lyophilized NMP-NLC powder, were gently consolidated in an aluminum holder. The detection conditions were as follows: the source of radiation was Cu-Kα tube and the wavelength was 1.5406 Å. The tube pressure was 40 kV and the current was 40 mA. The samples were scanned from 3° to 40° (2θ) at the scan rate of 1.2°/min at the step size of 0.02°. The X-ray diffractograms were assessed by DIFFRAC plus EVA (ver.9.0) diffraction software.
Fourier transform infrared spectroscopy (FT-IR)
The infrared absorption characteristics of NMP bulk drug, physical mixtures, and NMP-NLC lyophilized powder were compared by FT-IR (IRTracer-100; Shimadzu, Kyoto, Japan). The crystal form of the drug was detected by the changes of the characteristic absorption peaks or the hydrogen bond formation between the components. The sample was prepared by the KBr tablet method and the FT-IR spectra was recorded by an FT-IR analyzer.25 Briefly, samples were squeezed to a fine powder and added with anhydrous KBr at a mass ratio of 1:5 (sample:KBr), compressed to a thin pellet. The pressure was 5 tons for 3 minutes in a hydraulic press and the scanning range was 500–4,000 cm−1 at a resolution of 1.0 cm−1.
In situ single-pass intestinal perfusion (SPIP) study
The SPIP study was carried out with a slight modification of the previous studies.26 The sample preparation process was as follows: NMP-NLC and NMP solutions were diluted to 30 μg/mL in a Krebs Ringer’s buffer.27 All animal experiments were conducted under a protocol approved by the China Pharmaceutical University Ethics Committee and in accordance with the Guide for Care and Use of Laboratory Animals. The Sprague–Dawley rats (200–250 g; Qinglongshan Animal Center, Nanjing, China) were hocussed with 10% chloral hydrate (3 mL/kg) by intraperitoneal injection. Then they were fixed on the rat plate with infrared light for maintaining the body temperature. An abdominal cavity (length 3–4 cm) was opened at the midline of the rats’ abdomen. The intestine segments (small intestine and colon) were isolated. Then, the physiological saline at 37°C was pushed into the intestine segment to clean the intestinal contents. Silicone tubes were inserted into the intestine segment ends. Pre-weighted donor and receptor vials were placed on either side of the intestine segments. Before perfusion, the intestine was perfused for 15 minutes at 3 mL/min to achieve a steady state. Then, the perfusates (NMP-NLC and NMP solution, 30 μg/mL) were infused at 1 mL/min. The donor and receptor vials were quickly replaced at intervals of 15 minutes and the perfusate was collected at 60, 75, 90, 105, and 120 minutes. The samples were stored at –20°C before analysis. After the rats were executed, each intestine section was cut off with the length (l) and inner diameter (r) recorded. Finally, the dissolved samples were centrifuged at 12,000 rpm for 10 minutes. After diluting the supernatant with methanol, the samples (20 μL) were analyzed by HPLC.
The absorption rate (Ka) and apparent permeability (Papp) were calculated based on the following equations:
|
|
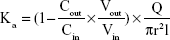
|
|
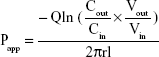
where Cout and Cin represent the drug concentrations in the outlet and inlet, respectively; Vout and Vin are the volume of perfusates in the receptor vial and the donor vial, respectively; Q is the flow rate (mL/min), l is the intestinal length (cm), and r is the intestinal radius (cm).
In vivo pharmacokinetic study
Animals and dosing
All animal experiments in this study were approved by the China Pharmaceutical University Ethics Committee and carried out in accordance with the National Institute of Health Guide for the Care and Use of Laboratory Animals. Male Sprague–Dawley rats of 200–250 g were raised at a temperature of 25°C±2°C and the relative humidity was 45%–50%. All rats were randomly divided into two groups (n=7), and fasted for 12 hours (free access to water) before the experiment. NMP suspension was prepared by dissolving NMP powder in a 0.5% CMC-Na solution.28 Then, NMP-NLC and NMP suspensions were administered orally to rats at a dose of 40 mg/kg, respectively. Blood samples (0.5 mL) were collected via the suborbital vein at 0, 0.25, 0.5, 1, 1.5, 2, 2.5 3, 3.5, 4, 8, 12, and 24 hours after administration. All the blood samples were instantly centrifuged at 5,000 rpm for 5 minutes to acquire the transparent plasma, followed by storing at −20°C before HPLC analysis.29
Sample preparation and determination
Diazepam solution (50 μL) was used as the internal standard (50 μg/mL) in this study and mixed a 100 μL plasma sample. After being vortexed for 1 minute, 500 μL of tert-butyl methyl ether were added into the mixture and vortexed for 5 minutes at room temperature to extract the drugs. Then, the mixture was centrifuged at 3,000 rpm for 10 minutes. The organic layer was transferred to a new vessel, followed by evaporation in the vacuum desiccator. The residue was dissolved with 200 μL of the mobile phase. The final mixture was centrifuged at 3,000 rpm for 5 minutes to obtain the supernatant for HPLC analysis.30
Data analysis processing
The main pharmacokinetic parameters were calculated by a pharmacokinetic program PK solver 2.0.31 The maximum concentration (Cmax) and time of Cmax (Tmax) were obtained by the experimental data. The area under the concentration–time curve (AUC) was calculated by the linear trapezoidal method. The relative bioavailability of NMP-NLC was calculated by using the following formula:
|
|
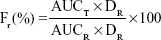
where Fr represents the relative bioavailability, AUC is the area under the blood concentration curve, D is the dosage of administration, T is the determinant formulation (NMP-NLC), and R is the reference formulation (NMP suspension).
Statistical analysis
All results were expressed as mean ± SD and the t-verification was carried out by SPSS 22.0 software. The numerical data were used to assess the significance of the differences and *P<0.05, **P<0.01, and ***P<0.001 were defined as statistically significant.
