Back to Archived Journals » Current Biomarker Findings » Volume 4
Predicting cetuximab efficacy in patients with advanced colorectal cancer
Received 11 September 2013
Accepted for publication 6 December 2013
Published 18 June 2014 Volume 2014:4 Pages 61—68
DOI https://doi.org/10.2147/CBF.S54310
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Ibrahim H Sahin, Christopher R Garrett
Department of Gastrointestinal Medical Oncology, University of Texas MD Anderson Cancer Center, Houston, TX, USA
Abstract: Cetuximab has demonstrated activity, both as monotherapy, and in combination with cytotoxic chemotherapy, albeit modest. Efforts over the last decade have focused on determining which patient populations are most likely to benefit from this chimeric monoclonal antibody therapy. As the antibody targets the epidermal growth factor receptor (EGFR), cell surface expression by immunohistochemistry was hypothesized to be a biomarker of clinical efficacy; subsequent clinical trials have shown that this was not the case. Tumor KRAS mutation (the most frequently observed site is at codon 12) has been shown to be a negative biomarker (ie, a marker of cetuximab resistance); since 2008, treatment of patients with cetuximab has been restricted to those whose tumors do not harbor a KRAS mutation. There is considerable heterogeneity of KRAS mutations, and studies are ongoing to determine whether cetuximab resistance extends to those patients whose tumors have a KRAS codon 13, 61, and 164, mutation. EGFR gene copy, or more precisely a lack of increase in EGFR gene copy number, has been demonstrated to be a negative biomarker of EGFR efficacy; currently, it is not in routine use as a clinical standard of care. Tumor BRAF status, NRAS status, and PIK3CA mutation status are being evaluated as additional potential negative biomarkers of treatment efficacy. High expression of the receptor ligands epiregulin and amphiregulin has been shown to be a positive biomarker for treatment efficacy and is continuing to be studied clinically. After almost 10 years following the widespread introduction of cetuximab into the clinic as a treatment for metastatic colorectal cancer, the story of identifying suitable biomarkers of efficacy is still evolving. The tremendous tumor heterogeneity at the molecular level and the cell complexity of signaling pathways suggest that there is still a significant amount of work to be done to optimally define those patients most likely to benefit from this form of therapy.
Keywords: biomarkers, epidermal growth factor receptor, EGFR, KRAS mutation
Introduction
In February 2004, cetuximab became the first monoclonal antibody targeting the epidermal growth factor receptor (EGFR) to be approved for the treatment of metastatic colorectal cancer (CRC) in the United States; it received initial accelerated approval based upon observed objective response rates in clinical trials.1 EGFR is a member of the receptor tyrosine kinase family and found in the membrane of the epithelial cells lining in the surface of many organs, including the gastrointestinal system.2 EGFR mediates transduction of growth signals and has oncogenic activity in certain cancers (such as lung cancer and CRC),3 and its overexpression is associated with poorly differentiated histology, more advanced clinical stage, and an adverse prognosis.4–6 Therefore, EGFR presented an attractive potential therapeutic target for the treatment of a broad range of malignancies.
Murine antibodies with high affinity to EGFR, both agonist and antagonist, were generated by Drs Gordon Sato and John Mendelsohn, in 1982.7 Initial human Phase I studies of the compound M225 were performed in patients with advanced squamous cell carcinoma of the lung but were associated with the development of human anti-mouse antibody responses, limiting its clinical utility. A chimeric version of M225 (C225, cetuximab) was developed for human use in order to avoid anti-mouse antibody production. Studies have demonstrated that these antibodies can bind to EGFR, with a higher affinity than the natural ligands, and therefore may interrupt EGFR-dependent cell signaling.8,9 Preclinical studies of M225 and C225 have demonstrated efficacy in human epithelial tumor xenograft models;10 additionally, cetuximab was shown to be synergistic with topoisomerase I inhibitors.11 These preclinical observations were borne out in Phase II clinical trials, where cetuximab was demonstrated to have monotherapy activity in patients with chemotherapy-refractory CRC;12 in patients with irinotecan-refractory disease, the addition of cetuximab to irinotecan was shown to overcome resistance.13
Despite the efficacy which led to accelerated approval (and full approval October 200714 following clinical evidence to support a survival advantage with its monotherapy use15), it was realized that the majority of CRC patients with advanced metastatic disease, treated with cetuximab as monotherapy, appear not to benefit from therapy. Better identification of biomarkers of cetuximab efficacy would allow for more effective clinical use; this manuscript seeks to review the last decade of research regarding biomarkers of cetuximab efficacy in patients with CRC.
