")
Back to Journals » International Journal of Nanomedicine » Volume 10 » Supplement 1 Challenges in biomaterials research
Preparation and characterization of cefditoren pivoxil-loaded liposomes for controlled in vitro and in vivo drug release
Authors Venugopala G, Lakshmipathy R, Sarada NC
Received 14 January 2015
Accepted for publication 18 March 2015
Published 1 October 2015 Volume 2015:10(Supplement 1 Challenges in biomaterials research) Pages 149—157
DOI https://doi.org/10.2147/IJN.S79994
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr Thomas Webster
Gojjala Venugopalarao,1 Rajasekhar Lakshmipathy,2 Nallani Chakravarthula Sarada1
1Environmental and Analytical Chemistry Division, School of Advanced Sciences, VIT University, Vellore, 2Centre for Material Science, KCG College of Technology, Chennai, India
Background: The application of antibiotics has been limited due to weak biodistribution and pharmacokinetics. Encapsulation of these drugs in lipid vesicles might be a good solution for obtaining the required properties. Liposomes are one of the most suitable drug-delivery systems to deliver the drug to the target organ and minimize the distribution of the drug to non-target tissues.
Objective: The study reported here aimed to develop cefditoren pivoxil liposomes by thin-film hydration, characterize them in terms of physical interactions, and undertake in vitro and in vivo release studies.
Methodology: The pre-formulation studies were carried out using Fourier-transform infrared spectroscopy and differential scanning calorimetry. Cefditoren pivoxil liposomal formulations were formulated by thin-film hydration using biomaterials ie, soya lecithin and cholesterol in different molar ratios. The best molar ratio was determined by in vitro studies such as entrapment efficacy, particle size distribution, and diffusion.
Results: From the in vitro release studies, it was found that the formulation that contained soya lecithin and cholesterol in a 1.0:0.6 molar ratio gave good entrapment of 72.33% and drug release of 92.5% at 36 hours. Further, the formulation’s zeta potential and surface morphology were examined and stability and in vivo studies were undertaken evaluating the pharmacokinetic parameters, which showed promising results.
Conclusion: Formulation CPL VI showed the maximum drug-loading capacity of 72.3% with good controlled release and acceptable stability when compared with the other formulations. In vivo studies in rabbits showed that the drug release from the liposomes was successfully retarded with good controlled release behavior which can be used to treat many bacterial infections with a minimal dose.
Keywords: biomaterials, liposomes, drug-delivery system, soya lecithin, cholesterol
Introduction
Liposomes are microscopic vesicles consisting of phospholipid bilayers that enclose aqueous compartments and are utilized as drug-delivery systems for both hydrophilic and lipophilic drugs.1 Liposomes are one of the most suitable drug-delivery systems to deliver a drug to the target organ and minimize the distribution of the drug to non-target tissues. The importance of liposomes as a drug-delivery vehicle is now becoming more established, and they deliver drugs in a controlled manner when compared with conventional dosage forms. These findings make it increasingly necessary to develop liposomes to satisfy pharmaceutical considerations.2–4
Cefditoren pivoxil (CP) is a semisynthetic, third-generation cephalosporin exhibiting bactericidal action by inhibiting cell-wall synthesis.5 CP is a prodrug which can be hydrolyzed by esterase during absorption to cefditoren as an active drug and is distributed in the blood. Cefditoren is used for the treatment of uncomplicated skin and skin structure infections.6,7 CP has a broad spectrum of activity against Gram-negative and Gram-positive bacterial infections, including strains of Staphylococcus pyogenes, Haemophilus influenza, Klebsiella pneumonia, and Staphylococcus aureus. CP is the most frequently used drug for the treatment of tonsillitis, pharyngitis, and acute exacerbations of chronic bronchitis.8–10
The application of some antibiotics in health care has been limited because of their toxicity or weak biodistribution and pharmacokinetics. Encapsulation of these drugs in lipid vesicles is a good solution for producing the required pharmacokinetic and pharmacodynamic properties and also to reduce their side effects.11,12
In the study reported here, we developed CP liposomes by the technique of thin-film hydration and analyzed the prepared liposomes in terms of physical interactions, entrapment efficacy, and particle size. We also undertook scanning electron microscopy (SEM), in vitro, and in vivo release studies of the prepared liposomes.
Materials and methods
Materials
The CP used in the study was a generous gift from Hetero Drugs (Hyderabad, India); phosphatidyl choline was a gift from Lipoid GmbH (Ludwigshafen am Rhein, Germany); while cholesterol, ethanol, methanol, chloroform, and all other solvents of analytical reagent grade were purchased from SD Fine-Chem Limited (Mumbai, India).
