Back to Journals » The Application of Clinical Genetics » Volume 19
Precise Reproductive Counseling Enabled by Long-Read Sequencing in a Case of a F8 Intron 1 Inversion and Duplication
Authors Yan Y, Jin P, Xu L, Ma Y, Dong M
Received 3 March 2026
Accepted for publication 8 June 2026
Published 17 June 2026 Volume 2026:19 606654
DOI https://doi.org/10.2147/TACG.S606654
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Martin Maurer
Yuying Yan,1 Pengzhen Jin,2 Lidan Xu,1 Ya Ma,1 Minyue Dong1,2
1Department of Reproductive Endocrinology, Women’s Hospital, Zhejiang University School of Medicine, Hangzhou, Zhejiang, 310000, People’s Republic of China; 2Department of Reproductive Genetics, Women’s Hospital, Zhejiang University School of Medicine, Hangzhou, Zhejiang, 310000, People’s Republic of China
Correspondence: Minyue Dong, Email [email protected]
Purpose: To demonstrate the clinical value of integrating next-generation sequencing (NGS) with long-read sequencing (LRS) for resolving complex F8 variants and guiding personalized reproductive strategies in Haemophilia A (HA).
Patients and Methods: A patient with a history of three adverse pregnancy outcomes underwent comprehensive preconception genetic evaluation. NGS-based carrier screening initially excluded common single-gene disorders but flagged complex variants in the F8 gene. Subsequent LRS was employed to characterize the specific structural variations.
Results: NGS screening excluded 155 single-gene disorders and normal FMR1 repeats. LRS confirmed F8 intron 1 inversion (Inv1) and a duplication variant, while ruling out intron 22 inversion. The patient was identified as an asymptomatic female carrier. Based on this diagnosis, reproductive counseling recommended spouse testing, preimplantation genetic testing for monogenic diseases (PGT-M) combined with aneuploidy screening (PGT-A), and prenatal diagnosis.
Conclusion: This case underscores that while NGS is an effective screening tool, its limitations in detecting structural variations necessitate a stepwise diagnostic approach. Integrating LRS was indispensable for resolving complex F8 variants, transforming ambiguous genetic signals into precise diagnoses. This precision serves as the cornerstone for accurate risk assessment and empowers couples with informed reproductive options, exemplifying a “personalized reproductive blueprint”.
Keywords: haemophilia A, F8 gene, long-read sequencing, preconception genetic screening, structural variant, reproductive strategy
Introduction
Haemophilia A (HA), also known as factor VIII (FVIII) deficiency, is an X-linked recessive bleeding disorder characterized by insufficient synthesis or functional impairment of coagulation factor VIII (FVIII) encoded by the F8 gene.1 With a reported prevalence of 1 in 5000–10,000 males worldwide, HA imposes a significant burden on affected individuals and their families.2 Clinically, patients present with spontaneous bleeding in joints, muscles, internal organs, and deep tissues, or prolonged bleeding following minor trauma, tooth extraction, or surgery.3,4 Delayed wound healing and recurrent bleeding episodes are also common manifestations, with the severity and frequency of bleeding closely correlating with the residual FVIII activity (FVIII:C). HA is classified into three subtypes: severe (FVIII:C < 1%), moderate (FVIII:C 1%–5%), and mild (FVIII:C 5%–25%).5 Notably, women are usually heterozygous carriers of pathogenic F8 variants and exhibit mild bleeding symptoms, rendering them symptomatic carriers.