Results and discussion
Preparation and optimization of NMP-NLC
Given that the lipid property was crucial in drug loading ability, stability, and even drug release capacity of NLC, a screening of the lipids was conducted to obtain the optimal nanostructured lipid systems. Hence, we first screened the solid lipids based on the different characteristic parameters. The related data, including particle size, polydispersity index (PDI), zeta potential and EE%, are listed in Table 1. It could be found that NLCs prepared with ATO5 presented the smallest particle size of 76.1±0.2 nm and the highest EE% of 87.5%±2.4%. In addition, a suitable liquid lipid played a crucial role in the NLCs for that it could form an incomplete lattice to store more drugs. Hence, the solubility of drugs in the proper liquid lipid was the key factor in the formulation screening. Therefore, the solubility study of the NMP in the liquid lipid was carried out. As shown in Table 2, the best solubility was achieved in Labrafac PG at 24.88±2.1 mg/g, which was notably higher than OA of 4.65±0.8 mg/g, IPM of 12.34±1.4 mg/g, EO of 13.58±1.2 mg/g, and MCT of 18.74±1.6 mg/g. Thus, Labrafac PG was chosen as the most appropriate liquid lipid. To study the optimal ratio of ATO5 and Labrafac PG, versatile ratios of mixtures were investigated (Figure 2A). When the proportion of the solid lipid and liquid lipid (g/g) was 7:3, no separation between the lipids was observed. NLCs prepared at this proportion possessed the smallest particle size of 86.1±0.2 nm and the highest EE% of 86.8%±2.8%. In addition, the effect of different drug content was evaluated by the changes of drug and lipid mass ratios (Figure 2B). In accordance with the results, the EE% began to decrease when the mass ratio (drug:lipid) was over 0.03:1. It was considered that when the dose was higher, the amount of the drug exceeded the space capacity of the lipid blends, resulting in a lower EE% due to the precipitated drugs. Thus, a mass ratio of 0.03:1 (drug: lipid) was considered as the most appropriate one.
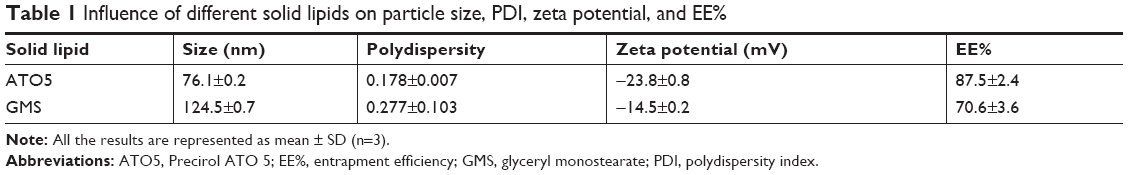 | Table 1 Influence of different solid lipids on particle size, PDI, zeta potential, and EE% |
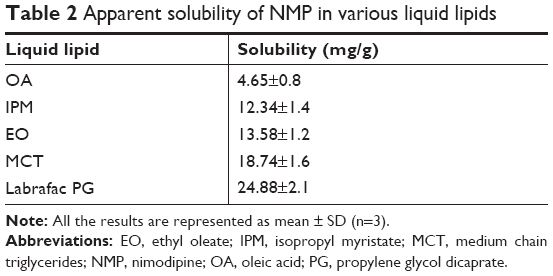 | Table 2 Apparent solubility of NMP in various liquid lipids |
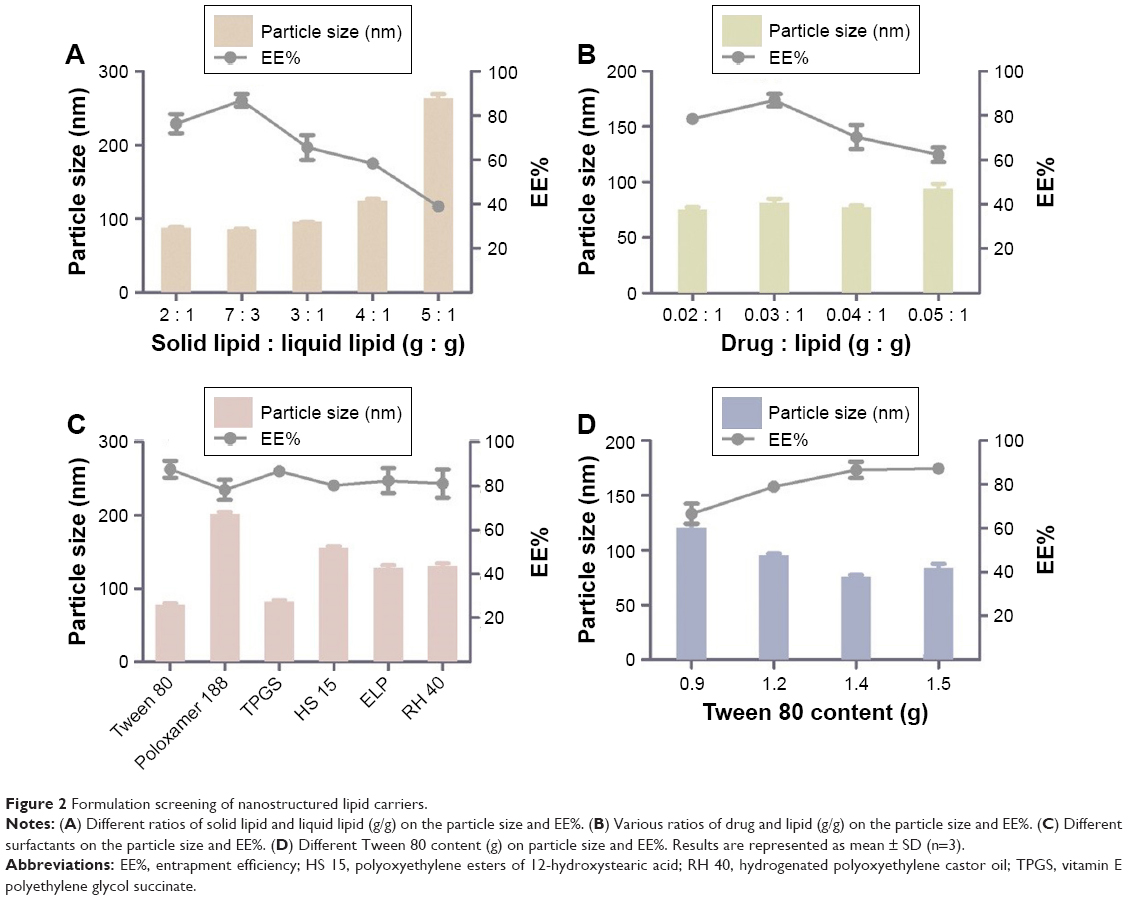 | Figure 2 Formulation screening of nanostructured lipid carriers. |
An emulsifier could effectively reduce the particle size and increase the physical stability of the lipid nanoparticles due to its ability in covering the surface of hydrophobic particles to reduce the surface tension between particles and the aqueous medium, sequentially preventing the aggregation of particles or the growth of nucleation.32 Thus, six kinds of nonionic surfactants, including Tween 80, Poloxamer 188, TPGS, Solutol HS-15, ELP, and RH 40, were first screened according to the particle size and EE%, which are listed in Figure 2C. Among all the surfactants, Tween 80 achieved the smallest particle size of 78.6±1.3 nm and the highest EE% of 87.7%±3.8%. On the other hand, the surfactant content also had a vital effect on the particle size and EE% (Figure 2D). When the Tween 80 content increased to over 1.4 g, the particle size and EE% increased. A higher content of Tween 80 would cause drug deposition and micelle aggregation, thus disturbing the stability of the system. Therefore, a Tween 80 content of 1.4 g was chosen as the optimum formulation.
