Back to Journals » OncoTargets and Therapy » Volume 13
Pre-mRNA Processing Factor 8 Accelerates the Progression of Hepatocellular Carcinoma by Regulating the PI3K/Akt Pathway
Authors Wang S, Wang M, Wang B, Chen J, Cheng X, Sun X ![]()
Received 5 December 2019
Accepted for publication 2 March 2020
Published 26 May 2020 Volume 2020:13 Pages 4717—4730
DOI https://doi.org/10.2147/OTT.S241214
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Nicola Silvestris
Shouhan Wang,1 Min Wang,2 Bin Wang,1 Jiaqi Chen,1 Xianbin Cheng,1 Xiaodan Sun3
1Department of Hepatopancreatobiliary Surgery, Cancer Hospital of Jilin Province, Changchun, People’s Republic of China; 2Department of Pathology, Cancer Hospital of Jilin Province, Changchun, People’s Republic of China; 3Department of 2nd Gynecologic Oncology Surgery, Cancer Hospital of Jilin Province, Changchun, People’s Republic of China
Correspondence: Xiaodan Sun
Department of 2nd Gynecologic Oncology Surgery, Cancer Hospital of Jilin Province, No. 1018, Huguang Road, Changchun, People’s Republic of China
Email [email protected]
Background: The specific function of pre-mRNA processing factors (Prps) in human malignancies has not been yet investigated. The aim of the present study was to determine the impacts of Prp8 in a common human malignancy, hepatocellular carcinoma (HCC).
Materials and Methods: RT-qPCR and Western blotting were performed to measure the expression levels of Prp8 in various HCC cell lines and HCC tissues. A hepatic astrocyte line was transfected with a eukaryotic expression plasmid to overexpress Prp8. In addition, the endogenous expression level of Prp8 in HCC cells was silenced using a short hairpin RNA method, and the role of Prp8 on cell proliferation and migration was examined by Cell Counting Kit-8, wound healing assay and Transwell assays following knockdown in HCC cells, and overexpression in astrocytes.
Results: Upregulation of Prp8 expression was found to be associated with poor clinical outcomes in patients with HCC. The upregulation of Prp8 promoted cell viability, metastasis and the activity of the PI3K/Akt pathway in hepatic astrocytes cells and HCC cells. Interestingly, loss of Prp8 had no obvious impact on cell viability and migration in hepatic astrocytes, but significantly inhibit the cell malignancy of HCC cells. Functionally, the inhibition of the PI3K/Akt pathway reversed the increased cell viability and migration of HCC cells induced by Prp8 via inhibiting EMT process.
Conclusion: Collectively, the present results suggested that Prp8 served as a tumor promoter in HCC by targeting and regulating the PI3K/Akt pathway.
Keywords: pre-mRNA processing factor 8, phosphatidylinositol 3-kinase, protein kinase B, hepatocellular carcinoma
Introduction
Pre-mRNA splicing is essential for gene expression in all eukaryotes.1 In higher eukaryotes, such as mammals, ~95% of the nucleotides in the primary transcript (pre-mRNA) of a protein-encoding gene are introns.2 These introns need to be removed precisely by splicing before the mRNA can be transported from the nucleus into the cytoplasm, where it can be translated.3 Alternative splicing greatly expands the gene coding capacity and >60% of human genes are alternatively spliced.4 It is also becoming increasingly clear that alternative splicing is a fundamental component of eukaryotic gene regulation, influencing cell differentiation, development and many processes in the nervous system.5 A typical intron contains a conserved 5ʹ splice site (5ʹ ss), a branch point sequence (BPS) followed by a polypyrimidine tract (PYT), and a 3ʹ ss.6 Introns are removed through two transesterification reactions catalyzed by the spliceosome.5 The spliceosome contains five smalls nuclear RNAs (snRNAs), such as U1, U2, U4, U5 and U6 snRNAs, which form five small nuclear ribonucleoproteins (snRNPs) with their associated proteins, in addition to numerous other protein splicing factors.7 Notably, the total number of proteins in the spliceosome is more than 