")
Back to Journals » International Journal of Nanomedicine » Volume 15
Preparation and Characterization of Anti-GPC3 Nanobody Against Hepatocellular Carcinoma
Authors Xia L, Teng Q, Chen Q, Zhang F
Received 17 October 2019
Accepted for publication 10 March 2020
Published 30 March 2020 Volume 2020:15 Pages 2197—2205
DOI https://doi.org/10.2147/IJN.S235058
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Mian Wang
Lijie Xia, Qiao Teng, Qi Chen, Fuchun Zhang
Xinjiang Key Laboratory of Biological Resources and Genetic Engineering, College of Life Science and Technology, Xinjiang University, Urumqi 830046, People’s Republic of China
Correspondence: Lijie Xia
Xinjiang Key Laboratory of Biological Resources and Genetic Engineering, College of Life Science and Technology, Xinjiang University, 666 Shengli Road, Urumqi, Xinjiang 830046, People’s Republic of China
Email [email protected]
Background: Glypican-3 (GPC3) is a newly identified target molecule for the early diagnosis of hepatocellular carcinoma (HCC), while targeted inhibition of GPC3 signaling may help to control the proliferation and metastasis of HCC cells. The purpose of this study was to prepare the anti-GPC3 nanobody and to investigate the affinity of the anti-GPC3 nanobodies in vitro and the anticancer effects on hepatocellular carcinoma in vivo.
Methods: To screen for unknown anti-GPC3 antibodies, we constructed an antibody phage display library. After three rounds of panning, positive phage clones were identified by enzyme-linked immunosorbent assay (ELISA). Further, the nanobody fusion protein was expressed in E. coli BL21 cells and purified by affinity chromatography. Competitive ELISA and flow cytometry were conducted to confirm the affinity of the anti-GPC3 nanobodies in vitro. The antitumor effects of VHHGPC3 were assessed in vivo.
Results: The results showed that the nanobody VHHGPC3 had specific high-affinity binding to His-GPC3 antigen. Moreover, VHHGPC3 exhibited specific binding to commercial human GPC3 and recognized the surface GPC3 protein of the hepatoma cell line HepG2. Importantly, in vivo study showed that GPC3 nanobody suppresses the growth of HepG2 and improves the survival rate of tumor mice.
Discussion: In summary, our new anti-GPC3 nanobody suggests a strong application potential for targeted therapy of liver cancer.
Keywords: GPC3, phage display library, selection, nanobody
Introduction
The incidence and overall mortality of hepatocellular carcinoma (HCC) are increasing annually.1 Traditional surgery can control liver cancer to some extent, but survival rate is still very low2 as HCC is prone to metastasis and relapse. Molecular targeting therapy,3 immunotherapy,4 and long non-coding RNA (IncRNA)-based therapy5 have been tested against HCC in preclinical studies, but these advances have not yet translated into more efficacious clinical treatments. Therefore, it is still vital to identify novel prognostic markers and treatment targets for HCC.6
Early diagnosis and treatment are keys to improving primary HCC outcome.7 The heparin sulfate proteoglycan glypican-3 (GPC3) is involved in the regulation of cell proliferation, adhesion, and migration.8,9 Moreover, elevated expression of GPC3 has been reported in HCC tissues.10 GPC3 also promoted HCC growth by stimulating the canonical wnt/β-catenin signaling pathway, while GPC3 gene silencing inhibited the proliferation of HCC cells and induced apoptosis.11 Pei et al found that autophagy suppressed the proliferation of HepG2 hepatoma cells in part by inhibiting GPC3/wnt/β-catenin signaling.12 Nobuhiro Tsuchiya and colleagues have developed a T cell therapy using T cell receptor (TCR) sequences obtained from GPC3 peptide-specific cytotoxic T lymphocyte (CTL) clones for improved efficacy in patients with advanced HCC.13 Hence, GPC3 can be used as a biomarker for the early diagnosis of hepatocellular carcinoma, while targeted inhibition of GPC3 signaling may help control the proliferation and metastasis of HCC cells.14
Camelids (dromedaries, llamas, alpacas, etc.) express unusual antibodies composed only of heavy chains without light chains (VHHs) referred to as nanobodies because diameter and length are in the nanometer range.15 Nanobodies possess several advantages for therapeutic applications such as tumor targeting, including low immunogenicity, high physicochemical stability and refolding capacity, and good tissue penetration,15 while antigen affinity is equal to that of conventional antibodies.16
The identification of new treatment targets has in many cases led to the development of novel treatment strategies. For instance, programmed death-1 receptor (PD-1)/programmed death ligand-1 (PD-L1) pathway blockade has proven effective and practical for cancer immunotherapy. So far, five drugs targeting the PD-1/PD-L1 pathway, Keytruda (pembrolizumab), Opdivo (nivolumab), Bavencio (avelumab), Tecentriq (atezolizumab), and Imfinzi (durvalumab) have been approved by the US Food and Drug Administration (FDA) for cancer treatment.17
Glypican-3 has the potential to differentiate between benign and malignant liver diseases, so antibodies and nanobodies may prove effective for drug targeting. To screen for unknown anti-GPC3 antibodies, we constructed a GPC3 phage display library. Three rounds of panning using competitive enzyme-linked immunosorbent assay (ELISA) and flow cytometry succeeded in isolating a specific high-affinity anti-GPC3 nanobody. The anti-GPC3 nanobody suppresses the growth of HepG2 in vivo and improves the survival rate of tumor mice, and may prove valuable for targeted therapy of liver cancer.