Methodology
Fourier-transform infrared spectroscopic studies
Fourier-transform infrared spectra of the pure drug, mixture of drugs, and polymers were recorded on a Thermo Nicolet™ 330 spectrometer (Thermo Electron Corporation, Madison, WI, USA) using a thin film supported on KBr pellets to check the compatibility between the drug and the polymers. Fourier-transform infrared spectra were recorded for the pure drug, a 6-month old physical mixture of the drug, and the ingredients. All the samples were scanned in the region of 4,000–400 cm−1.
Differential scanning calorimetry studies
Differential scanning calorimetry (DSC) studies were carried out to check compatibility between the drug, cholesterol, and the polymers. The studies were carried out on a Netzsch DSC 204 Thermal Analyzer (Netzsch-Gerätebau GmbH, Selb, Germany). Calorimetric measurements were made with an empty cell (high-purity alpha alumina disc) as a reference. The instrument was calibrated using high-purity indium metal as a standard. The dynamic scans were taken in a nitrogen atmosphere at a heating rate of 10°C/minute.
Preparation of CP liposomes
CP liposomal formulations of different molar ratios were prepared using the technique of thin-film hydration. The lipid components (soya lecithin and cholesterol either alone or mixed) in different molar ratios were dissolved in a chloroform:methanol mixture (2:1, v/v) in a round-bottomed flask. Then 20 mg of CP, dissolved in 5 mL of the same solvent mixture, was added to the lipid solution. The organic solvents were slowly removed under reduced pressure using a rotary evaporator at 45°C such that a very thin film of dry lipids formed on the inner surface of the flask. The dry lipid film was slowly hydrated with 10 mL of phosphate-buffered saline (PBS; pH 7.4). The resulting suspension was shaken mechanically for 1 hour at room temperature, which led to the formation of liposomes. The liposomal dispersion was left to mature overnight at 4°C to ensure full lipid hydration. All these steps were performed under aseptic conditions.
The CP liposomes were separated from the un-entrapped drug by centrifugation at 20,000 rpm for 30 minutes at −2°C using cooling ultracentrifuge. The cake thus formed was washed twice with 10 mL PBS and re-centrifuged again for 1 hour. All the formulations were replicated thrice and the best formulations achieved are reported in this paper.
SEM studies
SEM has been extensively employed to study the morphology and surface topography of liposomes. The morphology and surface topography of the selected liposomes were, therefore, examined by SEM (EVO® MA 15; Carl-Zeiss AG, Jena, Germany). The samples to be examined were mounted on the SEM sample stub using double-sided sticky tape. The samples mounted were coated with gold (200 A°) under reduced pressure (0.001 Torr) for 5 minutes. The gold-coated samples were observed under the scanning electron microscope at suitable magnifications.
Entrapment efficiency
The entrapped drug concentrations were determined after the lysis of prepared liposomes with absolute alcohol and sonication for 5 minutes. The concentration of CP in absolute alcohol was determined spectrophotometrically at 231 nm. The entrapment efficiency, expressed as entrapment percentage, was calculated by the following relationship13:
|

Different factors influencing the encapsulation of CP into the multilamellar liposomes prepared by thin-film hydration (such as cholesterol molar ratio, effect of hydration media, and effect of a surfactant) were studied to find out the conditions for optimal drug entrapment.
Particle size analysis and zeta potential
The mean particle size of the liposomes was determined by light scattering based on laser diffraction using a laser-diffraction particle-size analyzer (Malvern Mastersizer NS500; Malvern Instruments Ltd, Malvern, UK).
In vitro diffusion studies
The release of CP from all multilamellar liposomal formulations of different compositions was determined using the membrane-diffusion technique. In brief, an accurately measured amount of CP solution or CP liposomal formulation, equivalent to 20 mg of CP, was suspended in 1 mL of PBS (pH 7.4) in a glass cylinder 20 cm long with a diameter of 2.5 cm. This cylinder was fitted, before addition of the liposomal suspension, with a presoaked membrane (Spectra/Por® Membrane) and was placed in a beaker (200 mL) containing 50 mL of PBS (pH 7.4). The whole setup was placed on a magnetic stirrer adjusted to a constant speed (150 rpm). Samples were collected every hour over a period of 48 hours and assayed spectrophotometrically for drug content at 231 nm.The mean values of three runs were taken as the results, and the obtained results were subjected to kinetic treatment to determine the order of release pattern and release rate constant.