100.8 The formation of the E-complex involves the initial recognition of an intron by the spliceosome.5 The 5ʹ ss is recognized by U1 snRNP, whereas the BPS and PYT interact with other splicing factors. Subsequently, the U2 snRNP joins the spliceosome to form the a complex, which is followed by the recruitment of the U4/U6.U5 triple snRNP (tri-snRNP), forming the B complex.9 Extensive structural rearrangements occur at this stage to form the catalytically active B complex that mediated the first splicing step.10 After the first step reaction, the spliceosome repositions the substrate, allowing the second catalytic reaction and forming the C complex.11 The second reaction is followed by post-catalytic rearrangements to release the mature mRNA for the nuclear export, releasing the lariat intron, which will be degraded, and the snRNPs, which will be recycled.12
Errors in splicing contribute to >30% of human genetic disorders, including retinitis pigmentosa (RP), spinal muscular atrophy and myotonic dystrophy.13 RP is an autosomal dominant genetic disorder that leads to photoreceptor degeneration and vision impairment.14 Mutations or deletions of a number of splicing factors, including pre-mRNA processing factor 8 (Prp8), small nuclear ribonucleoprotein U5 subunit 200 (Brr2), Prp3 and Prp31, have been found to cause various subtypes of RP.15 These proteins are all components of the U4/U6.U5 tri-snRNP complex and are ubiquitously expressed in all tissues.16 Intriguingly, mutations or heterozygous deletion of these splicing factors affect primarily photoreceptors, which are one of the most metabolically active cell types in the body, and have no obvious effect on any other organs.17 Furthermore, a 90% reduction in the protein level of splicing factor 3b subunit 1 (SF3b1), a key component of the U2 snRNP complex, leads to developmental defects in very specific organs instead of lethality or widespread defect in many organs, highlighting the cell type-specific effects of inhibiting the basal splicing machinery.18 Therefore, the present study hypothesized that inhibiting the spliceosomal activity may selectively inhibit cancer cell growth or survival with limited effect on normal cells.16
Approaches aimed to control the metastasis and recurrence of hepatocellular carcinoma (HCC) has been found to be insufficient in the treatment of this disease, and no currently available treatments have been identified to be efficient against metastatic HCC.19 Our present study identified that Prp8 was upregulated in HCC; however, to the best of our knowledge, there are no studies investigating the impact of Prp8 on the tumorigenesis of malignant HCC. Therefore, the aim of the present study was to investigate the expression pattern of Prp8, its role and its underlying mechanisms in HCC malignancy. Importantly, the regulatory mechanism of the Prp8/PI3K/Akt axis in HCC remains unclear. Therefore, the dysregulation of Prp8 and its regulatory mechanism in HCC were examined in the present study. The present results may facilitate the development of a novel and efficient HCC treatment.
Materials and Methods
Experimental Sample
In the present study, HCC and normal hepatic specimens were obtained from 206 patients in Cancer Hospital of Jilin Province between May 2006 and September 2013. Before the experiment, written informed consent was collected from all the patients. The participants did not receive any treatment except for surgery. The present study was approved by The Institutional Ethics Committee of Cancer Hospital of Jilin Province.
Clinical Tissue Sample Collection
We collected 130 primary gastric cancer tissue samples and 108 paired adjacent normal tissues from patients who underwent surgery at the Hospital of Chengdu University of TCM (Chengdu, China). None of the patients received any anticancer therapy before tumor resection and diagnosed with any additional malignancies. Pathological staging was based on the UICC/AJCC TNM Classification (8th edition of 2016).