Materials and Methods
Materials and Reagents
Phagemid vector pCANTAB 5E and Helper phage M13KO7 were obtained from GE Healthcare (NJ, USA), Escherichia coli TG1, E. coli BL21, prokaryotic vector pET28a, and HepG2 cells from the Xinjiang Key Laboratory of Biological Resources and Genetic Engineering, Xinjiang University (Urumqi, Xinjiang, China), Taq DNA polymerase and deoxynucleotides (dNTPs) from Takara (Dalian, China), DAB (3, 3ʹ-diaminobenzidine), ampicillin, kanamycin sulfate, and isopropyl-D- thiogalactopyranoside (IPTG) from Solarbio Biotechnology (Beijing, China), anti-His and horseradish peroxidase (HRP)-conjugated anti-his monoclonal antibodies (mAbs) from Beyotime Biotechnology (Shanghai, China), anti-human GPC3 mAbs from ACRO Biosystems (Beijing, China), and Anti-6×His fluorescein isothiocyanate (FITC) from abcam (Cambridge, UK).
Camel Immunization and Ethics Statement
A Bactrian camel from Xinjiang Province, China, was immunized five times with 50 mg of GPC3 fusion protein.18 All animal experiments were approved by the Committee on the Ethics of Animal Experiments of Xinjiang Key Laboratory of Biological Resources and Genetic Engineering (BRGE-AE001) and performed under the guidelines of the Animal Care and Use Committee of College of Life Science and Technology, Xinjiang University. Serum antibody titers were determined by ELISA.
Phage Library Construction
Total mRNA was extracted from peripheral blood mononuclear cells (PBMCs) using Trizol reagent and examined using agarose gel electrophoresis. cDNA was generated using M-MLV Reverse Transcriptase. Specific primers were designed based on complementary sequences to two conserved regions in the heavy-chain variable region of camel nanobody. VHH genes were then amplified by PCR in a 25-μL reaction mixture containing 5 μL 5 × PS buffer (+Mg2+), 0.5 μL dNTP, 0.25 μL cDNA, 0.25 μL PrimeSTAR, and 0.5 μL forward primer and reverse primer. The PCR products and the pCANTAB-5E vector were digested sequentially with Sfi I and Not I, then ligated by T4 DNA ligase at 16°C overnight and electro-transformed into competent E. coli TG1 cells.19
Phage Amplification and Titering
The phage library (100 μL) was inoculated into 50 mL of LB medium containing 50 μL ampicillin at 37°C and grown until log phage (OD600 = 0.5−0.6). Helper phage M13KO7 at about 5 × 1012 colony-forming units (cfu)/mL was then added and the mixture incubated for 1 hr at 37°C. Bacteria were harvested by centrifugation for 10 mins at 2500 g and resuspended in 100 mL fresh LB medium with 100 μL ampicillin. The new suspension was incubated at 37°C overnight. The next day, bacterial cells were removed by centrifugation for 20 mins at 2500 g and phages were precipitated from the supernatant with 1/5 volume 20% polyethylene glycol(PEG)-NaCl 8000. The precipitated phages were resuspended in phosphate-buffered saline (PBS) for titer measurement and the next round of bio-panning. For titering, the phage solutions were diluted 1:10, 1:100, and 1:1000 in fresh LB medium. Serial dilutions of phage (100 μL) were added to 900 μL TG1 cells and the mixture incubated for 30 mins at 37°C. Then, a 100 μL volume of this cell suspension was coated onto solid LB culture medium and incubated at 30°C overnight. The colonies were counted and phage titer calculated as plaque-forming units (pfu).20,21
Screening of Special VHH Against GPC3
GPC3 protein in carbonate buffer [pH 9.6] (1, 5, or 10 μg/mL) was used to coat 96-well plates. Briefly, wells were filled with 100 μL GPC3 solution and incubated overnight at 4°C, then washed at least three times with PBST. A 200 μL volume of 5% (w/v) non-fat milk was added for 2 hrs at 37°C. After washing with PBST, phage library solution was added to each well and incubated for 2 h at 37°C. Wells were again washed with PBST to remove unbound phages. A 50-μL volume of 100 mM glycine-HCl (pH 2.2) was added for 10 min to elute target-bound phages and immediately neutralized with 7 μL of 2 M Tris-HCl (pH 9.1). The solution in each well was collected and inoculated into TG1 cell suspension, followed by incubation at 37°C for 30 mins. A portion of the infection solution was used for enrichment statistics, and the remaining volume for the next round of screening. Each round of library screening required amplification and titering as described.19–21 The schematic procedure for the production of phage nano-antibody library is shown in Figure 1.