Stability test
For all pharmaceutical dosage formulations, it is important to determine their stability. Stability studies were carried out for the most satisfactory formulation, per the International Conference on Harmonization guidelines. All the prepared formulations were sealed separately in an aluminum package and the formulations was assessed individually at two different temperature conditions ie, 4°C±2°C and 30°C±2°C for 3 months by using stability chambers.14
Lyophilization
Glucose dissolved in 7.4 PBS was used as cryoprotectant, along with the liposomal formulations and frozen overnight at −80°C, then later freeze-dried for 24 hours under vacuum at −50°C. The resulting lyophilized cakes were rehydrated to their original volume at room temperature with PBS and subjected to further investigation.15
In vivo studies
The objective of the in vivo studies was to determine the plasma-concentration profile of cefditoren. The experiments were approved by the Animal Ethical Committee at VIT University, Vellore, India (Committee for the Purpose of Control and Supervision of Experiments on Animals [CPCSEA] Reg No 1333/c/10CPCSEA, New Delhi, India). The study was carried out on New Zealand white rabbits. The formulation (CPL VI) was administered orally. Blood samples were collected from the marginal ear vein at regular intervals in heparinized centrifuge tubes and centrifuged immediately. The plasma separated was stored at −20°C until the time of analysis.16 The drug was estimated at 231 nm.
Extraction of plasma
As mentioned, the samples were centrifuged immediately, and the plasma separated was stored at −20°C until the time of analysis. The drug was extracted from the plasma. To 1 mL of plasma, 2 mL of acetonitrile was added in a stoppered test tube. This was vortexed for 5 minutes and centrifuged at 5,000 rpm for 10 minutes. The organic phase was collected separately then evaporated under nitrogen gas before being reconstituted with water. The drug concentration was determined using reversed-phase high-performance liquid chromatography (HPLC), as described following.
Preparation of mobile phase
The mobile phase was prepared by mixing 0.005 M ammonium acetate solution, acetonitrile, and triethylamine in the ratio 60.0:40.0:0.1 and the pH was adjusted to 3.0 with orthophosphoric acid. The solution was then filtered through a 0.45 μm membrane filter and degassed.
Preparation of standard stock solution
Five milligrams of the CP reference standard was weighed accurately and transferred to a 50 mL clean and dry volumetric flask. The volume was made up to the mark with the mobile phase after thorough shaking. Thus, a standard stock solution with concentration of 100 ppm of CP was prepared.
Preparation of standard solution
Standard stock solution (5 mL) was transferred to a 50 mL volumetric flask, and the volume was made up to the mark with the mobile phase and mixed well. This yielded a standard solution with a nominal concentration of 10 ppm of CP.
Analysis of sample
A Waters 2487 HPLC Peerless HT-C18 (1.8 μm, 10.0 cm × 4.6 mm, id) column was used as the stationary phase. The isocratic mobile phase, consisting of a mixture of 0.005 M ammonium acetate solution, acetonitrile, and triethyl amine in the ratio 60.0:40.0:0.1 the pH was adjusted to 3.0 with orthophosphoric acid and the resultant mixture was used throughout the analysis. The flow rate of the mobile phase was 1.0 mL/minute. The detector signal was monitored at a wavelength of 231 nm. The column temperature was kept ambient and the injection volume was 20 μL. The obtained chromatogram is shown in Figure 1.