It has been reported that NLCs could be prepared by high-pressure homogenization or ultrasonic dispersion.33 The high-pressure homogenization has been considered as a method for the large-scale production of NLCs, which can reduce the use of an organic solvent during the preparation and create a more homogeneous particle size distribution. Thus, the NMP-NLC was prepared by using high-pressure homogenization. Notably, before homogenization, a high-speed shearing of the coarse dispersion was used to smash big particles and avoid the blockage of the high-pressure homogenizer. Effects of different shear rates on the particle size distribution are shown in Figure 3A. When the shear rate was 15,000 rpm, no floating oil adhered to the surface of the dispersion. NLCs at this rate showed the smallest particle size of (71.9±0.9) nm and uniform distribution. Moreover, the pressure and numbers of homogenization cycles also influenced particle size and PDI. As shown in Figures 3B, C and 6, cycles at 100 MPa were affirmed as the final homogeneous condition to acquire the optimal formulation.
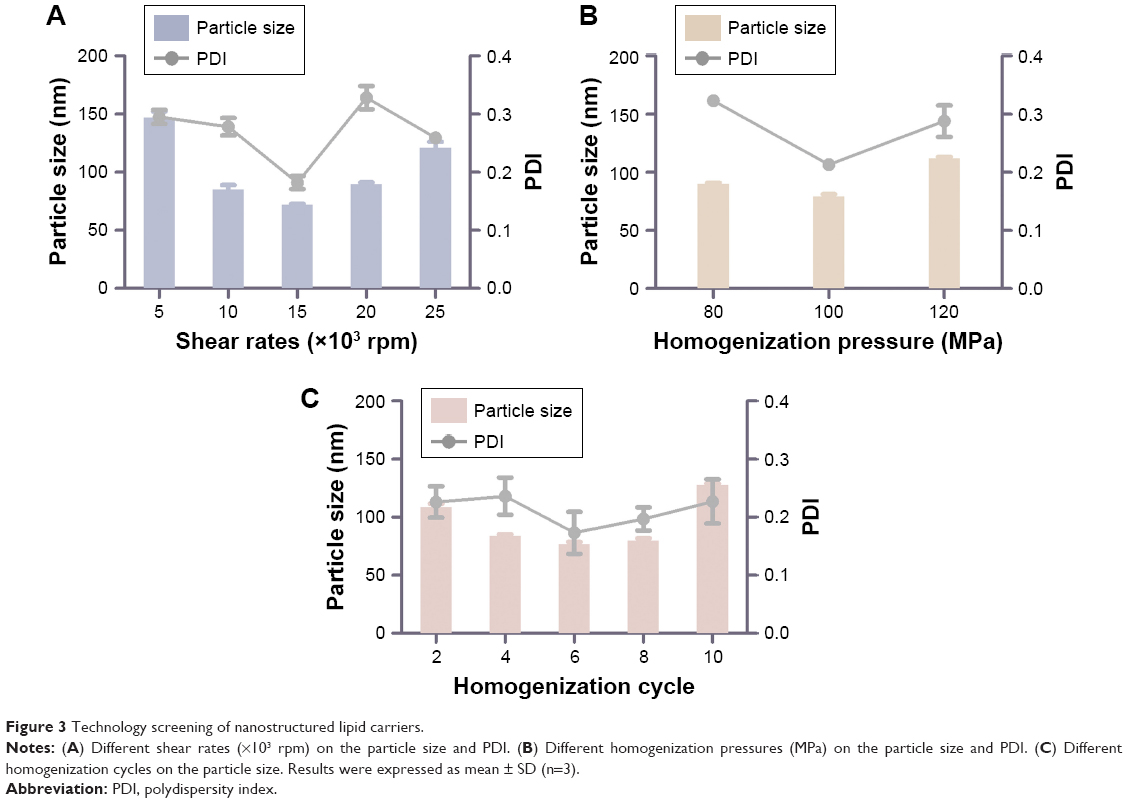 | Figure 3 Technology screening of nanostructured lipid carriers. |
Pharmaceutical characterization of nanoparticles
Particle size, zeta potential, EE%, and LE%
The obtained NMP-NLCs were in a light blue transparent solution without insoluble ingredients, agglomerates, or precipitation of drugs. Moreover, it achieved satisfactory flowability. The mean particle size of NMP-NLC was 71.0±1.3 nm as measured by DLS (Figure 4A), and the PDI was 0.057±0.027. Such a narrow and uniform particle size distribution could increase the surface area and wettability of the drug, which would lead to increased solubility of drugs in the gastrointestinal tract according to the Noyes–Whitney equation and promote the absorption of drugs. The negative zeta potential of NMP-NLC at −24.8±2.0 mV could guarantee the physical stability of the nanosystem, because it was generally considered that when the zeta potential of the nanoparticles was ~ ±30 mV, superfluous aggregation and agglomeration can be avoided by electrostatic barriers.34 Furthermore, the average LE% of the NMP-NLC was 3.2%±1.5%, and EE% was 86.8%±2.1%.