Clinical biomarkers of cetuximab efficacy
As with the receptor tyrosine kinase EGFR inhibitors gefinitinib and erlotinib,16 in patients with advanced lung cancer, the degree of skin toxicity was correlated with treatment efficacy in multiple CRC clinical trials.17 Clinical trials have tried to exploit this observation with dose escalation of cetuximab in those patients who do not develop grade 2 acneiform rash; in a randomized Phase II study where this “dose-escalation-to-rash” strategy was utilized, the dose escalation arm was associated with a higher response rate (30% versus 38%) but not a superior overall survival (OS).18 This pharmacodynamics clinical observation is an interesting phenomenon of limited clinical utility.19
EGFR mutation and cell surface expression
Initial clinical trials with cetuximab limited enrollment to those patients whose tumors demonstrated expression of EGFR by immunohistochemistry. Subsequent subset analysis of these studies demonstrated that the degree of EGFR expression by immunohistochemistry did not correlate with OS or objective response rate.20 Unlike non-small-cell lung cancer,21 de novo tumor EGFR mutations are uncommon in CRC (being present in less than 1% of the cases) and when present, usually do not represent activating mutations and are not predictors of response to cetuximab.22,23
EGFR gene copy number
In a small number of patients (31) treated with cetuximab or panitumumab (a fully humanized anti-EGFR monoclonal antibody24 that binds at a different, albeit partially overlapping, epitope than cetuximab25), it was noted that of the nine patients who responded to anti-EGFR monoclonal antibody therapy, eight had an increased EGFR copy number, as measured by fluorescent in situ hybridization (FISH) compared with only one of 21 of the nonresponders.26 A larger retrospective study evaluating 85 patients who received cetuximab monotherapy (5), or in combination with irinotecan (77) or oxaliplatin (3), and utilizing a scoring system (a positive score representing three or greater signals per nucleus or gene amplification), demonstrated that there was a statistical trend toward improved OS in time to progression and OS in those patients whose tumors were EGFR FISH positive.27 Limitations to the utility of EGFR gene copy number include significant within-sample and within-patient heterogeneity, and the lack of accepted cutoff values for increase gene copy number.21 Table 1 summarizes the studies evaluating EGFR gene copy number as a biomarker of anti-EGFR efficacy.
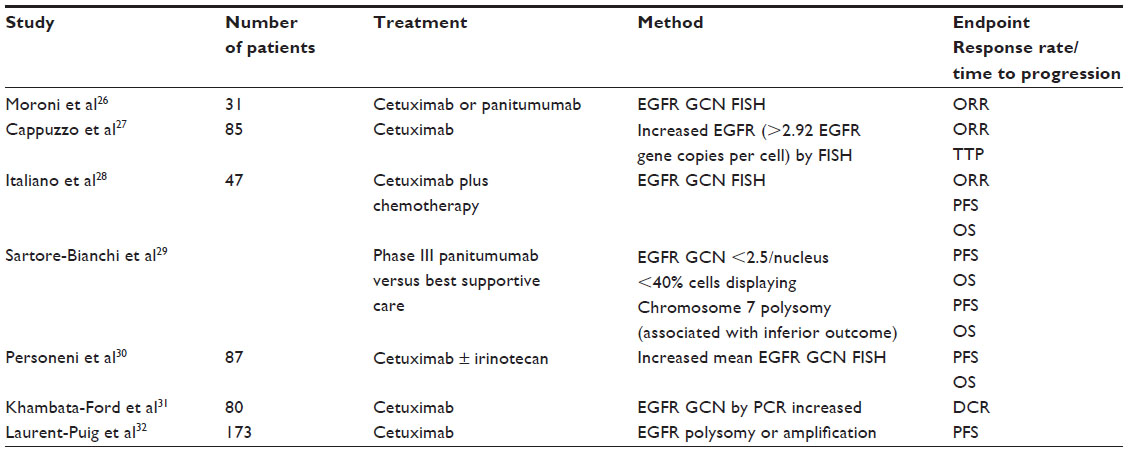 | Table 1 EGFR GCN by FISH as a biomarker of cetuximab sensitivity |
Whereas EGFR amplification is easily identified by FISH, and is associated with improved outcome with anti-EGFR antibodies, it occurs in less than 5% of patients. Rather than increased gene copy number of EGFR, the non-increase may be a more accurate negative biomarker (ie, predictor of lack of efficacy of anti-EGFR therapy), in that the majority of CRC patients who respond to anti-EGFR therapy have an increased EGFR copy number, but a minority of patients with tumors with an increased EGFR copy number respond to therapy. Patients whose tumors have a high EGFR copy number may not respond to anti-EGFR therapy due to downstream activation of KRAS, BRAF, or PIK3C2A.28 Tumor EGFR gene copy number was noted to vary among different centers in one study, demonstrating the need for standardization of testing.33
Tumor KRAS status
As mentioned above, constitutive activation of signaling pathways downstream of the EGFR may lead to resistance of anti-EGFR strategies.34,35 Tumor KRAS mutations are present in approximately 40% of CRC;36 in codon 12, the mutations most commonly seen are G12C, G12R, G12S, G12A, G12D, and G12V, and in codon 13, the most commonly identified mutations are G13C, G13R, G13S, G13A, G13D, and G13R. Consistent with the belief that downstream constitutive activation of EGFR signaling would confer resistance to anti-EGFR monoclonal antibody therapy, an initial study demonstrated that the presence of an activating tumor KRAS mutation was a