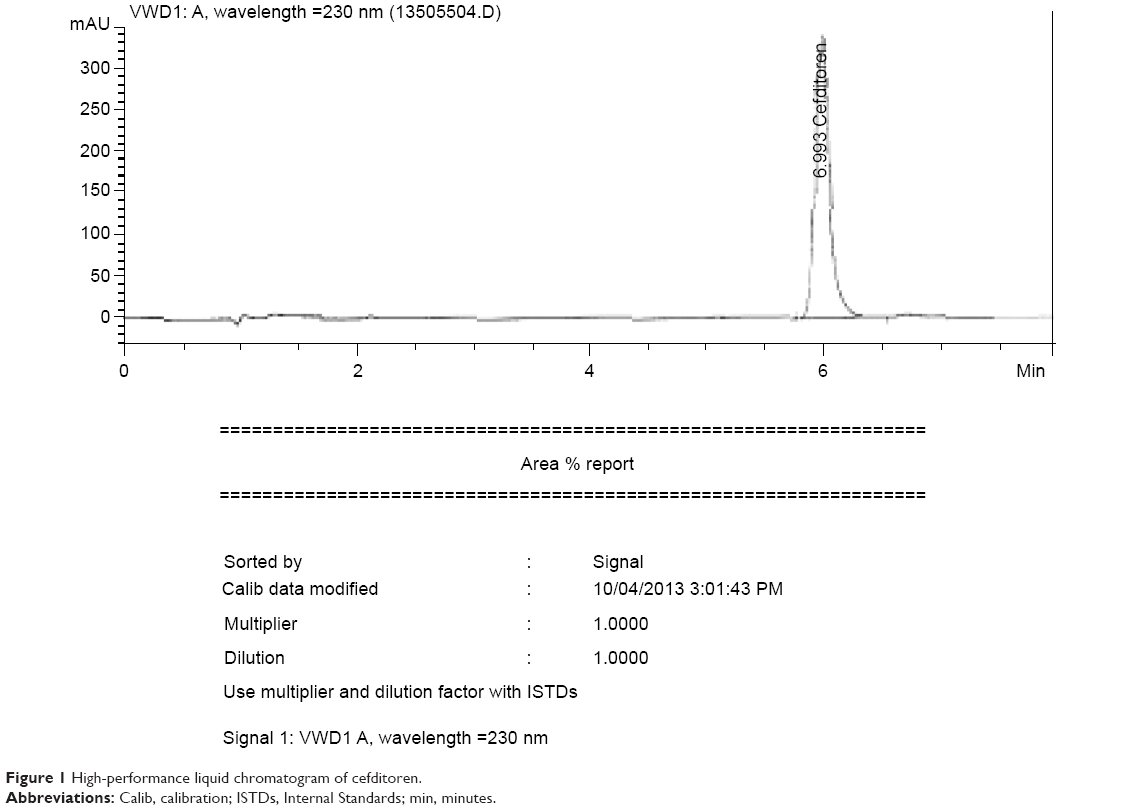 | Figure 1 High-performance liquid chromatogram of cefditoren. |
Results and discussion
Compatibility studies
Compatibility studies between the selected drug, cholesterol, and phosphatidylcholine were carried out for a period of 6 months using the infrared (IR) spectral-matching method. Characteristic IR peaks were identified for the drug, other ingredients, and the mixture, and observed for their appearance or disappearance from the original spectra. No significant changes in terms of peak shifting, or appearance or disappearance of peaks were noted with the drug, other ingredients, or mixture (Table 1). This confirms the absence of chemical interaction between the selected drug and the other ingredients.
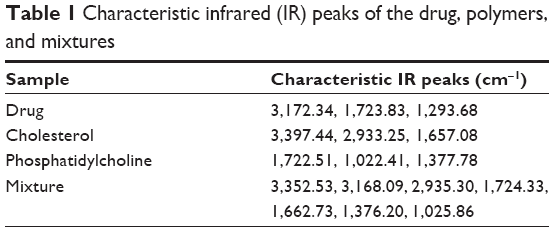 | Table 1 Characteristic infrared (IR) peaks of the drug, polymers, and mixtures |
DSC studies
The compatibility between the selected drug and the polymers was also confirmed by DSC pattern-matching. The DSC thermograms, given in Figure 2, reveal that there were no possible interactions between the selected drug and the polymers. The data disclose that the pure drug peak was at 209°C. The drug peak, when combined with cholesterol, showed at 202°C, and the drug peak was 194.8°C when combined with cholesterol phosphatidylcholine, which shows that there was a negligible shift in the peak and confirms that there were no interactions between the drug and the polymers.