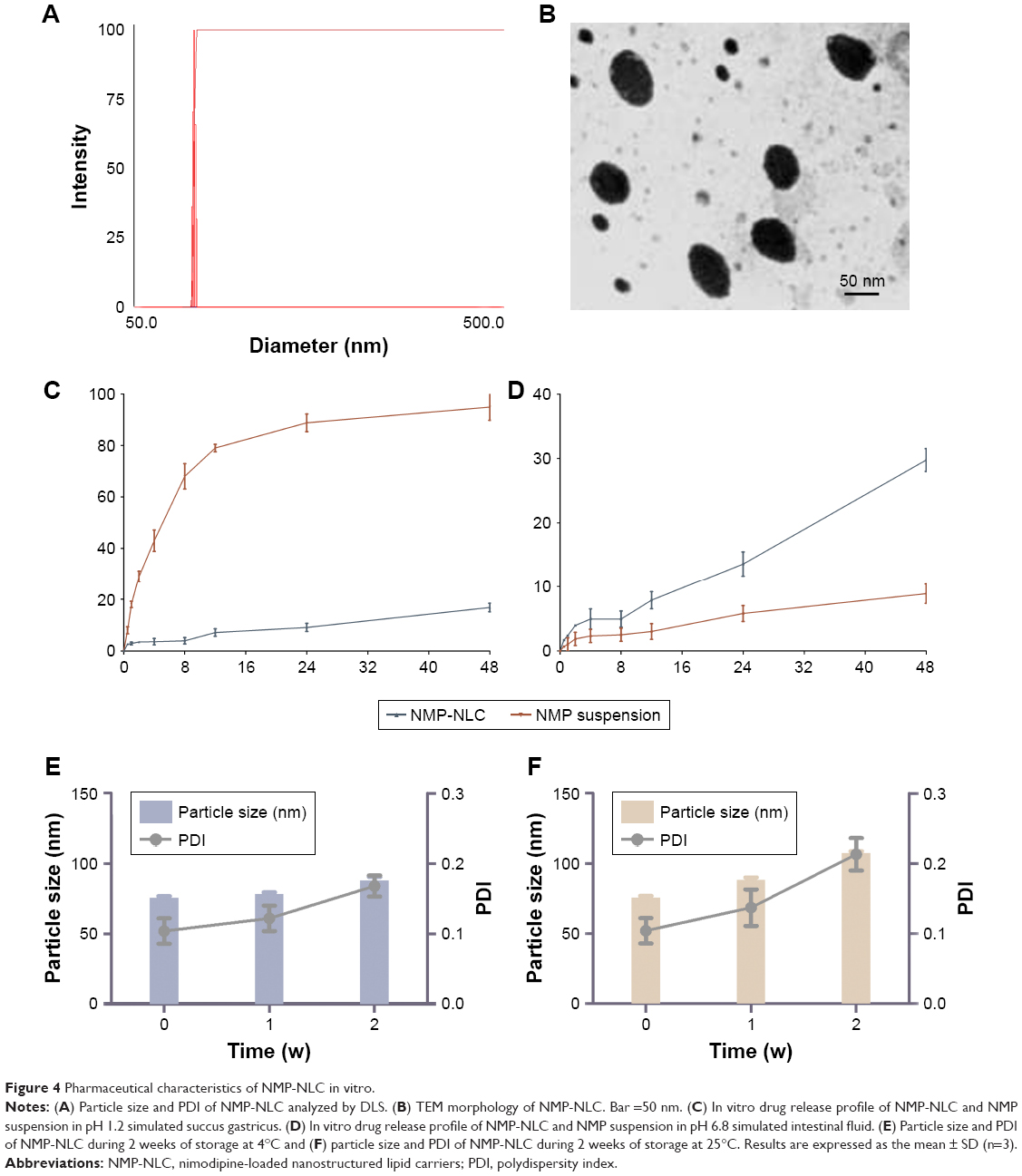 | Figure 4 Pharmaceutical characteristics of NMP-NLC in vitro. |
TEM
As shown in Figure 4B, the TEM image revealed that the particles were spherical in shape and monodispersed in diameter. Moreover, the mean size of NMP-NLC was 57.755±9.508 nm, which was smaller than that detected by DLS. It could be explained that the particle size measured by DLS was the hydrodynamic diameter, whereas TEM measured the true size of dried particles.35
In vitro release study
In this study, sink conditions are crucial in mimicking physiological environments.36 Thus, the saturated solubility of NMP in different media was measured. The saturation solubility in distilled water, 0.3% Tween 80 solution, 0.5% Tween 80 solution, 0.7% Tween 80 solution were 13.98, 74.45, 96.33, 132.56 μg/mL, respectively. Sink conditions were satisfied when the Tween 80 content was 0.5%, 0.7%, and 0.9%. However, given that the surfactant concentration could not be too high, a 0.5% Tween 80 solution was selected as the optimal surfactant concentration. A release study was performed in various media at different pH, which was designed to mimic the physiological pH gradient (pH 1.2 as succus gastricus and pH 6.8 as intestinal fluid). Comparing the release curves of NMP-NLC at pH 1.2 and pH 6.8, only 10% of the NMP was released in the first four hours, which indicated that the free drugs were attached on the surface of the particles. As presented in Figure 4C and D, 29.77% of NMP was released from NMP-NLC at pH 6.8 for 48 hours, which was higher than that released at pH 1.2 (20%). However, NMP suspensions in pH 1.2 tended to release 90% of NMP at 24 hours. The results suggested that the lipid carrier could encapsulate the NMP and protect NMP from gastric acid damage, followed by the delivery of NMP to the small intestinal. It could be absorbed by intestinal cells and transported to the circulation in vivo. On the other hand, the amorphous lattice of NMP in NMP-NLC could lead to a higher dissolution rate than the crystalline lattice under specific conditions.37
Stability study
Physical stability is crucial during the exploration of lipid carriers, as it is necessary to ensure that the nanoparticles were sterile and had a steady particle size. Herein, a short-term stability test was carried out to evaluate the physical stability of NMP-NLC at 4°C and 25°C. As shown in Figure 4E, during the 2 weeks, the particle size of NMP-NLC at 4°C increased slightly from 75.6±1.2 nm to 88.1±2.4 nm and PDI increased from 0.104±0.018 to 0.168±0.015. Similarly, an increase in particle size from 75.6±1.2 nm to 107.3±1.4 nm and PDI from 0.104±0.018 to 0.213±0.023 of the NMP-NLC was observed at 25°C (Figure 4F). The particle size of NMP-NLC stored at 25°C continued to increase after 1 week, while no significant change was observed at 4°C. Hence, the NMP-NLC was stable at 4°C and 25°C for a total period of 2 weeks and was more suitable to be stored at 4°C.