|
Figure 1 Schematic procedure for the production of phage nano-antibody library. |
Construction, Expression and Purification of the Nanobody
The PCR products of nanobody fragments and pET28a vector were digested with restriction enzymes EcoR I and Xho I, then ligated by T4 DNA ligase at 16°C overnight. Single colonies with good shape and size were identified by PCR and double enzyme digestion. Finally, the recombinant plasmid was sent to Sangon Biotech (Shanghai) Co., Ltd. for sequencing and identification. Further, the nanobody fusion protein was expressed in E. coli BL21 cells and purified by affinity chromatography. The fusion protein purity was measured by 12% SDS-PAGE electrophoresis.19
Affinity Measurements
Commercial human GPC3 mAb was coated onto 96-well plates overnight at 4°C. Plates were then washed with PBST and nanobody added to each well for 2 hrs at 37°C, followed by his-tag (mouse) antibody (1:3000) as the secondary antibody for 2 hrs at 37°C and HRP-conjugated anti-his tag antibody (1:5000) for 1 hr at 37°C. TMB substrate was then added to each well for 15 mins at 37°C. The reaction was stopped by addition of 50 μL/well H2SO4. Finally, the OD at 492 nm was measured.
Cell Binding Analysis by Flow Cytometry
HepG2 cells (5×105 per culture dish) were incubated with different concentrations of nanobody (experimental group), commercial rabbit anti-human GPC3 mAb (positive control group), or left untreated (negative control group) for 2 hrs at 4°C. Cultures were then washed with PBS and stained with FITC-conjugated anti-6×His (experimental group, negative control group) or FITC-conjugated goat anti-rabbit antibody (positive control group) for 30 mins. Finally, cell staining was analyzed by flow cytometry using Flow Jo 7.6 software.
Proliferation Analysis
The 3-(4, 5-dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide (MTT) (Sigma, St. Louis, MO, USA) assay was used to evaluate the growth inhibition of nanobody on HepG2 cells. Briefly, cells (5000 cells/well) were seeded in 96-well plates, and then treated with various doses of different concentrations of nanobody (experimental group), commercial rabbit anti-human GPC3 mAb (positive control group), or untreated (negative control group) for 24 hrs. Supernatant was discarded after centrifugation at 1200 rpm for 5 mins and 100 μL of MTT solution (0.5 mg/mL in PBS) was added to each well and incubated at 37°C for 3 hrs. The formed formazan crystals were dissolved in 200 μL DMSO. The OD490 values were measured by a 96-well microplate reader (Bio-Rad Laboratories, CA, USA). The relative cell viability was calculated according to the formula: Cell viability (%) = (ODtreated/ODuntreated) ×100%.
Establishment of the Tumor Models in Nude Mice
Female BALB/c-nu mice of SPF grade, 4–5 week-old, were purchased from Beijing laboratory animal research center (Beijing, China), and received pathogen-free water and food. The cells of logarithmic phase were washed twice by serum-free culture solution and resuspended to a concentration of about/mL. The HepG2 cells (1×107) in 0.1 mL of PBS were inoculated subcutaneously on the back of each nude mouse. After the inoculation, the animals were randomly divided into three groups (n=6): the negative control group (10mg/kg BSA), the positive control group (5 mg/kg cisplatin), the 10mg/kg GPC3 nanobody group. All animals were peritumorally injected drugs every 2 days which lasted for 10 times. The mice were weighed and the tumor growth in nude mice was monitored carefully once a day. Tumor sizes were measured using calipers and tumor volume was calculated according to the formula: tumor volume (mm3) = (length×width2)/2. The survival of tumor mice was monitored every day.