The F8 gene, located at Xq28, encodes FVIII and comprises 26 exons spread over a large genomic region, rendering it highly susceptible to a diverse spectrum of mutations. Its mutational landscape is highly heterogeneous, encompassing point mutations (missense, nonsense, splice-site), small insertions/deletions, and large structural rearrangements. Pathogenic variants associated with HA have been identified across nearly all exons of the F8 gene, with intron 22 inversion (Inv22) and intron 1 inversion (Inv1) being the most prevalent structural rearrangements.6 Inv22 accounts for 40%–50% of severe HA cases, while Inv1 contributes to 2%–5% of cases.7,8 Molecular genetic analysis of the F8 gene is crucial for familial risk assessment, carrier identification, and reproductive planning. Conventional detection methods, including Sanger sequencing of coding regions, Inv22/Inv1 screening, and next-generation sequencing (NGS), have identified numerous pathogenic F8 variants. However, approximately 1%–2% of severe HA patients remain genetically undiagnosed due to hidden pathogenic variants, such as deep intronic variants or complex structural variants (SVs) that evade conventional testing.9 This highlights the need for integrated molecular testing strategies to capture the high allelic heterogeneity of the F8 gene. The advent of long-read sequencing (LRS) technologies has revolutionized the detection of such variants by generating reads long enough to span repetitive regions and directly resolve complex genomic rearrangements.10 Consequently, a comprehensive diagnostic strategy that integrates broad carrier screening with advanced tools like LRS is becoming increasingly essential for precise genetic counseling.
For affected individuals and carriers, the absence of a definitive cure underscores the critical importance of accurate genetic diagnosis in guiding reproductive choices. Preconception carrier screening, coupled with a clear molecular diagnosis, forms the foundation for discussing options such as preimplantation genetic testing (PGT) to prevent the transmission of the pathogenic variant or prenatal diagnosis (PND) for informed pregnancy management. This report details the application of a tiered genomic approach—from expanded carrier screening to targeted LRS—in the case of a female carrier of a complex F8 structural variant, highlighting its indispensable role in crafting a precise and personalized reproductive roadmap.
This study focused on a 32-year-old female, who presented to our reproductive genetics clinic for pre-pregnancy consultation. Her primary concern stemmed from a history of three consecutive pregnancy losses, prompting a thorough evaluation. The first pregnancy ended in a medical abortion, and no embryonic genetic analysis was performed. The second pregnancy resulted in a missed miscarriage at 8 weeks of gestation; chromosomal analysis of the products of conception (POC) revealed a trisomy 14 karyotype. The third pregnancy also ended as a missed miscarriage at 9 weeks, with POC analysis identifying a triploid karyotype. Unfortunately, the sex of the three lost pregnancies was not determined. This patient reported no personal history of abnormal bleeding tendencies, such as prolonged bleeding after minor injuries, menorrhagia, or easy bruising. She had no known joint issues or other symptoms suggestive of a bleeding disorder. Crucially, there was no reported family history of bleeding disorders, haemophilia, or recurrent pregnancy loss across three generations, suggesting a sporadic presentation. As part of a comprehensive pre-pregnancy carrier screening panel, she was tested for 155 single-gene disorders, including haemophilia A, and for Fragile X syndrome. While the panel reported no pathogenic or likely pathogenic variants for the diseases screened, a technical note indicated the presence of a complex variant pattern in the F8 gene, necessitating further investigation.
Materials and Methods
Ethical Statement and Study Design
This was a retrospective case analysis conducted in accordance with the Declaration of Helsinki. The study protocol was reviewed and approved by the Institutional Review Board of Women’s Hospital, Zhejiang University School of Medicine (Approval No: IRB-20260082-R).
Carrier Screening for 155 Monogenic Disorders and Haemophilia A
Genomic DNA was extracted from peripheral blood leukocytes using the QIAamp DNA Blood Mini Kit (Qiagen, Hilden, Germany). Expanded carrier screening was performed using a targeted next-generation sequencing (NGS) panel. Briefly, a custom-designed set of oligonucleotide probes was used to capture the exonic and flanking intronic regions of 148 genes associated with 156 common autosomal recessive and X-linked conditions. The captured libraries were prepared according to the manufacturer’s protocol and sequenced on an NovaSeq 6000 platform to achieve a minimum mean coverage depth of 100x. Bioinformatic analysis involved alignment to the human reference genome (GRCh38), variant calling, and annotation. Variants were filtered and interpreted according to the American College of Medical Genetics and Genomics (ACMG) guidelines, focusing on the identification of pathogenic or likely pathogenic variants associated with the screened disorders.