Spectral analysis of NMP-NLC
DSC
DSC is a basic method of investigating the variation in phase transition temperature and energy to explore the lattice morphology of drugs in the mixed system.38 Thus, DSC was performed to detect the interactions between drugs and excipients. The DSC thermograms of NMP, physical mixtures, and lyophilized NMP-NLC powder are depicted in Figure 5A. The melting peak of NMP in the thermogram was about 124.9°C, which belonged to the crystalline nature of NMP. The peaks of the DSC curve of physical mixtures were at 50.9°C and 123.8°C. For the lyophilized NMP-NLC, there were two melting peaks at 43.0°C and 54.7°C, respectively, which belonged to the lipid mixture. This pattern could be explained by the melt-recrystallization during the heating process. Moreover, the peak of NMP at around 124.9°C disappeared in the spectrogram of the NMP-NLC, which indicated that the NMP was been entrapped into the NLCs and was presented as an amorphous state rather than a crystalline state. The results were consistent with the results of in vitro release experiments in which the drug achieved a reasonable release rate due to an amorphous state.
 | Figure 5 The crystal form of NMP-NLC. |
PXRD analysis
Independent components and physical mixtures in the PXRD pattern showed distinct peaks, which indicated different characteristic lattice forms. Herein, the diffraction pattern of free and encased NMP were detected by PXRD analysis. It was performed with NMP coarse powders, physical mixtures, and the lyophilized NMP-NLC powders. PXRD patterns are shown in Figure 5B. In the PXRD pattern of NMP coarse powders, remarkable intense and distinct diffraction peaks of NMP could be observed at two scattered angles of 6.556°, 12.368°, 12.912°, 17.387°, and 20.335°. The patterns of the physical mixtures showed decreased peak intensities of pure NMP coarse powder, which indicated that the NMP in physical mixtures was still in a crystalline state, evidently the presence of the lipids affected the peak intensities. However, there were two different diffraction peaks (5.524° and 22.951°) in the pattern of NMP-NLC compared with pure NMP, which was considered as the characteristic peaks of Precirol ATO 5. Furthermore, no sharp peaks in the NMP lattice were noticed in the NMP-NLC powder, suggesting that NMP in the NMP-NLC was in an unformed state rather than a crystal structure. These results were consistent with the DSC analysis.
FT-IR
FT-IR has been adopted to evaluate the molecular interactions between NMP and the lipid matrix. In the FT-IR spectrum (Figure 6), the characteristic bands of NMP including 3,296.9 cm−1, 1,309.5 cm−1 belong to the stretching vibration peak of N-H. The wavenumbers at 3,097.6 cm−1, 1,621.6 cm−1, 1,495.9 cm−1, and 809.4 cm−1 represent a benzene ring. The ester group bands were at 1,695.3 cm−1, 1,270.7 cm−1, and 1,022.6 cm−1, -NO2 was at 1,523.3 cm−1 and 1,346.2 cm−1, C-O-C was at 1,098.9 cm−1, C-H was at 2,982.3 cm−1, 2,933.9 cm−1, 1,454.9 cm−1, and 1,381.7 cm−1. These results were greatly in accordance with the Chinese Pharmacopoeia 2015 edition and previous studies.39 For the FT-IR data of physical mixtures and NMP-NLC, the new characteristic absorption bands of 3,461.4 cm−1, 3,306.9 cm−1, 2,916.1 cm−1, 2,849.8 cm−1, 1,737.9 cm−1, and 1,528.6 cm−1 were considered as the lipid matrix. However, the intensity of the characteristic bands of NMP were observably decreased in the physical mixture spectrum, and for the NMP-NLC, the peaks even disappeared, which suggested that there were no chemical interactions between the NMP and lipids. The entrapment of NMP in NLCs depended only on the dissolution between drugs and lipids. All the results in this experiment were consistent with a previous study.40
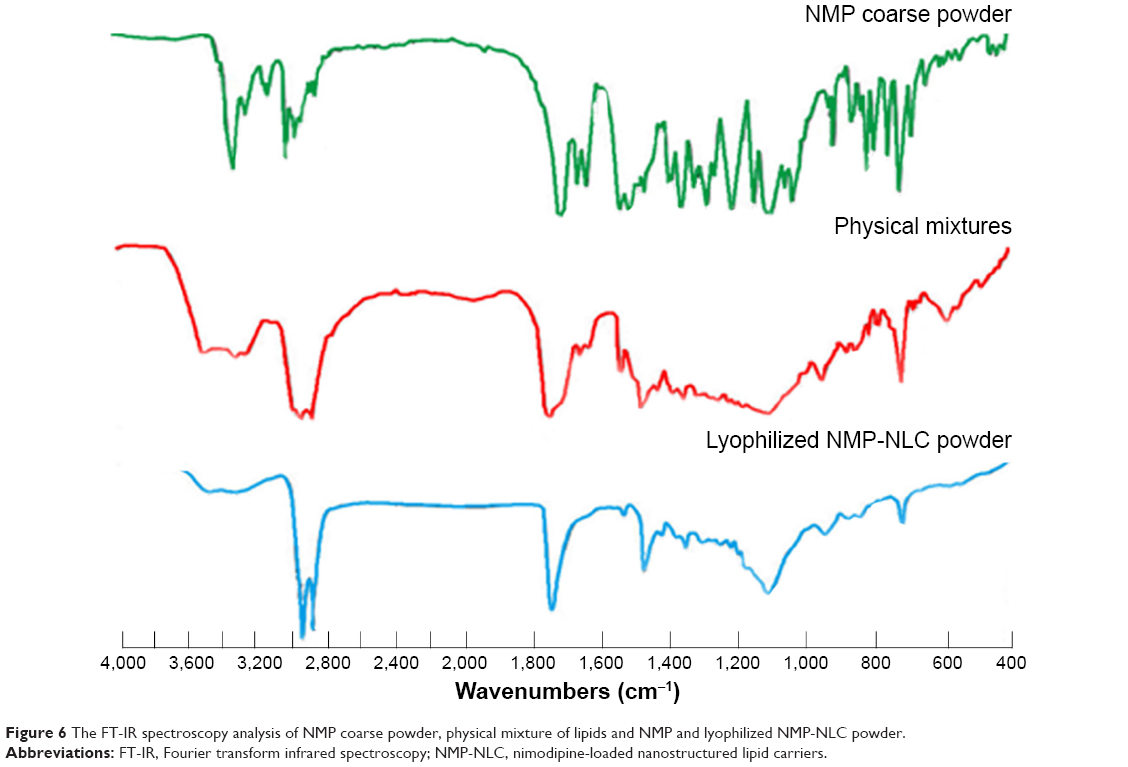 | Figure 6 The FT-IR spectroscopy analysis of NMP coarse powder, physical mixture of lipids and NMP and lyophilized NMP-NLC powder. |
In situ SPIP study
Previous studies have shown that NMP was stable in the perfusates before the perfusion.45 In general, the gravimetric method and phenol red labeling method were used to indicate volume changes. The present study adopted the gravimetric method to calibrate the volume changes of the perfusates. Because phenol red would be partially absorbed in the intestine, which could interfere with the original degree of poorly soluble drugs absorbed or transported in the intestinal perfusion of rats.41 The absorption rate constant (Ka) and apparent effective permeability coefficient (Papp) obtained in the SPIP study are illustrated in Figure 7. It was demonstrated that the order of Ka for NMP-NLC was small intestine > colon (P<0.05), and Papp value of NMP-NLC in the small intestine was obviously higher than that in the colon (P<0.05). Furthermore, the Ka of NMP-NLC in the small intestine increased from (2.99±0.08) ×10−3 min−1 to (5.98±0.20) ×10−3 min−1 (twofold higher), as compared to the NMP solution. The Papp of NMP-NLC in the small intestine was also increased by twofold