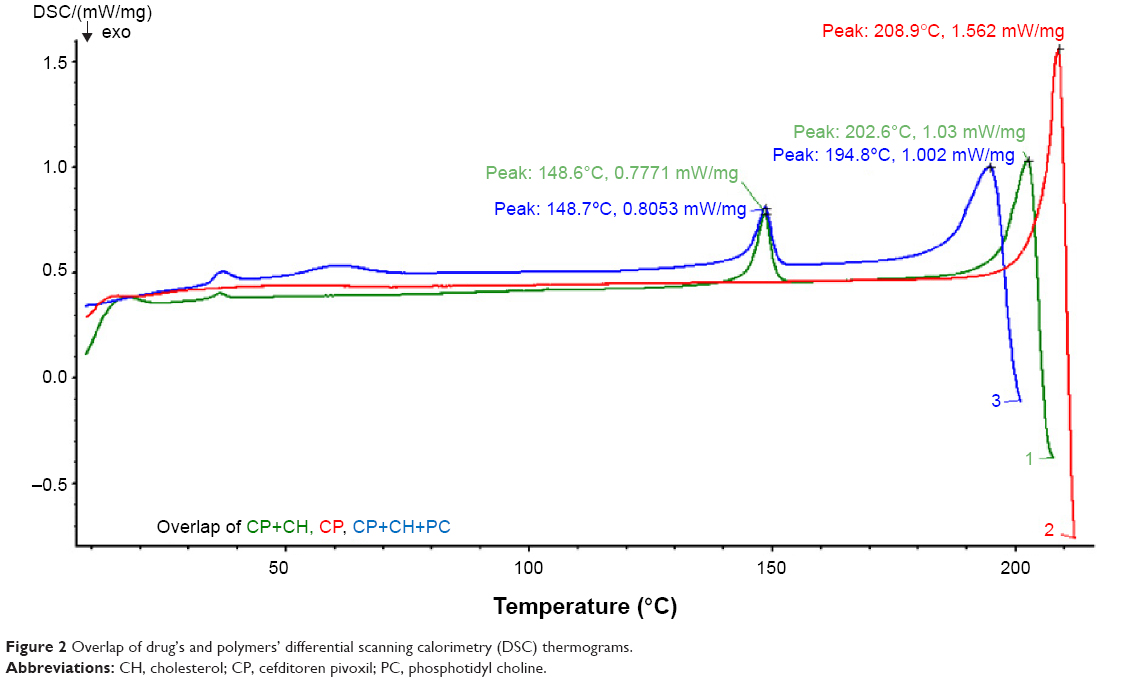 | Figure 2 Overlap of drug’s and polymers’ differential scanning calorimetry (DSC) thermograms. |
Drug entrapment efficiency
Table 2 shows that CP entrapment efficiency varied with lipid composition and cholesterol content. The results show that the percentage entrapment efficiency of CP increased by increasing the cholesterol content. The percentage entrapment efficiency of CP in multilamellar liposomes containing phosphatidylcholine alone was 10.85%, whereas the percentage was higher (72.33%) in liposomes prepared with phosphatidylcholine:cholesterol in the ratio 1.0:0.6. Further increase in entrapment efficiency was not recorded for liposomes composed of phosphatidylcholine:cholesterol in the ratio 1:1. When surfactants were added and the hydration medium changed to produce the final robust formulation (1.0:0.6 M of phosphatidylcholine:cholesterol), there was no significant increase in the entrapment efficiency of CP into the liposomes. Table 3 indicates that the change in hydration medium also improved the entrapment efficiency of CP into multilamellar liposomes.
 | Table 2 The effect of cholesterol on the encapsulation efficiency (EE) of cefditoren pivoxil (CP) into liposomes |
 | Table 3 The effect of hydration medium on the encapsulation efficiency (EE) of cefditoren pivoxil (CP) |
In vitro drug-release studies
The in vitro drug-release study results are depicted in Figure 3. Formulation CPL I showed maximum release of the drug (72.80%) at 12 hours, formulation CPL II showed maximum release of the drug (78.90%) at 12 hours, formulation CPL III showed maximum release of the drug (81.20%) at 18 hours, formulation CPL IV showed maximum release of the drug (81.45%) at 24 hours, formulation CPL V showed maximum release of the drug (86.82%) at 24 hours, formulation CPL VI showed maximum release of the drug (93.58%) at 36 hours, formulation CPL VII showed maximum release of the drug (88.72%) at 36 hours, formulation CPL VIII showed maximum release of the drug (81.28%) at 36 hours, formulation CPL IX showed maximum release of the drug (74.72%) at 36 hours, and formulation CPL X showed maximum release of the drug (69.20%) at 36 hours. Formulation CPL VI showed maximum controlled release of the drug (93.58%) at 36 hours, when compared with all other formulations. Thus, formulation CPL VI was considered optimal and used for further studies.