A Fragile X Syndrome (FMR1 Gene) Carrier Screening
Analysis of the CGG trinucleotide repeat expansion in the 5′ untranslated region of the FMR1 gene was performed to assess the risk for Fragile X syndrome. The assay employed Triplet Repeat Primed Polymerase Chain Reaction (TP-PCR) combined with capillary electrophoresis. Briefly, genomic DNA was amplified using a fluorescently labeled primer set specifically designed to anneal within and flanking the CGG repeat region. The PCR products were then separated and sized by capillary electrophoresis on an ABI 3500xl Genetic Analyzer (Applied Biosystems, USA). The number of CGG repeats was determined by comparison with internal size standards. Alleles were classified as normal (5–44 repeats), intermediate/grey zone (45–54 repeats), premutation (55–200 repeats), or full mutation (>200 repeats) based on established clinical thresholds.
Analysis of F8 Gene Intronic Inversions and Duplication Validation
Targeted long-read sequencing using the CycloneSEQ nanopore platform for comprehensive structural variant analysis. We used a targeted long-read sequencing assay based on the CycloneSEQ nanopore sequencing platform (CycloneSEQ G100-ER) to comprehensively detect all variant types in the F8 gene, including single nucleotide variants (SNVs), deletions, and large structural variants (SVs) such as inversions and duplications. This approach employs a long-read indexed PCR method using gene-specific long-range PCR primers to amplify the F8 gene regions of interest (eg, intron 1 homologous region and exon 14) The resulting amplicons were subjected to nanopore sequencing on the CycloneSEQ G100-ER platform, enabling simultaneous detection of all classes of variants, including copy number variants (CNVs) and complex structural rearrangements. The performance of this method was assessed through orthogonal validation using Sanger sequencing of breakpoint-spanning products.
The presence of the common F8 gene intron 1 (Inv1) and intron 22 (Inv22) inversions was investigated using an established long-range PCR (LR-PCR) protocol. For Inv22 detection, a set of primers was used to amplify characteristic fragment patterns from the int22h homologous regions, allowing discrimination between normal, inverted, and carrier states. For Inv1 detection, a separate LR-PCR assay was performed using primers spanning the int1h homologs. PCR products were analyzed by agarose gel electrophoresis (0.8–1.0%), and fragment sizes were compared to positive and negative controls to determine the inversion status.
Following the identification of a potential complex structural variant from the initial screening, a specific PCR-based assay was designed for orthogonal validation. Primers were designed to flank the putative duplicated region (using primers H1F, H1R, H2/3R, H3F or a custom set). PCR amplification was performed, and the products were analyzed by agarose gel electrophoresis. The presence of an additional or abnormally sized fragment, compared to a wild-type control, confirmed the existence of the duplication.
Results
The Value and Limitations of Broad-Spectrum Screening: The “Scout” Role of Next-Generation Sequencing
Second-generation sequencing (NGS)-based expanded carrier screening serves as a high-efficiency, broad-spectrum reconnaissance tool in reproductive planning. It simultaneously assays both partners for pathogenic mutations associated with hundreds of autosomal recessive and X-linked conditions, providing a crucial pre-conception risk assessment. In the case of the patient, this panel effectively ruled out her and her partner as carriers for the 155 targeted monogenic disorders, significantly lowering their risk of conceiving a child affected by these conditions. Additionally, screening for Fragile X syndrome showed a normal result (CGG repeat number of 30/30 in the FMR1 gene), excluding this common genetic cause of intellectual disability and premature ovarian insufficiency (Figure 1). These “negative” findings are of substantial value, offering reassurance for a wide array of potential hereditary risks.