when compared to the NMP solution (Table 3). This indicated intuitively that NMP-NLC could be mainly absorbed in the small intestine. The increased absorption of NMP-NLC in rats could be explained by some possible reasons. First, the nanoscale particle size created a sharp increase of drug surface area, which significantly increased the apparent solubility and dissolution rate of NMP from NMP-NLC. Second, the lipid carrier was mainly digested with lipase to form micelles or vesicles after oral administration. These newly formed drug carriers were easily absorbed by the small intestine through the static water layer.42 The lipid component in the carrier had a certain similarity with the composition of the intestinal epithelial cell membrane, which could increase the affinity with the cell membrane and facilitate the absorption or transport of the drug. Furthermore, it has been reported that p-glycoprotein (p-gp) was widely expressed in cells such as intestinal epithelial cells, hepatocytes, and renal tubular epithelial cells. However, the Tween 80 in the NMP-NLC formulation was a p-glycoprotein inhibitor that could reduce p-gp activity and reduce the efflux of the drug, acted as an absorption enhancer. Finally, it could also be related to the physiological structure of the small intestine. The wrinkling and fluff of the small intestine greatly increased the surface area of the intestinal lumen, which provided a possibility for the effective absorption of the drug, and was beneficial to the absorption of the drug by the intestinal mucosa.
 | Figure 7 The in situ absorption of NMP-NLC in rat intestinal segments compared with NMP solution. |
 | Table 3 Absorption parameters of NMP solution and NMP-NLC in different intestinal segments |
In vivo pharmacokinetic study
The plasma concentration–time profiles after a single intragastric administration of NMP-NLC and NMP suspension are shown in Figure 8. Twenty-four hours after the oral administration, the NMP plasma concentration from the NMP-NLC group was still 0.89 μg/mL, whereas the drug was even undetected in the NMP suspension group. Table 4 presents the mean parameters calculated by non-compartmental analysis. It was found that NMP-NLC showed higher Cmax (32.516±3.151 μg/mL vs 11.887±3.464 μg/mL, P<0.05) and faster Tmax (0.250±0.500 h vs 3.000±0.540 h, P<0.01). The data indicated the higher absorption of NMP when loaded in the NLCs. The AUC0–∞ after oral administration of NMP-NLC was 73.546±8.933 μg/mL*h, which was roughly 1.6-fold higher than that of NMP suspension 45.691±7.667 μg/mL*h. Thus, the relative bioavailability of NMP-NLC was 160.96% compared with the NMP suspension. It was demonstrated that the systemic absorption of NMP could be significantly enhanced by NMP-NLC. The NLCs showed a great possibility for the oral bioavailability enhancement of poorly water-soluble drugs. It could be explained by the enhancement of oral bioavailability of NMP-NLC by various factors. First, on the basis of the Noyes–Whitney equation, the rate of drug dissolution is positively related to the surface area. The particle size of the NMP-NLCs were <200 nm, which could hold a huge specific surface area to incur faster dissolution of the drug.31 Moreover, smaller particles are also benefit the ability of mucosal attachment on gastrointestinal surfaces, which ensure enough retention time to promote oral bioavailability. Second, the lipids in the NLCs could be enzymatically degraded in the digestive tract to form emulsifiers such as monoacylglycerols and fatty acids, and enter into the duodenum. Meanwhile, bile salts, cholesterol, and pancreatic juice were stimulated and discharged from the gallbladder to the intestinal cavity. The NMP-NLC mixed with bile salt to form micelles and promoted the transport to the lymphatic circulation, and thus protected it from the liver’s first pass effect.43,44 Alternatively, the oral administration of nanoparticles were absorbed in Peyer’s patch by granular cells. After the nanoparticles were phagocytosed, they would be transported to the M cell basal cavity by vesicle transport. The nanoparticles were in an unbound state or in a state of being phagocytosed by macrophages and transported from the lymphatic circulation into the blood circulation. This process also avoided the first pass effect of the liver. These results were in accordance with the former research that both showed enough absorption of nanoparticles and the lymphatic transport pathway facilitated the enhancement of oral bioavailability.32 Moreover, the results in the intestinal perfusion study indicated that Tween 80 could inhibit drugs pumped out by p-gp efflux as a p-gp inhibitor, which could result in the improvement of Cmax and AUC0–∞. In addition, it has also been reported that NLCs could improve the fluidity of cell membranes to increase intracellular transport or to open the tight junctions on the intestinal mucosal epithelial cells to promote intercellular transfer of ionized drugs and hydrophilic macromolecules.