Statistical Analysis
All data were expressed as mean ± standard deviation (S.D.). Statistical analysis was conducted using one-way analysis of variance (ANOVA) among the treatment and control groups. p<0.05 was considered statistically significant.
Results
Construction of a Phage Display Library
A Bactrian camel was immunized five times with GPC3 protein and serum extracted for antibody detection, which revealed substantially elevated titers (Figure 2A). Total PBMCs were isolated for total RNA extraction and PCR amplification of VHH genes. Gene fragments of ~500 bp obtained by nested PCR were successfully cloned into the phagemid vector pCANTAB5E (Figure 2B). Phagemids were then used to transform TG1 E. coli, which yielded 268 single colonies on solid medium. Of these colonies, 20 were randomly selected for PCR validation. There were 12 positive clones, indicating a recombination rate of 60% and storage capacity of 1.608 × 106 cfu/mL. The titers of the helper phage and phage library were 5.321 × 1016 pfu/mL and 6.5 × 1013 pfu/mL, respectively.
|
Figure 2 Construction of a phage display library. (A) Anti-GPC3 serum titer of Bactrian Camel. (B) Identification of VHH gene amplification. M: DNA Marker DL 2000; Lanes 1–8: Positive clones. |
Screening of Special VHH Against GPC3
The phage display library was panned for three consecutive rounds using 96-well plates coated with GPC3 protein. The number of single colonies decreased in each round. Randomly selected colonies harboring the expected 500-bp fragment were identified by PCR (Figure 3), and the enrichment in each round of bio-panning was calculated (Table 1).
|
Figure 3 VHHGPC3 positive clones were identified by PCR. M: DNA Marker DL2000. -: Negative control. Lanes 1–15: Positive clones. |
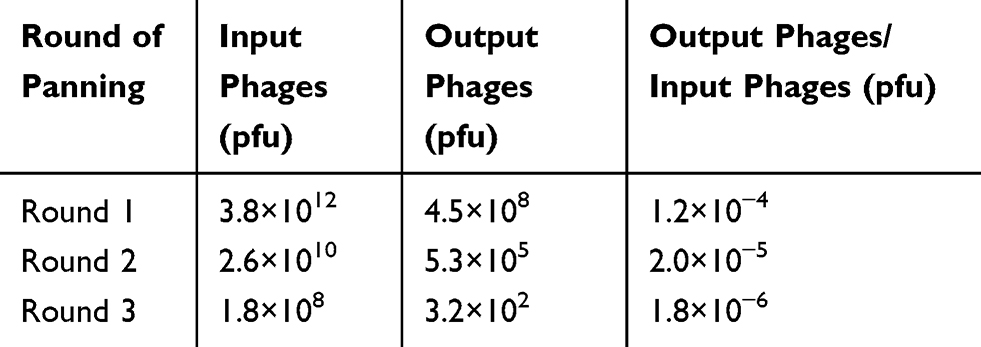 |
Table 1 Enrichment of Specific Phages During Subsequent Rounds of Bio-Panning |
Construction, Expression, and Purification of the Nanobody
After three rounds, we successfully constructed the pET28a-VHHGPC3 prokaryotic expression vector. It was expressed in E. coli BL21 cells after induction by IPTG. The molecular weight of the product nanobody was determined to be 13 kDa by 12% SDS-PAGE and Western Blot analysis (Figure 4A−C).
|
Figure 4 Expression and purification of VHHGPC3 antibody. (A) Expression of pET28a-VHHGPC3 fusion protein analyzed by SDS-PAGE. M: protein marker (10–170 kDa); Lane 1: Transformed bacterium carrying pET28a empty vector without induction, Lane 2: Transformed bacterium carrying pET28a empty vector induced with IPTG; Lane 3: pET28a-VHHGPC3 recombinant plasmid without induction; Lane4: pET28a-VHHGPC3 recombinant plasmid induced with 0.5mmol/L IPTG; Lanes5–6: pET28a-VHHGPC3 recombinant plasmid expressed in the supernatant and precipitation, respectively. (B) Expression of pET28a-VHHGPC3 fusion protein analyzed by Western blot. Lane1: pET28a-VHHGPC3 recombinant plasmid without induction; Lane 2:pET28a-VHHGPC3 recombinant plasmid induced with 0.5mmol/L IPTG; Lanes 3–4: pET28a-VHHGPC3 recombinant plasmid expressed in the supernatant and precipitation, respectively. (C) Purification of VHHGPC3 antibody. Lane 1: Purified his-VHHGPC3 fusion protein in the supernatant. |
Affinity Measurements
The affinity of VHHGPC3 was assessed by sandwich ELISA on 96-well plates coated with commercial human GPC3 protein (experimental group) or skim milk protein (negative control). A commercial anti-human GPC3 mAb served as the positive control. Optical density measurement indicated that VHHGPC3 bound specifically to the commercial human GPC3 but not milk protein (Figure 5).