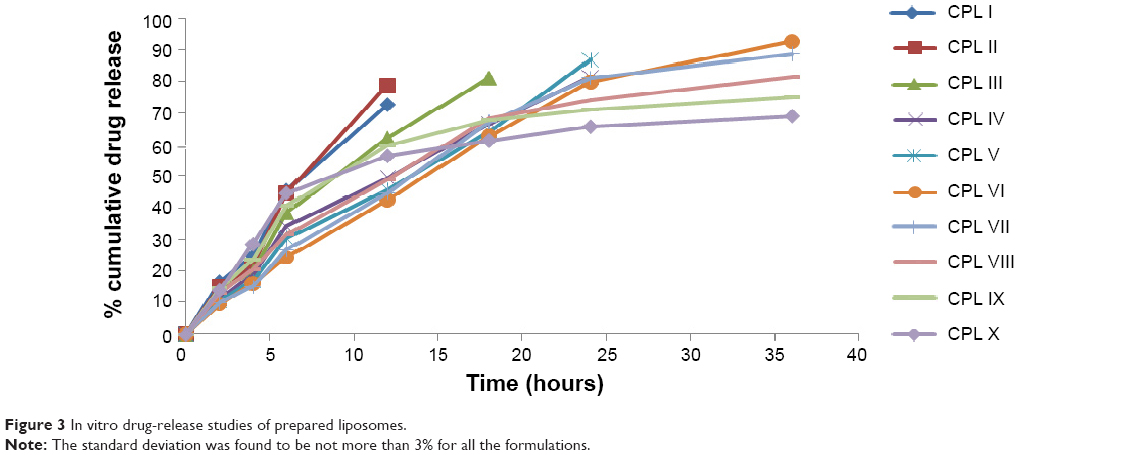 | Figure 3 In vitro drug-release studies of prepared liposomes. |
Particle size analysis and zeta potential studies
The particle size measurements of the freshly prepared multilamellar liposomal dispersion with lipid component in the molar ratio of 1.0:0.6 are presented in Figure 4A. The particle size distribution of the tested liposomal formulation showed unimodal normal symmetrical frequency distribution patterns. Figure 4A and B show that the mean particle diameter of the multilamellar vesicles was found to be 417.3 nm and the zeta potential was found to be −4.64, respectively, indicating low electrolytic reactions, which in turn improves the stability of liposomal formulations. The standard deviations were found to be not more than 7%.
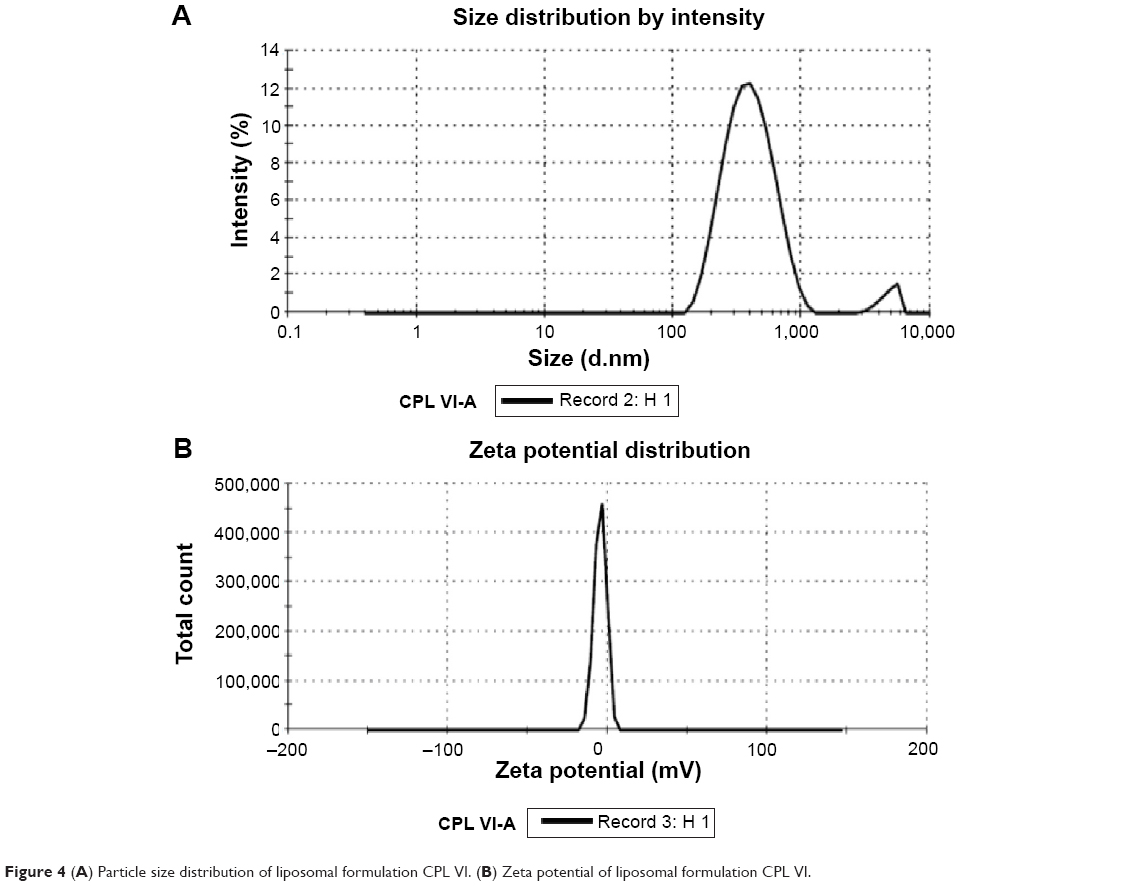 | Figure 4 (A) Particle size distribution of liposomal formulation CPL VI. (B) Zeta potential of liposomal formulation CPL VI. |
Lyophilization of liposomes
The optimization of the cryoprotectant glucose at 4 g/1 g lipid content concentration was found to be effective in retaining the entrapment of CP (81.432%±0.428%) in the liposomes during lyophilization (Table 4).