negative biomarker of cetuximab efficacy.37 Multiple large tissue samples correlated with tumor response and outcome with anti-EGFR therapy have confirmed this observation (Table 2).31,37–44 Although never confirmed in a prospective randomized trial, all of these retrospective tissue studies suggest that tumors with a KRAS codon 12 mutation are resistant to anti-EGFR therapy, particularly in comparison with KRAS wild-type tumors. Consistent with this data, use of cetuximab and panitumumab, by the manufacturer’s label, is restricted to patients with wild-type tumors. The data are less clear when patients who have a KRAS codon 13 mutation are analyzed separately. One analysis of patients treated with cetuximab suggests there is a statistical trend toward an OS and progression-free survival (PFS) benefit when cetuximab is added to chemotherapy (when compared with chemotherapy alone).45 A pooled analysis of three randomized studies of patients treated with panitumumab (either as monotherapy compared with best supportive care, or combined with FOLFIRI [folinic acid, 5-fluorouracil, and irinotecan] OR FOLFOX-4 [oxaliplatin, folinic acid, and 5-fluorouracil]) failed to demonstrate any panitumumab efficacy for patients with KRAS codon 13 tumors.46 At this time, the data are insufficient to recommend the use of anti-EGFR therapies in patients with KRAS codon 13 specific mutations. Most tissue analysis evaluating KRAS mutations by polymerase chain reaction (PCR) interrogate the tissue for codon 12 and 13 mutations; more recently, codon 61 and 146 KRAS mutations in CRC have been described. At this time, the prognostic significance, as well as the potential anti-EGFR predictive implications of KRAS codon 61 and 146 mutations are not known.47
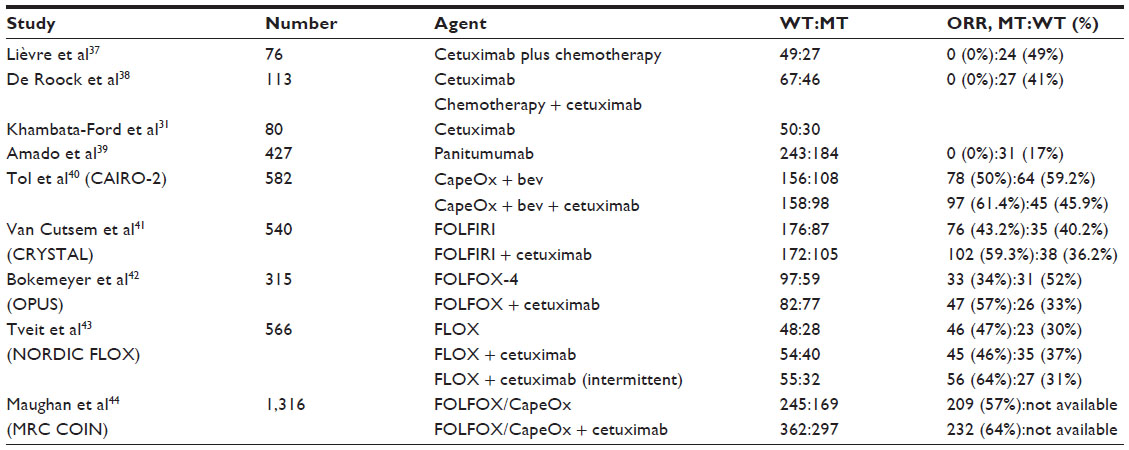 | Table 2 Studies demonstrating tumor KRAS mutation as a negative predictor of anti-EGFR response |
Newer technologies, with higher sensitivity to detecting KRAS mutations, are being evaluated, which may improve the predictive efficacy of KRAS as a negative biomarker. Evaluation of tumor KRAS codon 12 and 13 mutation by direct sequencing, in addition to MALDI TOF (matrix-assisted laser desorption/ionization-time-of-flight) mass spectrometry, mutant-enriched PCR, and engineered mutant-enriched PCR, in 111 patients identified an additional 13 patients with KRAS mutations beyond the initial 43 patients identified by direct sequencing.48 Tumor KRAS mutations have also been detected in plasma cell-free DNA from CRC patients;49 these studies suggest that if validated as a biomarker, it may be possible in the future to perform tumor KRAS analysis on plasma. Studies of plasma mutant KRAS levels, evaluated by quantitative PCR, suggest evaluation of tumor mutations in plasma may be a viable source of tissue for KRAS and BRAF biomarker analysis evaluation.50
Tumor BRAF mutation
Similar to KRAS, BRAF is a proto-oncogene, which is located downstream of EGFR receptor; BRAF mutations are present in approximately 10% of CRC cases, most commonly the V600E point mutation.51,52 Therefore, the impact of this mutant signal molecule on cetuximab sensitivity has also been investigated in patients with wild-type KRAS CRC. A retrospective analysis of 79 KRAS wild-type patients showed that BRAF protocol-oncogene (BRAF V600E) was associated with resistance to cetuximab treatment in treatment refractory metastatic CRC patients.53 Another retrospective analysis also demonstrated the resistance to cetuximab treatment in tumors with BRAF V600E mutation and wild-type KRAS.54 Despite these preliminary studies supporting the fact that the presence of a BRAF mutation was a negative predictor of response to cetuximab, a large randomized Phase III clinical trial (the “COIN” trial), which evaluated the addition of the cetuximab to conventional chemotherapy (CapeOx [capecitabine plus oxaliplatin]) as a first-line treatment of advanced stage CRC patients, did not show any relationship between OS and PFS dependent on tumor BRAF mutation status.44 The difference observed in these clinical trials could be due to different treatment settings (palliative treatment compared with first-line treatment). Thus, the role of the BRAF mutation as a negative predictor of cetuxmab efficacy is continuing to be studied.