 |
Figure 1 TP PCR results of the siblings. TP PCR and fluorescence capillary electrophoresis. The number of CGG repeats in the 5-prime non-coding region of the FMR1 gene of the subject is 30/30, corresponding to the normal type. The green box marks the main peak position at 300.9 bp, corresponding to the normal allele with 30 CGG repeats. |
However, the screening report included a critical technical note: the detection of a complex variant pattern in the F8 gene, which encodes coagulation factor VIII. This finding required careful interpretation. While F8 variants are causative for Hemophilia A, an X-linked recessive bleeding disorder, the patient’s status as a heterozygous female carrier typically does not confer a personal bleeding phenotype. The principal reproductive implication is the 50% risk of transmitting the variant to her offspring: sons inheriting the variant would be affected, and daughters would be carriers. It is vital to emphasize that this X-linked variant has no direct causal relationship with the aneuploidies (trisomy 14 and triploidy) identified in her prior pregnancy losses, underscoring the necessity of precise, phenotype-guided variant interpretation.
This case highlights a key technical limitation of standard NGS panels: they are often inadequate for reliably detecting and resolving large structural variations (SVs), such as inversions, duplications, or deletions. The NGS result acted as an alert—signaling an anomaly in F8—but could not delineate the exact structural nature, precise breakpoints, or orientation of the variant. It was akin to a scout reporting unidentified activity without providing a detailed description. This ambiguity necessitated a definitive diagnostic follow-up to characterize the variant fully and assess its clinical significance for her reproductive journey.
Delving into the Genomic Architecture: Precise Resolution by Third-Generation Long-Read Sequencing
When next-generation sequencing identifies ambiguous complex variants, third-generation long-read sequencing (LR-sequencing), such as PCR combined with single-molecule sequencing, emerges as an indispensable “molecular microscope” to resolve structural ambiguities. Unlike short-read NGS, LR-sequencing generates DNA fragments spanning tens to hundreds of kilobase pairs, enabling direct visualization of large structural variants (SVs) such as inversions, duplications, and deletions with precise breakpoint mapping and orientation determination—capabilities critical for clarifying the nature of complex genetic alterations.
In this case, this advanced technical approach yielded definitive molecular insights (Figure 2A and B). PCR amplification combined with single-molecule LR-sequencing clearly confirmed two distinct SVs in the F8 gene: an intron 1 inversion (Inv1) in the heterozygous state and a duplication (DUP) variant, while ruling out the presence of the intron 22 inversion (Inv22). Using the LR-PCR amplicon coordinates mapped to GRCh38, the inversion breakpoints were delineated to two distinct regions within intron 1 of F8: one spanning chrX:154,880,332–154,890,316 (amplified by H1F-H1R, ∼9.98 kb) and another spanning chrX:155,376,441–155,388,707 (amplified by H2F-H23R, ∼12.27 kb). The duplication involved a segment estimated to be approximately 23.5 kb, based on the average read length of the H1F-H23R amplicon (∼9.35 kb) and the observation of two discontinuous alignment intervals (chrX:154,880,815–154,890,316 and chrX:155,454,662–155,464,272) in the long-read sequencing data, indicating a duplicated region with high sequence homology. The LR-sequencing data provided unambiguous evidence of the Inv1 breakpoint locations and the extent of the F8 gene duplication, eliminating the ambiguity inherent in NGS results. This step was irreplaceable: it delivered molecular diagnostic-grade precision that serves as the cornerstone for interpreting variant pathogenicity, defining inheritance patterns, and guiding subsequent genetic counseling. Without such precise characterization, all risk assessments would remain speculative, lacking a solid scientific foundation.