 | Table 4 Retention of cefditoren pivoxil (CP) in freeze-dried liposomes as a function of cryoprotectants |
SEM
The SEM image (Figure 5A) of freshly prepared CPL VI shows spherical vesicles have formed, while the SEM image (Figure 5B) of the freeze-dried formulation shows a cryoprotectant layer has formed, masking the spherical-shaped multilamellar vesicles.
 | Figure 5 Scanning electron microscopy studies of (A) freshly prepared CPL VI liposomes and (B) lyophilized CPL VI liposomes. |
Stability studies
Results of the stability studies of the selected formulation CPL VI are shown in Table 5. There were no physical changes observed up to the 60th day of stability study at the storage condition of 4°C. However, when stored at 25°C for 60 days, there was an observable change in drug content (93.200±0.418 compared with 98.9±0.258% at Day 0), indicating that the prepared liposomal formulation was more stable under refrigeration. In addition, at 4°C, the CPL VI formulation showed good physical properties and in vitro drug content and drug release at 60 days. Therefore, formulation CPL VI was selected as the final optimized formulation and further kinetic studies and in vivo animal studies were carried out on this formulation.
 | Table 5 Stability studies of the selected liposomal formulation |
In vivo studies
The drug concentrations in rabbit plasma were characterized by HPLC. Figure 6 shows the time course of changes in the drug concentrations after oral administration to rabbits. From the in vivo studies it was found that concentration (maximum concentration after administration [Cmax]) of the formulation was 12.95 μg/mL, the time to Cmax (Tmax) of the formulation was 36 hours, the elimination half-life (t1/2) of the formulation was 15 hours, the elimination rate constant (ke) of the formulation was 0.046 h−1, and area under the curve of the formulation was 489.4 μg/mL·h. These findings show that the CPL VI liposomal formulation had good controlled-release behavior.
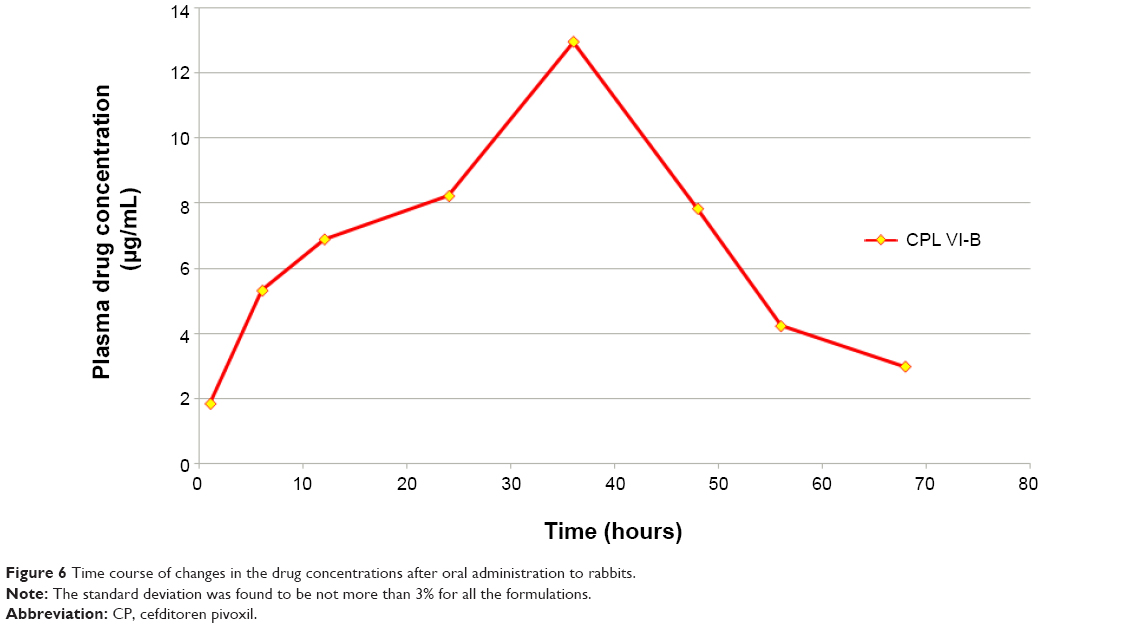 | Figure 6 Time course of changes in the drug concentrations after oral administration to rabbits. |
Conclusion
With the aim of developing controlled release formulation for CP, the study reported here was carried out using phosphatidylcholine and cholesterol. The liposomes, prepared by thin-film hydration, were found to have acceptable formulation parameters. The CPL VI (1.0:0.6 molar ratio of phsphatidylcholine:cholesterol) formulation showed the highest drug-loading capacity of 72.3% and showed a good controlled-release behavior with good stability when compared with the other formulations. The plasma concentration of cefditoren in rabbits administered with CPLVI formulation showed good controlled release behavior when compared to other formulations. However, further work is needed to establish the in vivo safety and efficacy of the prepared liposomes, which could be used to treat many bacterial infections.