|
Figure 5 Determination of affinity of VHHGPC3 and antigen protein. The 96-well plates were coated with 2μg/mL antigen, incubated with 5μg/mL VHHGPC3. Skim milk was used as negative control. Commercial anti-human GPC3 mAb was used as positive control. |
Cell Binding Analysis by Flow Cytometry
Flow cytometric analysis was performed to assess the specific binding activity of the nanobody to human GPC3 expressed on the surface of HepG2 hepatoma cells. Indeed, exposure to VHHGPC3 induced a substantial shift in fluorescence intensity of HepG2 cells similar to that observed using a commercial human anti-GPC3 (Figure 6).
|
Figure 6 Detection of VHHGPC3 binding with GPC3 expressed on HepG2 by FCM. (A) Not incubated by VHHGPC3 was used as negative group with black line. Commercial anti-human GPC3 mAbs was used as positive with green line. 35 μg and 45μg of VHHGPC3 were red and blue lines, respectively. (B) Statistical analysis showed a FITC intensity significant increase in HepG2 as compared to the negative control. |
GPC3 Nanobody Suppresses the Growth of HepG2 in vivo and Improves the Survival Rate of Tumor Mice
To investigate whether GPC3 nanobody could suppress the growth of HepG2 cells in vivo, mice were injected with HepG2 cells and treated with BSA (control group), cisplatin or GPC3 nanobody. After 7 days of HepG2 cell injection, tumor mice were peritumorally injected with GPC3 nanobody every 2 days which lasted for 10 times. The body weight of mice and tumor sizes were monitored. As shown in Figure 7A, body weight of the mice group treated with GPC3 nanobody showed no significant difference while body weight of the mice group treated with cisplatin sharply decreased, suggesting that the selected doses of GPC3 nanobody have no obvious side effect. Furthermore, the tumor growth in mice treated with 10 mg/kg of GPC3 nanobody was significantly inhibited (Figure 7B). Moreover, the GPC3 nanobody treatment greatly improved the survival of tumor mice (4/6) compared with control group (0/6) and cisplatin group (3/6) by the end of the experiment (Figure 7C).
|
Figure 7 GPC3 nanobody inhibits tumor growth in nude BALB/c mice. Nude mice (n=6) were inoculated with HepG2 cells subcutaneously on the back of each nude mouse. After 7 days, tumor mice were treated with 10 mg/kg GPC3 nanobody, BSA (control) and cisplatin. Body weight of mice (A), tumor sizes (B), and the survival rates (C) were monitored at the indicated time points. |
Discussion
Liver cancer incidence is rising in both men and women across all age groups. In addition to traditional treatments (chemotherapy, radiotherapy), targeted immunotherapies are rapidly evolving, offering new hope to liver cancer patients.22 However, objective molecular markers are urgently needed to help standardize the histological diagnosis of early-stage HCC and guide appropriate treatment plans.23,24
Yukihiro Haruyam et al demonstrated that GPC3 is a prognostic factor and potential immunotherapeutic target for HCC treatment.8 In contrast, serum alpha-fetoprotein (AFP) has a relatively low sensitivity, and so is of limited value as a serum marker for hepatoblastoma.25 Glypican-3 expression rate is substantially elevated in HCC tissue and gradually increases in parallel with clinical upstaging.26 One study reported that the combined use of AFP and GPC3 can enhance diagnostic sensitivity compared to each marker alone.27 Moreover, concomitant use of heat-shock protein 70, glutamine synthetase, and GPC3 demonstrated even greater specificity and sensitivity for the diagnosis of hepatitis B virus (HBV)-related HCC.28,29 Montalbano et al proposed that GPC3 is not merely a biomarker but is central to HCC pathogenesis.30 Thus, elucidating signaling pathways and tumorigenic processes involving GPC3 may reveal important aspects of disease development and identify novel potential treatment targets.30
Most currently available GPC3 antibodies are polyclonal or monoclonal antibodies from mouse hybridoma cells. While these antibodies can specifically identify GPC3 proteins in cells and tissues,31,32 the high molecular weight of these molecules is not conducive to rapid clearance in vivo or effective penetration into tumor tissue. Most studies on improved antibody applicability for immunotherapy have focused on the modification of conventional antibody molecules, such as single-chain antibodies, Fab antibodies, and Fv single domain antibodies.33,34 However, these antibodies have many shortcomings such as cumbersome preparation processes, poor water solubility, and easy aggregation. Therefore, it is necessary to obtain more advantageous GPC3 antibodies to improve diagnostic sensitivity, targeting and metabolism.