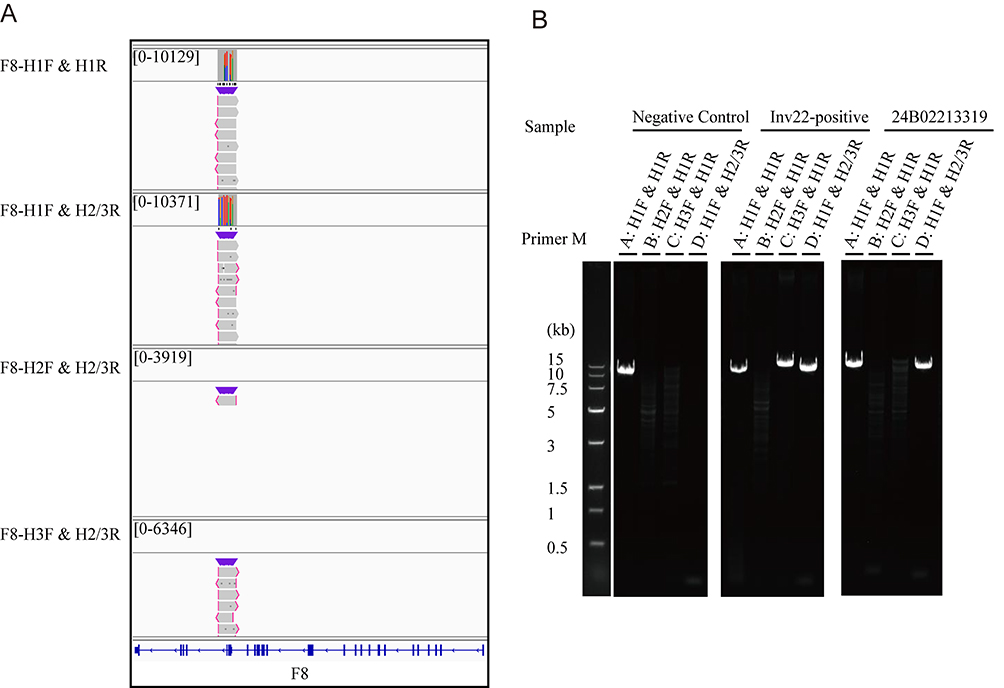 |
Figure 2 Molecular confirmation of the F8 structural variants. (A) An IGV chart depicting a segment of an inversion region (the 1st intron) identified through long-read sequencing. (B) PCR amplification and agarose gel electrophoresis are used to verify the repetitive variations, indicating that the sample contained the DUP variant type of the F8 gene. |
Clinical Significance of the F8 Variant and Reproductive Counseling
The F8 gene is well-recognized as the pathogenic driver of haemophilia A (HA), an X-linked recessive bleeding disorder. For this proband, as a female heterozygous carrier of F8 gene variants (Inv1 heterozygosity and duplication), her clinical status aligns with the typical inheritance pattern of X-linked recessive disorders: she exhibits no abnormal bleeding history (eg, menorrhagia, prolonged bleeding after surgery or trauma), indicating that the variants do not impair her own coagulation function This absence of symptomatic manifestations further supports the preliminary judgment that the F8 gene variants are not clinically impactful for her personal health, consistent with the observation that most female HA carriers remain asymptomatic or have mild, unrecognized bleeding tendencies. Notably, long-read sequencing results for patient’s parents were negative for the identified F8 gene variants (Figure 3), suggesting that these variants may be de novo, which is a key consideration in genetic risk assessment.
 |
Figure 3 Family pedigree. Family diagram. The symbols and labels are defined as: I-1 (father of the proband), I-2 (mother of the proband), and II-1 (the affected proband, index patient). The dot within the symbol denotes a carrier of the FMR1 gene variant. |
The detection of a complex F8 variant (Inv1 heterozygous + Duplication) in this case carries distinct clinical implications that diverge from her miscarriage phenotype and directly inform her future reproductive planning. The cornerstone of an accurate risk assessment was targeted F8 gene testing for the male partner. This step was critical for defining the offspring’s genetic risk profile. Two scenarios were outlined in genetic counseling: 1) If the partner carries no pathogenic F8 variant, the inheritance follows an X-linked pattern. 2) Although statistically remote, the possibility of the partner carrying a different F8 variant (eg, a missense mutation) necessitated exclusion, as this would confer a significantly different and more complex risk of compound heterozygosity in any female offspring.