Tumor NRAS mutation
Following the investigation of the biomarkers described above, the impact of other EGFR-related genes such NRAS was evaluated in clinical trials to understand its role in predicting survival. NRAS was found to be mutated in approximately 2.6%–4.0% of CRC cases.55 A retrospective analysis of 773 tumor samples from multiple centers showed that NRAS mutation was a negative predictor for the cetuximab response in patients treated with cetuximab plus chemotherapy.56 Another prospective study did not find any survival relationship between NRAS or KRAS mutations and cetuximab treatment.32 Because of the lack of convincing data in the literature, the precise biomarker implications of the presence of a tumor NRAS on cetuximab response is not known at this time.
PIK3CA mutation
PIK3CA is also a signal molecule of EGFR, and a mutant form has been suggested to be involved in carcinogenesis in many tumors.57 Since PIK3CA mutation may also change the signal transduction of EGFR, cetuximab efficacy was also investigated in metastatic CRC patients with PIK3CA mutation. Similar to KRAS, activating mutation of PIK3CA was found to be resistant to anti-EGFR treatment. A cohort of the 110 patients showed that none of the patients with PI3KCA mutation had objective response to either panitumumab or cetuximab.58 In another study, the authors reported that PIK3CA exon 9 mutation showed significantly less response to cetuximab and shorter PFS compared with wild-type PIK3CA in CRC patients.54 Although current available data suggest that both PIK3CA mutation and decreased expression of PTEN cause resistance to cetuximab therapy, larger randomized prospective clinical trials are necessary to assess possible biomarker utility of these genes.
Immunoglobulin G Fcγ receptor polymorphisms
In vitro studies have demonstrated that cetuximab can induce antibody-dependent cellular cytotoxicity by activating immune system elements in various tumors regardless of KRAS mutation status.57 Initial clinical study results have pointed to a possible relationship between immunoglobulin G Fcγ receptor polymorphisms and cetuximab response;58 and recent large pooled data failed to demonstrated any survival association in metastatic CRC patients.59 In the latter study, the authors reported marginally better disease control in patients with KRAS-mutated tumors with favorable polymorphism, suggesting that the antibody-dependent cellular cytotoxicity occurring in the macrometastatic disease has very limited effect on the efficacy of cetuximab therapy.
Tumor amphiregulin and epiregulin gene expression
Since the anti-EGFR therapy targets the growth signaling in cancer cells, the impact of the external growth signal mediators has been investigated in CRC; when secreted in a paracrine fashion by tumor cells, it plays a role in promoting tumor progression. A study of 110 patients tested the role of epiregulin and amphiregulin in metastatic CRC patients who were treated with cetuximab monotherapy.31 Patients with metastatic CRC that have high expression of epiregulin and amphiregulin in their tumors showed better response to cetuximab therapy (P=0.000015 and P=0.000025, respectively), suggesting that local growth signals are also involved in the disease progression, and this paracrine and autocrine process can be reversed by blocking the EGFR via monoclonal antibodies to induce tumor regression.31 Another study also investigated these two ligands of EGFR in metastatic colorectal patients by measuring the gene expression in primary tumors by stratifying according to the KRAS mutation status.60 This study also demonstrated the correlation between higher growth hormone ligands and better PFS, but only in patients with wild-type KRAS mutation. There was no similar benefit in patients whose tumor was mutant-KRAS, indicating that KRAS-mutant CRC cells do not depend on local growth signals to survive and progress.