Camelids produce unusual antibodies composed only of heavy chains. The variable domain is designated VHH for Camelid heavy-chain antibodies (hcAbs) or more generally, nanobodies or single-domain antibodies. These nanobodies have additional advantages, such as low production cost, good water solubility, strong stability, weak immunogenicity, and good tissue penetration, while retaining high affinity for the ligand-binding site.35 Indeed, nanobodies are already widely used as research tools, diagnostic tools, and therapeutics.36 However, how to improve the affinity of the nano-antibody and prolong the half-life has been a bottleneck in the application of the nanobodies.
The YP7 mAb, HN3 human single domain antibody, and HS20 human single-chain Fv antibody have been demonstrated to bind GPC3 with both high specificity and affinity.37–39 Although human mAbs against tumor antigens are widely recognized as rational immunotherapeutic tools for cancer treatment, but many mAbs have shown limited efficacy in human clinical trials due to high immunogenicity.23 Rituximab and daratumumab can trigger cytokine release syndrome and even cause opportunistic virus infection.40,41 Alternatively, Homayouni et al successfully prepared a functional anti-human TIM-3-specific nanobody with high affinity and anti-proliferative activity on an AML cell line.42 These attributes make nanobodies, in particular those engineered from the variable heavy-chain fragment (VHH gene) found in Camelidae heavy-chain antibodies (or IgG2 and IgG3), are the smallest fragments that retain the full antigen-binding capacity of the antibody with advantageous properties as diagnostic and therapeutic applications.
In our study, we chose to immunize Bactrian camels rather than llamas or alpacas as these animals are native to central Asia, including Xinjiang Province, China. The VHH gene fragment was inserted into the capsid vector of a filamentous phage in vitro by phage display technology, and the phage display library was successfully constructed. Through three rounds of selection, we successfully screened the phage for a nanobody that could specifically bind to His-GPC3 antigen. The statistics of the enrichment indicated a progressive increase with round. Also, specificity increased with lower antigen coating. The VHHGPC3 obtained was not only able to specifically bind commercial human GPC3 mAbs but also recognized the surface of HepG2 cells.
Early diagnosis and treatment of HCC are critical for the outcome. The generation of high-affinity nanobodies by phage display library screening may facilitate the development of new targeted immunotherapies for hepatocellular carcinoma.
Data Sharing Statement
We declared that materials described in the manuscript, including all relevant raw data, will be freely available to any scientist wishing to use them for non-commercial purposes, without breaching participant confidentiality.
Ethics and Consent Statement
All animal experiments were approved by the Committee on the Ethics of Animal Experiments of Xinjiang Key Laboratory of Biological Resources and Genetic Engineering (BRGE-AE001) and performed under the guidelines of the Animal Care and Use Committee of College of Life Science and Technology, Xinjiang University.
Consent for Publication
All authors consent for publication.
Author Contributions
All authors contributed to data analysis, drafting and revising the article, gave final approval of the version to be published, and agree to be accountable for all aspects of the work.
Funding
This work was supported by the National Natural Science Foundation of China (no. 31500752), the “Tianshan Youth Project” Young Ph.D. Science and Technology talents Project (no. 2017Q077) and the Natural Science Foundation of Xinjiang (no. 2016D01C078).
Disclosure
The authors declare no conflict of interest in this work.
References
1. Forner A, Llovet J, Bruix J. Hepatocellular carcinoma. Lancet. 2012;379(9822):1245–1255. doi:10.1016/S0140-6736(11)61347-0