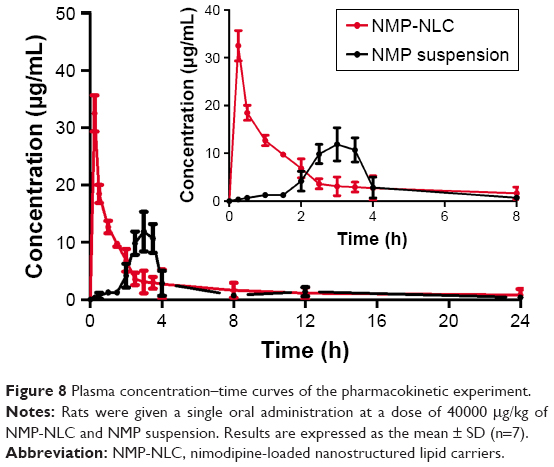 | Figure 8 Plasma concentration–time curves of the pharmacokinetic experiment. |
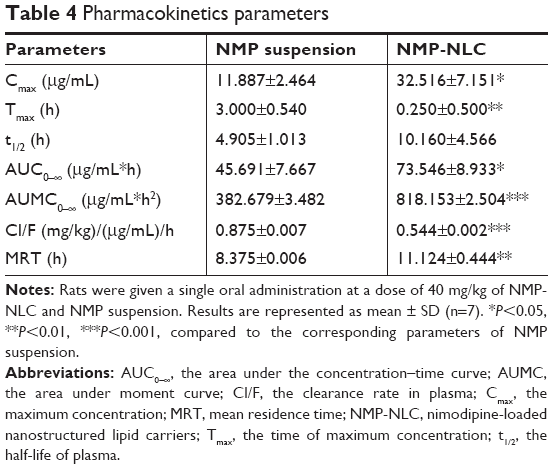 | Table 4 Pharmacokinetics parameters |
Conclusion
In summary, we developed NLCs to entrap clinical drug NMP. It was prepared by a high-pressure homogenization method. The obtained NMP-NLC showed nanoscale, evenly distributed and excellent entrapment ability. Moreover, in vitro release research revealed that NMP-NLC showed an extended release capacity of NMP without a distinct initial burst release consequence. The in vivo study of NMP-NLC indicated that the small intestine was the main absorption site and possessed a markedly improved oral bioavailability in rats. Overall, the NLCs exhibited a tremendous application prospect for the oral delivery of low water dissolved drugs.
Acknowledgments
The authors gratefully acknowledge the financial support from the National Natural Science Foundation of China (No 81573379 and 81501582), Natural Science Foundation of Jiangsu Province (No BK20171390), and National Key Research and Development Program (2017YFD0501403). This study was also supported by the Project of State Key Laboratory of Natural Medicines, China Pharmaceutical University (No SKLNMZZCX201816), the National Science and Technology Major Project (2017ZX09101001), the Postgraduate Research & Practice Innovation Program of Jiangsu Province (KYCX18_0762), and the financial support from Development Funds for Priority Academic Programs in Jiangsu Higher Education Institutions. The authors thank the Public Platform of State Key Laboratory of Natural Medicines for assistance with HPLC.
Disclosure
The authors report no conflicts of interest in this work.
References
Zhao Y, Xin T, Ye T, Yang X, Pan W. Solid dispersion in the development of a nimodipine delayed-release tablet formulation. Asian J Pharm Sci. 2014;9(1):35–41. | ||
Fu Q, Sun J, Ai X, et al. Nimodipine nanocrystals for oral bioavailability improvement: role of mesenteric lymph transport in the oral absorption. Int J Pharm. 2013;448(1):290–297. | ||
Moreno LC, da Silva Oliveira GZ, Cavalcanti IM, Santos-Magalhães NS, Rolim HM, de Freitas RM. Development and evaluation of liposomal formulation containing nimodipine on anxiolytic activity in mice. Pharmacol Biochem Behav. 2014;116:64–68. | ||
Hu FQ, Zhang Y, du YZ, Yuan H, Fq H, Yz D. Nimodipine loaded lipid nanospheres prepared by solvent diffusion method in a drug saturated aqueous system. Int J Pharm. 2008;348(1–2):146–152. | ||
Guan T, Miao Y, Xu L, et al. Injectable nimodipine-loaded nanoliposomes: preparation, lyophilization and characteristics. Int J Pharm. 2011;410(1–2):180–187. | ||
Garg NK, Tyagi RK, Singh B, et al. Nanostructured lipid carrier mediates effective delivery of methotrexate to induce apoptosis of rheumatoid arthritis via NF-κB and FOXO1. Int J Pharm. 2016;499(1–2):301–320. | ||
Guada M, Lasa-Saracíbar B, Lana H, Dios-Viéitez MC, Blanco-Prieto MJ. Lipid nanoparticles enhance the absorption of cyclosporine A through the gastrointestinal barrier: In vitro and in vivo studies. Int J Pharm. 2016;500(1–2):154–161. | ||
Beloqui A, Solinís MÁ, Rodríguez-Gascón A, Almeida AJ, Préat V. Nanostructured lipid carriers: Promising drug delivery systems for future clinics. Nanomedicine. 2016;12(1):143–161. | ||
Tapeinos C, Battaglini M, Ciofani G. Advances in the design of solid lipid nanoparticles and nanostructured lipid carriers for targeting brain diseases. J Control Release. 2017;264:306–332. | ||
Sánchez-López E, Espina M, Doktorovova S, Souto EB, García ML. Lipid nanoparticles (SLN, NLC): Overcoming the anatomical and physiological barriers of the eye – Part I – Barriers and determining factors in ocular delivery. Eur J Pharm Biopharm. 2017;110:70–75. | ||
Park SJ, Garcia CV, Shin GH, Kim JT. Improvement of curcuminoid bioaccessibility from turmeric by a nanostructured lipid carrier system. Food Chem. 2018;251:51–57. | ||
Ali Khan A, Mudassir J, Mohtar N, Darwis Y. Advanced drug delivery to the lymphatic system: lipid-based nanoformulations. Int J Nanomedicine. 2013;8:2733–2744. | ||
Chalikwar SS, Belgamwar VS, Talele VR, Surana SJ, Patil MU. Formulation and evaluation of Nimodipine-loaded solid lipid nanoparticles delivered via lymphatic transport system. Colloids Surf B Biointerfaces. 2012;97:109–116. | ||
Beg S, Sharma G, Thanki K, Jain S, Katare OP, Singh B. Positively charged self-nanoemulsifying oily formulations of olmesartan medoxomil: Systematic development, in vitro, ex vivo and in vivo evaluation. Int J Pharm. 2015;493(1–2):466–482. | ||
Kaithwas V, Dora CP, Kushwah V, Jain S. Nanostructured lipid carriers of olmesartan medoxomil with enhanced oral bioavailability. Colloids Surf B Biointerfaces. 2017;154:10–20. | ||
Liu K, Sun J, Wang Y, He Y, Gao K, He Z. Preparation and characterization of 10-hydroxycamptothecin loaded nanostructured lipid carriers. Drug Dev Ind Pharm. 2008;34(5):465–471. | ||
Esposito E, Drechsler M, Cortesi R, Nastruzzi C. Encapsulation of cannabinoid drugs in nanostructured lipid carriers. Eur J Pharm Biopharm. 2016;102:87–91. | ||
Zhuang CY, Li N, Wang M, et al. Preparation and characterization of vinpocetine loaded nanostructured lipid carriers (NLC) for improved oral bioavailability. Int J Pharm. 2010;394(1–2):179–185. | ||