Further considerations
Recent studies have suggested that analysis of multiple mutations in the RAS/RAF kinase pathway may be more sensitive in determining those patients likely to be resistant to anti-EGFR therapy. In the Fire-3 (AIO KRKI-0306) Phase III trial, comparing FOLFIRI combined with either cetuximab or bevacizumab, patients underwent analysis of KRAS, NRAS, and BRAF mutations; in patients who received FOLFIRI plus cetuximab, the RAS wild-type subgroup (KRAS, BRAF, and NRAS), the objective response rate was 76% compared with 42.9%.61 These studies suggest that multiple biomarkers of the RAS pathway may be necessary to optimally identify patients unlikely to benefit from anti-EGFR therapy. Furthermore, additional studies have demonstrated that acquired resistance to anti-EGFR therapy may be secondary to clonal selection of tumors with mutations in the EGFR receptor ectodomain.62,63 KRAS amplification may also contribute to primary resistance to anti-EGFR monoclonal antibodies.64
Conclusion
The current evidence from preclinical and clinical studies indicates that cetuximab as a monoclonal antibody has a potential reversing effect on EGFR-mediated growth signals in CRC cells and induces tumor suppression, and as a consequence, an increase in the survival of metastatic CRC patients. However, these benefits are observed in a limited number of patients with advance stage disease and certain pathological properties. Patients with KRAS mutations, specifically with KRAS codon 12 mutation, are unlikely to benefit from cetuximab treatment. While the data for KRAS p.G13D mutation is controversial, current guidelines do not recommend the use of this agent in patients whose tumor harbors this mutation. Accumulating evidence also suggests that mutations in other signal transduction molecules involved in EGFR signaling might also have an impact on cetuximab therapy outcomes, yet more evidence is required to establish the biomarker utility of these molecules. Available data also suggest that metastatic CRC patients with higher expression of local growth factor, such as amphiregulin and epiregulin and increased EGFR gene copy number, most probably benefit from monoclonal targeting in the absence of KRAS mutation.
Since molecularly targeting treatment by monoclonal antibodies results in a highly expensive economic burden, defining useful biomarkers to target the correct patient population is mandatory. The selection of patients who may potentially benefit from this agent not only decreases health costs but also avoids the side effects resulting from the use of cetuximab in patients who would not receive benefit from such therapy. Although molecular testing itself is associated with high health costs, the use of molecular biomarkers, by more carefully selecting patients for therapy, with higher certainty of efficacy, may lead to more cost-effective medical care.
Future studies may also look at different molecular subtypes of CRC, as identified by tumor gene expression profiling, and determine response to anti-EGFR monoclonal antibody therapy, as well as other pathway-specific inhibitors.65
Overall, cetuximab has been shown to be effective in metastatic CRC patients with irinotecan-resistance and has superior outcomes as a single agent compared with best supportive care. The addition of cetuximab to conventional chemotherapy in adjuvant settings in metastatic CRC patients could also potentially provide survival benefits. Combining with other targeting agents which inhibit signal transducing molecules involved in EGFR signaling will likely increase the efficacy of cetuximab treatment. However, the recently discovered diversity of active network signaling pathways in cancer cells appears to be a challenge for scientists in cancer research. Studies of biomarker exploration will have to take into account interactions beyond the tumor and incorporate knowledge of the tumor microenvironment and the immune response to therapy into biomarker discovery.
Disclosure
The authors report no conflicts of interest in this work.