2. Cao H, Phan H, Yang L. Improved chemotherapy for hepatocellular carcinoma. Anticancer Res. 2012;32(4):1379–1386.
3. Giannelli G, Rani B, Dituri F, et al. Moving towards personalised therapy in patients with hepatocellular carcinoma: the role of the microenvironment. Gut. 2014;63(10):1668–1676. doi:10.1136/gutjnl-2014-307323
4. Greten TF, Wang XW, Korangy F. Current concepts of immune based treatments for patients with HCC: from basic science to novel treatment approaches. Gut. 2015;64(5):842–848. doi:10.1136/gutjnl-2014-307990
5. Xiaoting Z, Jihang Y, Tengteng Z, et al. Long noncoding RNA glypican 3 (GPC3) antisense transcript1 promotes hepatocellular carcinoma progression via epigenetically activating GPC3. FEBS J. 2016;283:3739–3754. doi:10.1111/febs.13839
6. Sersté T, Barrau V, Ozenne V, et al. Accuracy and disagreement of computed tomography and magnetic resonance imaging for the diagnosis of small hepatocellular carcinoma and dysplastic nodules: role of biopsy. Hepatology. 2012;55(3):800–806. doi:10.1002/hep.24746
7. Kudo M. Emerging strategies for the management of hepatocellular carcinoma. Dig Dis. 2014;32(6):655–657. doi:10.1159/000367981
8. Haruyama Y, Kataoka H. Glypican-3 is a prognostic factor and an immunotherapeutic target in hepatocellular carcinoma. World J Gastroenterol. 2016;22(1):275–283. doi:10.3748/wjg.v22.i1.275
9. Sung YK, Hwang SY, Park MK, et al. Glypican-3 is overexpressed in human hepatocellular carcinoma. Cancer Sci. 2003;94:259–262. doi:10.1111/j.1349-7006.2003.tb01430.x
10. Tsuchiya N, Sawada Y, Nakatsura T. Biomarkers for the early diagnosis of hepatocellular carcinoma. World J Gastroenterol. 2015;21(37):10573–10583. doi:10.3748/wjg.v21.i37.10573
11. Capurro MI, Xiang YY, Lobe C, Filmus J. Glypican-3 promotes the growth of hepatocellular carcinoma by stimulating canonical Wnt signaling. Cancer Res. 2005;65(14):6245–6254. doi:10.1158/0008-5472.CAN-04-4244
12. Pei H, Bin C, Yulin H, et al. Autophagy suppresses proliferation of HepG2cells via inhibiting glypican-3/wnt/β-catenin signaling. Onco Targets Ther. 2018;11:193–200. doi:10.2147/OTT.S150520
13. Tsuchiya N, Yoshikawa T, Fujinami N, et al. Immunological efficacy of glypican-3 peptide vaccine in patients with advanced hepatocellular carcinoma. Oncoimmunology. 2017;6(10):e1346764. doi:10.1080/2162402X.2017.1346764
14. Hippo Y, Watanabe K, Watanabe A, et al. Identification of solubleNH2-terminal fragment of glypican-3 as a serological marker for early-stage hepatocellular carcinoma. Cancer Res. 2004;64(7):2418–2423. doi:10.1158/0008-5472.CAN-03-2191
15. Krah S, Schröter C, Zielonka S, et al. Single-domain antibodies for biomedical applications. Immunopharmacol Immunotoxicol. 2016;38(1):21–28. doi:10.3109/08923973.2015.1102934
16. van der Linden RH, Frenken LG, de Geus B, et al. Comparison of physical chemical properties of llama VHH antibody fragments and mouse monoclonal antibodies. Biochim Biophys Acta. 1999;1431(1):37–46. doi:10.1016/S0167-4838(99)00030-8
17. Jia L, Zhang Q, Zhang R. PD-1/PD-L1 pathway blockade works as an effective and practical therapy for cancer immunotherapy. Cancer Biol Med. 2018;15(2):117–122.
18. Teng Q, Xia LJ, Zhang FC. Bioinformatics prediction of GPC3 gene structure and function. Genomics Appl Biol. 2018;37(3):1131–1136.
19. Tiansen L, Huang M, Xiao H, et al. Selection and characterization of specific nanobody against bovine virus diarrhea virus(BVDV) E2 protein. PLoS One. 2017;12(6):e0178469. doi:10.1371/journal.pone.0178469
20. Bagheri S, Yousefi M, Safaie Qamsari E, et al. Selection of single chain antibody fragments binding to the extracellular domain of 4-1BB receptor by phage display technology. Tumor Biol. 2017;5:1–13.
21. Qiang X, Sun K, Xing L, et al. Discovery of a polystyrene binding peptide isolated from phage display library and its application in peptide immobilization. Sci Rep. 2017;7(1):2673. doi:10.1038/s41598-017-02891-x
22. Shimizu Y, Suzuki T, Yoshikawa T. Cancer immunotherapy-targeted glypican-3 or neoantigens. Cancer Sci. 2018;109:531–541. doi:10.1111/cas.13485
23. Dong Z, Yao M, Wang L, et al. Down-regulating glypican-3 expression: molecular-targeted therapy for hepatocellular carcinoma. Mini Rev MedChem. 2014;14(14):1183–1193. doi:10.2174/1389557515666150101105135
24. Sun B, Huang Z, Wang B, et al. Significance of glypican-3(GPC3) expression in hepatocellular cancer diagnosis. Med Sci Monit. 2017;23:850–855. doi:10.12659/MSM.899198
25. Attallah AM, El-Far M, Omran MM, et al. GPC-HCC model: a combination of glypican-3 with other routine parameters improves the diagnostic efficacy in hepatocellular carcinoma. Tumor Biol. 2016;37:12571–12577. doi:10.1007/s13277-016-5127-6
26. Tunissiolli NM, Castanhole-Nunes MMU, Biselli-Chicote PM, et al. Hepatocellular carcinoma: a comprehensive review of biomarkers, clinical aspects, and therapy. Asian Pac J Cancer Prev. 2017;18(4):863–872.