Wolf M, Klang V, Halper M, et al. Monoacyl-phospatidylcholine nanostructured lipid carriers: Influence of lipid and surfactant content on in vitro skin permeation of flufenamic acid and fluconazole. J Drug Deliv Sci Technol. 2017;41:419–430. | ||
Okonogi S, Riangjanapatee P. Physicochemical characterization of lycopene-loaded nanostructured lipid carrier formulations for topical administration. Int J Pharm. 2015;478(2):726–735. | ||
Yostawonkul J, Surassmo S, Iempridee T, et al. Surface modification of nanostructure lipid carrier (NLC) by oleoyl-quaternized-chitosan as a mucoadhesive nanocarrier. Colloids Surf B Biointerfaces. 2017;149:301–311. | ||
Liu X, Wang S, Chai L, et al. A two-step strategy to design high bioavailable controlled-release nimodipine tablets: the push-pull osmotic pump in combination with the micronization/solid dispersion techniques. Int J Pharm. 2014;461(1–2):529–539. | ||
Nnamani PO, Hansen S, Windbergs M, Lehr CM. Development of artemether-loaded nanostructured lipid carrier (NLC) formulation for topical application. Int J Pharm. 2014;477(1–2):208–217. | ||
Kumbhar DD, Pokharkar VB. Engineering of a nanostructured lipid carrier for the poorly water-soluble drug, bicalutamide: Physicochemical investigations. Colloids Surf A Physicochem Eng Asp. 2013;416(1):32–42. | ||
He J, Han Y, Xu G, et al. Preparation and evaluation of celecoxib nanosuspensions for bioavailability enhancement. RSC Adv. 2017;7(22):13053–13064. | ||
Luo Y, Chen D, Ren L, Zhao X, Qin J. Solid lipid nanoparticles for enhancing vinpocetine’s oral bioavailability. J Control Release. 2006;114(1):53–59. | ||
Singh B, Singh R, Bandyopadhyay S, Kapil R, Garg B. Optimized nanoemulsifying systems with enhanced bioavailability of carvedilol. Colloids Surf B Biointerfaces. 2013;101(1):465–474. | ||
Moreno L, Rolim HML, Freitas RM, Santos-Magalhães NS. Antidepressant-like activity of liposomal formulation containing nimodipine treatment in the tail suspension test, forced swim test and MAOB activity in mice. Brain Res. 2016;1646:235–240. | ||
Pokharkar V, Patil-Gadhe A, Kaur G. Physicochemical and pharmacokinetic evaluation of rosuvastatin loaded nanostructured lipid carriers: influence of long- and medium-chain fatty acid mixture. J Pharm Invest. 2018;48(4):465–476. | ||
Sun C, Wang J, Liu J, Qiu L, Zhang W, Zhang L. Liquid proliposomes of nimodipine drug delivery system: preparation, characterization, and pharmacokinetics. AAPS Pharm Sci Tech. 2013;14(1):332–338. | ||
Zhang H, Chen W, Zhao Z, et al. Lyophilized Nanosuspensions for Oral Bioavailability Improvement of Insoluble Drugs: Preparation, Characterization, and Pharmacokinetic Studies. J Pharm Innov. 2017;12(3):271–280. | ||
Fathi HA, Allam A, Elsabahy M, Fetih G, El-Badry M. Nanostructured lipid carriers for improved oral delivery and prolonged antihyperlipidemic effect of simvastatin. Colloids Surf B Biointerfaces. 2018;162:236–245. | ||
Garg NK, Sharma G, Singh B, et al. Quality by Design (QbD)-enabled development of aceclofenac loaded-nano structured lipid carriers (NLCs): An improved dermatokinetic profile for inflammatory disorder(s). Int J Pharm. 2017;517(1–2):413–431. | ||
Jacobs C, Kayser O, Müller RH. Nanosuspensions as a new approach for the formulation for the poorly soluble drug tarazepide. Int J Pharm. 2000;196(2):161–164. | ||
Neves AR, Queiroz JF, Reis S. Brain-targeted delivery of resveratrol using solid lipid nanoparticles functionalized with apolipoprotein E. J Nanobiotechnol. 2016;14:27. | ||
Flanner H, Moore JW. Mathematical comparison of curves with an emphasis on dissolution profiles. Pharm Technol. 1996;20(6):64–74. | ||
Fang G, Tang B, Chao Y, et al. Cysteine-Functionalized Nanostructured Lipid Carriers for Oral Delivery of Docetaxel: A Permeability and Pharmacokinetic Study. Mol Pharm. 2015;12(7):2384–2395. | ||
Cavalli R, Caputo O, Carlotti ME, Trotta M, Scarnecchia C, Gasco MR. Sterilization and freeze-drying of drug-free and drug-loaded solid lipid nanoparticles. Int J Pharm. 1997;148(1):47–54. | ||
Lee HJ, Kim JY, Park SH, Rhee YS, Park CW, Park ES. Controlled-release oral dosage forms containing nimodipine solid dispersion and hydrophilic carriers. J Drug Deliv Sci Technol. 2017;37:28–37. | ||
Seyed Yagoubi A, Shahidi F, Mohebbi M, Varidi M, Golmohammadzadeh S. Preparation, characterization and evaluation of physicochemical properties of phycocyanin-loaded solid lipid nanoparticles and nanostructured lipid carriers. J Food Meas Characterization. 2018;12(1):378–385. | ||
Zhang Z, Bu H, Gao Z, Huang Y, Gao F, Li Y. The characteristics and mechanism of simvastatin loaded lipid nanoparticles to increase oral bioavailability in rats. Int J Pharm. 2010;394(1–2):147–153. | ||
Deng C, Jia M, Wei G, et al. Inducing Optimal Antitumor Immune Response through Coadministering iRGD with Pirarubicin Loaded Nanostructured Lipid Carriers for Breast Cancer Therapy. Mol Pharm. 2017;14(1):296–309. | ||
Courant T, Bayon E, Reynaud-Dougier HL, et al. Tailoring nanostructured lipid carriers for the delivery of protein antigens: Physicochemical properties versus immunogenicity studies. Biomaterials. 2017;136:29–42. | ||
Yang S, Zhu J, Lu Y, Liang B, Yang C. Body distribution of camptothecin solid lipid nanoparticles after oral administration. Pharm Res. 1999;16(5):751–757. | ||
Li J, Fu Q, Liu X, Li M, Wang Y. Formulation of nimodipine nanocrystals for oral administration. Arch Pharm Res. 2016;39(2):202–212. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.