References
Goldberg RM. Cetuximab. Nat Rev Drug Discov. 2005;Suppl:S10–S11. | |
Normanno N, De Luca A, Bianco C, et al. Epidermal growth factor receptor signaling in cancer. Gene. 2006;366(1):2–16. | |
Scaltriti M, Baselga J. The epidermal growth factor receptor pathway: a model for targeted therapy. Clin Cancer Res. 2006;12(18):5268–5272. | |
Nicholson RI, Gee JM, Harper ME. EGFR and cancer prognosis. Eur J Cancer. 200;37 Suppl 4:S9–S15. | |
Resnick MB, Routhier J, Konkin T, Sabo E, Pricolo VE. Epidermal growth factor receptor, c-MET, beta-catenin, and p53 expression as prognostic indicators in stage II colon cancer: a tissue microarray study. Clin Cancer Res. 2004;10(9):3069–3075. | |
Lockhart AC, Berlin JD. The epidermal growth factor as a receptor target for colorectal cancer therapy. Semin Oncol. 2005;32:52–60. | |
Mendelsohn J. Targeting the epidermal growth factor receptor for cancer therapy. J Clin Oncol. 2002;20(Suppl 18):1S–13S. | |
Sato JD, Kawamoto, Le AD, et al. Biologic effects in vitro of monoclonal antibodies to human epidermal growth factor receptors. Mol Biol Med. 1983;1:511–529. | |
Kawamoto TK, Sato JD, Le A, et al. Growth stimulation of A431 cells by epidermal growth factor: identification of high-affinity receptors for epidermal growth factor by an anti-receptor monoclonal antibody. Proc Natl Acad Sci U S A. 1983;80:1337–1341. | |
Goldstein NI, Prewett M, Zuklys K, Rockwell P, Mendelsohn J. Biological efficacy of a chimeric antibody to the epidermal growth factor receptor in a human tumor xenograft model. Clin Cancer Res. 1995;1(11):1311–1318. | |
Prewett M, Hooper A, Bassi R, et al. Enhanced antitumor activity of anti-epidermal growth factor receptor monoclonal antibody IMC-C225 in combination with irinotecan (CPT-11) against human colorectal tumor xenografts. Clin Cancer Res. 2002;8:994–1003. | |
Saltz LB, Meropol NJ, Loehrer PJ Sr, et al. Phase II trial of cetuximab in patients with refractory colorectal cancer that express the epidermal growth factor receptor. J Clin Oncol. 2004;22(7):1201–1208. | |
Cunningham D, Humblett Y, Siena S, et al. Cetuximab monotherapy and cetuximab plus irinotecan in irinotecan-refractory metastatic colorectal cancer. N Eng J Med. 2004;351:337–345. | |
US Food and Drug Administration [homepage on the Internet]. Cetuximab. Silver Spring, MD: US Food and Drug Administration; 2007. Available from: http://www.fda.gov/AboutFDA/CentersOffices/OfficeofMedicalProductsandTobacco/CDER/ucm129241.htm. Accessed November 26, 2013. | |
Jonker DJ, O’Callaghan CJ, Karapetis CS, et al. Cetuximab for the treatment of colorectal cancer. N Engl J Med. 2007;357(20):2040–2048. | |
Clark G, Perez-Soler R, Siu L, et al. Rash severity is predictive of increased survival with erlotinib HCI. Proc Am Soc Clin Oncol. 2003;22:196 (abstract). | |
Saltz L, Rubin MS, Hochster H, et al. Acne-like rash predicts response in patients treated with cetuximab plus irinotecan in CPT-11 refractory colorectal cancer that expresses epidermal growth factor receptor. Clin Cancer Res. 2001;7:3766s (abstract). | |
Van Cutsem E, Tejpar S, Vanbeckevoort D, et al. Intrapatient cetuximab dose escalation in metastatic colorectal cancer according to the grade of early skin reactions: the randomized EVEREST study. J Clin Oncol. 2012;30(23):2861–2868. | |
Stintzing S, Lenz HJ. Targeted therapies: cetuximab dosing by rash – is the scaling of EVEREST meaningful? Nat Rev Clin Oncol. 2012;9:554–556. | |
Pippas AW, Lenz HJ, Mayer RJ, et al. Analysis of EGFR status in metastatic colorectal cancer patients treated with cetuximab monotherapy. J Clin Oncol. 2005;23:16S (abstract 3595). | |
Paez JG, Jänne PA, Lee JC, et al. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science. 2004;304(5676):1497–1500. | |
Ogino S, Meyerhardt JA, Cantor M, et al. Molecular alterations in tumors and response to combination chemotherapy with gefitinib for advanced colorectal cancer. Clin Cancer Res. 2005;11(18):6650–6656. | |
Metzger B, Chambeau L, Begon DY, et al. The human epidermal growth factor receptor gene in European patients with advanced colorectal cancer harbors infrequent mutations in its tyrosine kinase domain. BMC Med Genet. 2011;12:144. | |
Hoda D, Simon GR, Garrett CR. Targeting colorectal cancer with anti-epidermal growth factor receptor antibodies: focus on panitumumab. Ther Clin Risk Manag. 2008;4(6):1221–1227. | |