27. Huang TS, Shyu YC, Turner R, et al. Diagnostic performance of alpha-fetoprotein, lens culinaris agglutinin-reactive alpha-fetoprotein, des-gamma carboxyprothrombin, and glypican-3 for the detection of hepatocellular carcinoma: a systematic review and meta-analysis protocol. Syst Rev. 2013;2:37. doi:10.1186/2046-4053-2-37
28. Moudi B, Heidari Z, Mahmoudzadeh-Sagheb H, et al. Concomitant use of heat-shock protein 70, glutamine synthetase and glypican-3 is useful in diagnosis of HBV-related hepatocellular carcinoma with higher specificity and sensitivity. Eur J Histochem. 2018;62(2859):42–50.
29. Shen Q, Nam SW. SF3B4 as an early-stage diagnostic marker and driver of hepatocellular carcinoma. BMB Rep. 2018;51(2):57–58. doi:10.5483/BMBRep.2018.51.2.021
30. Montalbano M, Georgiadis J, Masterson AL, et al. Biology and function of glypican-3 as a candidate for early cancerous transformation of hepatocytes in hepatocellular carcinoma (Review). Oncol Rep. 2017;37:1291–1300. doi:10.3892/or.2017.5387
31. Feng M, Ho M. Glypican-3 antibodies: a new therapeutic target for liver cancer. FEBS Lett. 2014;588:377–382. doi:10.1016/j.febslet.2013.10.002
32. Yang X, Liu H, Sun CK, et al. Imaging of hepatocellular carcinoma patient-derived xenografts using Zr-labeled anti-glypican-3 monoclonal antibody. Biomaterials. 2014;35:6964–6971. doi:10.1016/j.biomaterials.2014.04.089
33. Li Y, Siegel DL, Scholler N, Kaplan DE. Validation of glypican-3-specific scFv isolated from paired display/secretory yeast display library. BMC Biotechnol. 2012;12(1):23. doi:10.1186/1472-6750-12-23
34. Sham JG, Kievit FM, Grierson JR, et al. Glypican-3-targeting F(ab’)2 for 89Zr PET of hepatocellular carcinoma. J Nucl Med. 2014;55(12):2032–2037. doi:10.2967/jnumed.114.145102
35. Wesolowski J, Alzogaray V, Reyelt J, et al. Single domain antibodies: promising experimental and therapeutic tools in infection and immunity. Med Microbiol Immunol. 2009;198(3):157–174. doi:10.1007/s00430-009-0116-7
36. Muyldermans S. Nanobodies: natural single-domain antibodies. Annu Rev Biochem. 2013;82(1):775–797. doi:10.1146/annurev-biochem-063011-092449
37. Gao W, Tang Z, Zhang YF, et al. Immunotoxin targeting glypican-3 regresses liver cancer via dual inhibition of Wnt signalling and protein synthesis. Nat Commun. 2015;11(6):6536. doi:10.1038/ncomms7536
38. Gao H, Li K, Tu H, et al. Development of T cells redirected to glypican-3 for the treatment of hepatocellular carcinoma. Clin Cancer Res. 2014;20(24):6418–6428. doi:10.1158/1078-0432.CCR-14-1170
39. Fleming BD, Mitchell H. Glypican-3 targeting immunotoxins for the treatment of liver cancer. Toxins. 2016;8(274):1–13. doi:10.3390/toxins8100274
40. Smith MR. Rituximab (monoclonal anti-CD20 antibody): mechanisms of action and resistance. Oncogene. 2003;22(47):7359–7368. doi:10.1038/sj.onc.1206939
41. Xu XS, Yan X, Puchalski T, et al. Clinical implications of complex pharmacokinetics for daratumumab dose regimen in patients with relapsed/refractory multiple myeloma. Clin Pharmacol Ther. 2017;101(6):721–724. doi:10.1002/cpt.v101.6
42. Homayouni V, Ganjalikhani-hakemi M, Rezaei A, et al. Preparation and characterization of a novel nanobody against T-cell immunoglobulin and mucin-3 (TIM-3). Iran J Basic Med Sci. 2016;19(11):1201–1208.
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.