Based on this refined risk, a multi-tiered reproductive strategy was formulated. The most proactive option presented was Preimplantation Genetic Testing for Monogenic Disorders (PGT-M). This involves in vitro fertilization (IVF) followed by biopsy of developing embryos. The biopsied cells are analyzed for the specific maternal F8 Inv1/DUP variant, allowing for the selective transfer of embryos that did not inherit this pathogenic allele, thereby preventing its transmission to the next generation. This approach directly addresses the genetic risk before pregnancy is established.
Concurrently, the indispensable role of prenatal diagnosis (PND) was emphasized as a complementary or alternative safeguard. It was counseled that in the event of a natural conception, an invasive diagnostic procedure such as chorionic villus sampling (CVS) or amniocentesis would be recommended. Genetic analysis of the fetal DNA would provide a definitive diagnosis regarding the inheritance status of the F8 variant, enabling informed decision-making during pregnancy. This serves as the diagnostic “gold standard” and a critical safety net.
This comprehensive reproductive plan was framed within the context of the proband’s completed comprehensive pre-conception screening. The normal results from the expanded carrier screening (155 genes) and Fragile X syndrome testing provided valuable, broader contextual reassurance, allowing the clinical team and the couple to concentrate management efforts specifically on the identified F8-related risk. This integrated approach underscores the modern paradigm of reproductive medicine, where layered genetic screening, precise diagnostics, and tailored interventions converge to enable informed family planning.
Discussion
In the present study, we utilized a combination of molecular diagnostic techniques to elucidate the molecular abnormalities associated with a complex F8 structural variant in a female proband presenting for pre-pregnancy genetic counseling. Our investigation successfully identified a compound heterozygous variant consisting of an Intron 1 inversion (Inv1) and a concurrent duplication (DUP) within the F8 gene.
Inversions represent a prevalent class of structural rearrangements in the human genome. While many are benign, specific inversions involving critical genes are well-established causes of disease, such as in hemophilia A (HA), Hunter syndrome, and mucopolysaccharidosis type II.11,12 The pathogenic mechanism often involves the disruption of gene coding sequences or critical regulatory elements. In this case, the identified Inv1 is a known but less common cause of severe HA compared to the frequent Intron 22 inversion (Inv22), which accounts for approximately 40–50% of severe cases.12 The co-occurrence of a DUP variant alongside Inv1 presents a rare and complex mutational landscape. The F8 gene, located at Xq28, exhibits high allelic heterogeneity, with reported pathogenic variants including large deletions, intronic inversions (Inv1 and Inv22), missense, nonsense, splice-site, and frameshift mutations.13 The complexity of this variant underscores the challenge in achieving a complete genetic diagnosis using conventional methods alone.
It is estimated that 1–2% of patients with severe HA lack a genetic diagnosis following conventional testing pipelines.14 In such cases, cryptic pathogenic variants, such as deep intronic mutations or complex structural variations (SVs) in F8, are likely contributors. For the precise characterization of these elusive variants, long-read sequencing (LRS) technology offers distinct advantages over traditional short-read sequencing. LRS generates reads spanning tens to hundreds of kilobases, enabling the accurate phasing of alleles, resolution of repetitive genomic regions, and direct detection of large structural variants.15,16 As demonstrated in our proband, the definitive characterization of the compound Inv1/DUP variant was achieved through targeted LRS, moving the diagnosis from a generic “complex variant” signal to a precise molecular definition. This finding aligns with the work of Yuan et al, who employed LRS to resolve novel inversions and large deletions in F8 for families with severe HA where prior methodologies had failed17 The de novo origin of the variant in our proband, inferred from its absence in both parents, is a critical finding that confines the recurrence risk to her own offspring and significantly alters genetic counseling for the extended family. The proband and her partner are not consanguineous, excluding the possibility of increased recessive disorder risks from shared ancestral alleles.