Voigt M, Braig F, Göthel M, et al. Functional dissection of the epidermal growth factor receptor epitopes targeted by panitumumab and cetuximab. Neoplasia. 2012;14(11):1023–1031. | |
Moroni M, Veronese S, Benvenuti S, et al. Gene copy number for epidermal growth factor receptor and clinical response to anti-EGFR treatment in colorectal cancer: a cohort study. Lancet Oncol. 2005;6(5):279–286. | |
Cappuzzo F, Finocchiaro G, Rossi E, et al. EGFR FISH assay predicts for response to cetuximab in chemotherapy refractory colorectal cancer patients. Ann Oncol. 2008;19(4):717–723. | |
Italiano A, Follana P, Caroli FX, et al. Cetuximab shows activity in colorectal cancer patients with tumors for which FISH analysis does not detect an increase in EGFR gene copy number. Ann Surg Oncol. 2008;15(2):649–654. | |
Sartore-Bianchi A, Moroni M, Veronese S, et al. Epidermal growth factor receptor gene copy number and clinical outcome of metastatic colorectal cancer treated with panitumumab. J Clin Oncol. 2007;25(22):3238–3245. | |
Personeni N, Fieuws S, Piessevaux H, et al. Clinical usefulness of EGFR gene copy number as a predictive marker in colorectal cancer patients treated with cetuximab: a fluorescent in situ hybridization study. Clin Cancer Res. 2008;14(18):5869–5876. | |
Khambata-Ford S, Garrett CR, Meropol NJ, et al. Expression of epiregulin and amphiregulin and K-ras mutation status predict disease control in metastatic colorectal cancer patients treated with cetuximab. J Clin Oncol. 2007;25(22):3230–3237. | |
Laurent-Puig P, Cayre A, Manceau G, et al. Analysis of PTEN, BRAF, and EGFR status in determining benefit from cetuximab therapy in wild-type KRAS metastatic colon cancer. J Clin Oncol. 2009;27(35):5924–5930. | |
Sartore-Bianchi A, Fieuws S, Veronese S, et al. Standardisation of EGFR FISH in colorectal cancer: results of an international interlaboratory reproducibility ring study. J Clin Pathol. 2012;65(3):218–223. | |
Chen J, Huang XF, Katsifis A. Activation of signal pathways and the resistance to anti-EGFR treatment in colorectal cancer. J Cell Biochem. 2010;111(5):1082–1086. | |
Vaughn CP, Zobell SD, Furtado LV, Baker CL, Samowitz WS. Frequency of KRAS, BRAF, and NRAS mutations in colorectal cancer. Genes Chromosomes Cancer. 2011;50(5):307–312. | |
Benvenuti S, Sartore-Bianchi A, Di Nicolantonio F, et al. Oncogenic activation of the RAS/RAF signaling pathway impairs the response of metastatic colorectal cancers to anti-epidermal growth factor receptor antibody therapies. Cancer Res. 2007;67(6):2643–2648. | |
Lièvre A, Bachet JB, Le Corre D, et al. KRAS mutation status is predictive of response to cetuximab therapy in colorectal cancer. Cancer Res. 2006;66(8):3992–3995. | |
De Roock W, Piessevaux H, De Schutter J, et al. KRAS wild-type state predicts survival and is associated to early radiological response in metastatic colorectal cancer treated with cetuximab. Ann Oncol. 2008;19(3):508–515. | |
Amado RG, Wolf M, Peeters M, et al. Wild-type KRAS is required for panitumumab efficacy in patients with metastatic colorectal cancer. J Clin Oncol. 2008;26(10):1626–1634. | |
Tol J, Koopman M, Cats A, et al. Chemotherapy, bevacizumab, and cetuximab in metastatic colorectal cancer. N Engl J Med. 2009;360(6):563–572. | |
Van Cutsem E, Köhne CH, Hitre E, et al. Cetuximab and chemotherapy as initial treatment for metastatic colorectal cancer. N Engl J Med. 2009;360(14):1408–1417. | |
Bokemeyer C, Bondarenko I, Hartmann JT, et al. Efficacy according to biomarker status of cetuximab plus FOLFOX-4 as first-line treatment for metastatic colorectal cancer: the OPUS study. Ann Oncol. 2011;22(7):1535–1546. | |
Tveit KM, Guren T, Glimelius B, et al. Phase III trial of cetuximab with continuous or intermittent fluorouracil, leucovorin, and oxaliplatin (Nordic FLOX) versus FLOX alone in first-line treatment of metastatic colorectal cancer: the NORDIC-VII study. J Clin Oncol. 2012;30(15):1755–1762. | |
Maughan TS, Seymour MT, Wilson RH, et al. Addition of cetuximab to oxaliplatin-based first-line combination chemotherapy for treatment of advanced colorectal cancer: results of the randomised phase 3 MRC COIN trial. Lancet. 2011;377(9783):2103–2114. | |
De Roock W, Jonker DJ, Di Nicolantonio F, et al. Association of KRAS p.G13D mutation with outcome in patients with chemotherapy-refractory metastatic colorectal cancer treated with cetuximab. JAMA. 2010;304(16):1812–1820. | |