Acknowledgments
The authors thank VIT University for providing necessary infrastructure and support to carry out the present work. Authors also wish to thank Johnson and Johnson for their support.
Disclosure
The authors declare no conflicts of interest in this work.
References
Clares B, Gallardo V, Medina MM, Ruiz MA. Multilamellar liposomes of triamcinolone acetonide:preparation, stability, and characterization. J Liposme Res. 2009;19(3):197–206. | ||
Jia Y, Joly H, Omri A. Liposomes as a carrier for gentamicin delivery: development and evaluation of the physicochemical properties. Int J Pharm. 2008;359(1–2):254–263. | ||
Hathout RM, Mansour S, Mortada ND, Guinedi AS. Liposomes as an ocular delivery system for acetazolamide: in vitro and in vivo studies. AAPS Pharm Sci Tech. 2007;8(1):1. | ||
Agarwal R, Katare OP, Vyas SP. Preparation and in vitro evaluation of liposomal/niosomal delivery systems for antipsoriatic drug dithranol. Int J Pharm. 2001;228(1–2):43–52. | ||
Karlowsky JA, Jones ME, Draghi DC, Critchley IA, Thornsberry C, Sahm DF. In vitro susceptibility of recent clinical isolates of pneumococci to the investigational cephalosporin cefditoren. Diagn Microbiol Infect Dis. 2002;42(1):59–64. | ||
Clark CL, Nagai K, Dewasse BE, et al. Activity of cefditoren against respiratory pathogens. J Antimicrob Chemother. 2002;50(1):33–41. | ||
Kuti JL, Quintiliani R. Cefditoren pivoxil: a novel broad-spectrum oral cephalosporin. Formulary. 2001;36:271–275. | ||
Johnson DM, Biedenbach DJ, Beach ML, Pfaller MA, Jones RN. Antimicrobial activity and in vitro susceptibility test development for cefditoren against Haemophilus influenzae, Moraxella catarrhalis, and Streptococcus species. Diagn Microbiol Infect Dis. 2004;37(2):99–105. | ||
Nishi J, Yoshinaga M, Tokuda K, et al. Oral antimicrobial susceptibilities of Streptococcus pneumoniae, Haemophilus influenzae, and Moraxella catarrhalis isolates from Japanese children. Int J Antimicrob Agents. 2002;20(2):130–135. | ||
Venugopalarao G, Sreeniva Gowtham M, Sarada NC. Formulation evaluation and stability studies of hydrogel tablets containing Cefditoren Pivoxil. Journal of Pharmacy Research. 2013;7(3):230–234. | ||
Jain S, Jain N, Bhadra D, Tiwary AK, Jain NK. Transdermal delivery of an analgesic agent using elastic liposomes: preparation, characterization and performance evaluation. Curr Drug Deliv. 2005;2(3):223–233. | ||
Shabbits JA, Chiu GN, Mayer LD. Development of an in vitro drug release assay that accurately predicts in vivo drug retention for liposome-based delivery systems. J Control Release. 2002;84(3):161–170. | ||
Gulati M, Grover M, Singh M, Singh S. Study of azathioprine encapsulation into liposomes. J Microencapsul. 1998;15(4):485–494. | ||
Armengol X, Estelrich J. Physical stability of different liposome compositions obtained by extrusion method. J Microencapsul. 1995;12(5):525–535. | ||
El-Nesr OH, Yahiya SA, El-Gazayerly ON. Effect of formulation design and freeze-drying on properties of fluconazole multilamellar liposomes. Saudi Pharm J. 2010;18(4):217–224. | ||
Gomez-Hens A, Fernandez-Romero JM. Analytical methods for the control of liposomal delivery systems. Trends Anal Chem. 2006;25(2):167–178. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.