For the asymptomatic female proband, the clinical implications of this finding are primarily reproductive. The absence of a personal bleeding history is consistent with the X-linked recessive inheritance pattern and the common occurrence of skewed X-chromosome inactivation in female carriers, which can mitigate phenotypic expression. However, the primary clinical imperative shifts to quantifying and managing the transmission risk. Each male offspring has a 50% risk of inheriting the variant and manifesting severe HA, while each female offspring has a 50% chance of being a carrier. It is crucial to distinguish this Mendelian risk from the proband’s history of euploid pregnancy losses (trisomy 14 and triploidy). These aneuploidies are typically sporadic events resulting from errors in gametogenesis or early post-zygotic division and are not etiologically linked to the F8 variant. This distinction is vital for comprehensive counseling, as it informs a reproductive strategy that must address two independent risks: the inheritance of the monogenic disorder and the background risk for fetal aneuploidy.
Consequently, a multifaceted reproductive management plan was formulated. Preimplantation genetic testing for monogenic disorders (PGT-M) was presented as a proactive strategy to prevent the vertical transmission of the F8 variant by selecting unaffected embryos for transfer.18 Furthermore, combining PGT-M with preimplantation genetic testing for aneuploidy (PGT-A) could simultaneously mitigate both identified risks, potentially improving the likelihood of a successful and healthy pregnancy. For any natural conception, vigilant prenatal diagnosis via chorionic villus sampling or amniocentesis remains the definitive “gold standard” for fetal genotyping, offering an opportunity for concurrent assessment of chromosomal copy number.
In conclusion, this case exemplifies the transformative impact of a tiered genomic diagnostic approach on reproductive counseling. It demonstrates that while expanded carrier screening serves as a vital, broad-spectrum sentinel, advanced tools like LRS are often indispensable for the definitive resolution of complex variants flagged by initial screens. This precise molecular diagnosis forms the essential foundation for accurate risk assessment and empowers couples with a spectrum of informed reproductive choices, from preimplantation interventions to targeted prenatal diagnostics. It affirms that contemporary reproductive medicine is evolving towards the creation of a “personalized reproductive roadmap”, where integrated genetic technologies translate complex genomic information into actionable and empowered family planning decisions.
Conclusion
This study underscores the critical value of integrating next-generation sequencing (NGS) and long-read sequencing (LRS) in addressing the challenges posed by F8 gene complex structural variants in preconception care. The successful identification of a complex F8 inversion and duplication via long-read sequencing not only resolved a previously undiagnosed case of hemophilia A but also fundamentally altered the reproductive counseling approach. By combining PGT-M for the F8 mutation with PGT-A for aneuploidy screening, we demonstrate a comprehensive, evidence-based strategy that addresses both monogenic disease risk and chromosomal stability—critical for patients with a history of recurrent pregnancy loss. This dual-mode approach facilitates informed decision-making, optimizes the likelihood of a healthy pregnancy, and underscores the necessity of personalized, technology-driven care in modern genetic counseling.
Abbreviations
F8, Coagulation factor VIII; HA, Haemophilia A; Inv1, Intron 1 inversion; NGS, Next-generation sequencing; PGT-A, Preimplantation genetic testing for aneuploidies; PGT-M, Preimplantation genetic testing for monogenic diseases; SVs, Structural variants.
Ethical Approval
Necessary ethical approval was obtained from the Ethics Committee of Women’s Hospital, Zhejiang University School of Medicine (No. IRB-20260082-R).
Acknowledgments
We extend our deepest gratitude to all participants and their families. We thank the team of Women’s Hospital, Zhejiang University School of Medicine.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This work was supported by the Medical and Health Science Program of Zhejiang Province (Grant No.2024KY1154).