Peeters M, Douillard JY, Van Cutsem E, et al. Mutant KRAS codon 12 and 13 alleles in patients with metastatic colorectal cancer: assessment as prognostic and predictive biomarkers of response to panitumumab. J Clin Oncol. 2013;31(6):759–765. | |
Sahin IH, Garrett CR. The heterogeneity of KRAS mutations in colorectal cancer and its biomarker implications: an ever-evolving story. Transl Gastrointest Cancer. 2013;2(3):1–4. | |
Molinari F, Felicioni L, Buscarino M, et al. Increased detection sensitivity for KRAS mutations enhances the prediction of anti-EGFR monoclonal antibody resistance in metastatic colorectal cancer. Clin Cancer Res. 2011;17(14):4901–4914. | |
Spindler KG, Appelt AL, Pallisgaard N, Andersen RF, Jakobsen A. KRAS-mutated plasma DNA as predictor of outcome from irinotecan monotherapy in metastatic colorectal cancer. Br J Cancer. Epub November 21, 2013. | |
Spindler KL, Pallisgaard N, Vogelius I, Jakobsen A. Quantitative cell-free DNA, KRAS, and BRAF mutations in plasma from patients with metastatic colorectal cancer during treatment with cetuximab and irinotecan. Clin Cancer Res. 2012;18(4):1177–1185. | |
Fransén K, Klintenäs M, Osterström A, et al. Mutation analysis of the BRAF, ARAF and RAF-1 genes in human colorectal adenocarcinomas. Carcinogenesis. 2004;25(4):527–533. | |
Nagasaka T, Sasamoto H, Notohara K, et al. Colorectal cancer with mutation in BRAF, KRAS, and wild-type with respect to both oncogenes showing different patterns of DNA methylation. J Clin Oncol. 2004;15(22):4584–4594. | |
Di Nicolantonio F, Martini M, Molinari F, et al. Wild-type BRAF is required for response to panitumumab or cetuximab in metastatic colorectal cancer. J Clin Oncol. 2008;26(35):5705–5712. | |
De Roock W, Claes B, Bernasconi D, et al. Effects of KRAS, BRAF, NRAS, and PIK3CA mutations on the efficacy of cetuximab plus chemotherapy in chemotherapy-refractory metastatic colorectal cancer: a retrospective consortium analysis. Lancet Oncol. 2010;11(8):753–762. | |
Samuels Y, Velculescu VE. Oncogenic mutations of PIK3CA in human cancers. Cell Cycle. 2004;3(10):1221–1224. | |
Sartore-Bianchi A, Martini M, Molinari F, et al. PIK3CA mutations in colorectal cancer are associated with clinical resistance to EGFR-targeted monoclonal antibodies. Cancer Res. 2009;69(5):1851–1857. | |
Kimura H, Sakai K, Arao T, Shimoyama T, Tamura T, Nishio K. Antibody-dependent cellular cytotoxicity of cetuximab against tumor cells with wild-type or mutant epidermal growth factor receptor. Cancer Sci. 2007;98(8):1275–1280. | |
Zhang W, Gordon M, Schultheis AM, et al. FCGR2A and FCGR3A polymorphisms associated with clinical outcome of epidermal growth factor receptor expressing metastatic colorectal cancer patients treated with single-agent cetuximab. J Clin Oncol. 2007;25(24):3712–3718. | |
Geva R, Jensen B, Fountzilas G, et al. An international consortium study in chemorefractory metastatic colorectal cancer patients to assess the impact of FcGR polymorphisms on cetuximab efficacy. J Clin Oncol. 2011;29:3528 (abstract). | |
Jacobs B, De Roock W, Piessevaux H, et al. Amphiregulin and epiregulin mRNA expression in primary tumors predicts outcome in metastatic colorectal cancer treated with cetuximab. J Clin Oncol. 2009;27:5086–5074. | |
Stintzing S, Jung A, Rossius L, et al. Analysis of KRAS/NRAS and BRAF mutations in FIRE-3: a randomized phase III study of FOLFIRI plus cetuximab or bevacizumab as first-line treatment for wild-type KRAS (exon 2) metastatic colorectal cancer patients. European Cancer Congress 2013;LBA17 (abstract). | |
Misale S, Misale S, Yaeger R, et al. Emergence of KRAS mutations and acquired resistance to anti-EGFR therapy in colorectal cancer. Nature. 2012;486(7404):532–536. | |
Diaz LA Jr, Williams RT, Wu J, et al. The molecular evolution of acquired resistance to targeted EGFR blockade in colorectal cancers. Nature. 2012;486(7404):537–540. | |
Valtorta E, Misale S, Sartore-Bianchi A, et al. KRAS gene amplification in colorectal cancer and impact on response to EGFR-targeted therapy. Int J Cancer. 2013;133(5):1259–1265. | |
Sadanandam A, Lyssiotis CA, Homicsko K, et al. A colorectal cancer classification system that associates cellular phenotype and responses to therapy. Nat Med. 2013;19(5):619–625. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.