Disclosure
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
1. Gao K, Kumar P, Cortez-Toledo E, et al. Potential long-term treatment of hemophilia A by neonatal co-transplantation of cord blood-derived endothelial colony-forming cells and placental mesenchymal stromal cells. Stem Cell Res Ther. 2019;10(1):34. doi:10.1186/s13287-019-1138-8
2. Bolton-Maggs PH, Pasi KJ. Haemophilias A and B. Lancet. 2003;361(9371):1801–9. doi:10.1016/S0140-6736(03)13405-8
3. Chowdary P, Carcao M, Kenet G, Pipe SW. Haemophilia. Lancet. 2025;405(10480):736–750. doi:10.1016/S0140-6736(24)02139-1
4. Mannucci PM, Tuddenham EG. The hemophilias--from royal genes to gene therapy. N Engl J Med. 2001;344(23):1773–1779. doi:10.1056/NEJM200106073442307
5. Srivastava A, Brewer AK, Mauser-Bunschoten EP, et al. Guidelines for the management of hemophilia. Haemophilia. 2013;19(1):e1–e47. doi:10.1111/j.1365-2516.2012.02909.x
6. Shi M, Ma Y, Peng X, et al. Clinical validation and application of targeted long-range polymerase chain reaction and long-read sequencing-based analysis for hemophilia: experience from a hemophilia treatment center in China. J Thromb Haemost. 2024;22(12):3431–3447. doi:10.1016/j.jtha.2024.08.013
7. Wang X, Hu W, Gao Y, Li D, Lu Y. Rapid genotyping of F8 intron 22 inversion by nested PCR based on long-distance PCR. J Thromb Thrombolysis. 2020;49(4):591–601. doi:10.1007/s11239-020-02043-5
8. Li L, Luo W, Huang L, et al. Application of capillary gel electrophoresis in detection of Factor VIII gene intron 22 inversion of hemophilia A. J Hematop. 2025;18(1):40. doi:10.1007/s12308-025-00655-5
9. Dericquebourg A, Jourdy Y, Fretigny M, et al. Identification of new F8 deep intronic variations in patients with haemophilia A. Haemophilia. 2020;26(5):847–854. doi:10.1111/hae.14134
10. Yang Y, Wang Y, Gao J. Genetic diagnosis and prenatal diagnosis of a rare FVIII family with haemophilia A. J Cell Mol Med. 2024;28(23):e70275. doi:10.1111/jcmm.70275
11. Bondeson ML, Dahl N, Malmgren H, et al. Inversion of the IDS gene resulting from recombination with IDS-related sequences is a common cause of the Hunter syndrome. Hum Mol Genet. 1995;4(4):615–621. doi:10.1093/hmg/4.4.615
12. Bagnall RD, Waseem N, Green PM, Giannelli F. Recurrent inversion breaking intron 1 of the factor VIII gene is a frequent cause of severe hemophilia A. Blood. 2002;99(1):168–174. doi:10.1182/blood.v99.1.168
13. Johnsen JM, Fletcher SN, Huston H, et al. Novel approach to genetic analysis and results in 3000 hemophilia patients enrolled in the my life, our future initiative. Blood Adv. 2017;1(13):824–834. doi:10.1182/bloodadvances.2016002923
14. Swystun LL, James PD. Genetic diagnosis in hemophilia and von Willebrand disease. Blood Rev. 2017;31(1):47–56. doi:10.1016/j.blre.2016.08.003
15. Ling X, Pan L, Li L, et al. Detection of hemophilia A genetic variants using third-generation long-read sequencing. Clin Chim Acta. 2024;562:119884. doi:10.1016/j.cca.2024.119884
16. Miller DE, Galey M, Fletcher SN, et al. Long-read sequencing resolves complex structural variants and identifies missing disease-causing variants in unsolved cases of hemophilia. Blood. 2022;2(10):e556–e564. doi:10.1182/blood-2022-169355
17. Yuan S, Hu L, Zhong J, et al. Genetic analysis and reproductive interventions for two rare families affected by severe haemophilia A. Haemophilia. 2025;31(1):148–155. doi:10.1111/hae.15140
18. Bui TMP, Tran VK, Nguyen TTH, et al. Preimplantation genetic testing (PGT) for hemophilia A: experience from one center. Taiwan J Obstet Gynecol. 2022;61(6):1009–1014. doi:10.1016/j.tjog.2021.12.007
 © 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.