Immunohistochemistry Analysis
Routine hematoxylin and eosin staining were performed prior to immunohistochemistry analysis. Briefly, paraffin-embedded samples were cut into 3-μm sections, and then dewaxed with xylene and rehydrated for peroxidase (DAB) immunohistochemistry staining. For antigen retrieval, sections were heated at 97 ° C for 20 min. Following a brief proteolytic digestion and a peroxidase blocking, the sections were incubated with corresponding primary antibody Prp8 (cat. no. # ab79237, Abcam) overnight at 4 ° C, and HRP/Fab polymer conjugate (Cat. PV-6000-D, Zhongshan Goldenbridge Biotechnology Co. Ltd., China) was applied as secondary antibody. Finally, the sections were stained with diaminobenzidine substrate and counter-stained with hematoxylin. Two independent investigators semi quantitatively evaluated PRP8 positivity without prior knowledge of clinicopathologic data. The final immunoreactivity scores (IRS) were assessed according to scores of the percentage of positive stained cells (0 point, 0–5% positive cells; 1 point, 6–25%; 2 point, 26–50%; 3 point, 51–75%; 4 point, 76–100%) plus staining intensity scores (0 point, no staining; 1 point, weak staining; 2 point, moderate staining; 3 point, strong staining). The final IRS more than 4 were accepted as strong positivity, while, others were weak positivity.
Cell Lines and Transfection
Human hepatic astrocyte line LX-2, and MHCC97H Hep3B, Huh1 HCC cell lines were obtained from American Type Culture Collection (ATCC; Rockville, Md.) and seeded in RPMI-1640 medium containing 10% FBS. All cells were cultured at 37°C in 5% CO2. Prp8 vector and control vector were bought from Shanghai Genechem Co., Ltd. Prp8 vectors were transfected into HCC cells and using Lipofectamine 2000 (Invitrogen; Thermo Fisher Scientific, Inc.) following the manufacturer’s instructions and G418 (Sigma-Aldrich; Merck KGaA) was used to expand G418-resistant clones in culture as a monoclonal population.
PI3K Inhibitor Treatment
The PI3K inhibitor LY294002 was diluted to a final concentration of 40 µM in DMSO and stored at −20°C, cells were subsequently treated for 24 h at 20 nM in order to efficiently inhibit PI3K. Cells treated with the same volume of DMSO served as the control group.
RNA Extraction and Reverse Transcription-Quantitative PCR (RT-qPCR)
Total RNA was extracted using TRIzol reagent (Invitrogen; Thermo Fisher Scientific, Inc.) as previously described.20 The cDNA was synthesized by PrimeScript RT reagent (Takara Bio, Inc.). RT-qPCR was performed using SYBR Green Master Mix II (Takara Bio, Inc.) according to the manufacturer’s instructions. The expression levels of Prp8 and Prp31 were normalized to U6 or GAPDH. The expression levels of the genes investigated were calculated using the 2−∆∆Cq method. The primers used in the present work were as follows: Prp8 forward, 5ʹ- ATGAAGAGCAATCCATTCTGGTGGAC-3ʹ and reverse, 5ʹ- TTGGCACGGTTGATGGTAGGTGACCACCA-3ʹ; Prp31 forward, 5ʹ- GGATCCATGTCTCTGGCAGATGAGCTCTTA-3ʹ and reverse, 5ʹ- CCGCGGTCAGGTGGACATAAGGCCACTCTT-3ʹ; GAPDH forward, 5ʹ-ACATCGCTCAGACACCATG-3ʹ and reverse, 5ʹ-TGTAGTTGAGGTCAATGAAGGG-3ʹ.
Western Blot Analysis
Cells were lysed using RIPA buffer (Beyotime Institute of Biotechnology). Then, the supernatant containing the total protein was collected as previously described.21 The protein was separated by 10% SDS-PAGE. The protein was blocked using 5% non-fat milk for 1 h. The membranes were incubated with the following primary antibodies: Prp8 (cat. no. # ab79237, Abcam.), Prp31 (1:1000 dilution; cat. no. #ab188577, Abcam), PI3 Kinase p110α (cat. no.#4255, Cell Signaling Technology, Inc.), Akt (cat. no. #2920, Cell Signaling Technology, Inc.), anti-pAkt (Thr308) (cat. no. #13038, Cell Signaling Technology, Inc.), N-Cadherin (cat. no. #13116, Cell Signaling Technology, Inc.), E-Cadherin (cat. no. #14472, Cell Signaling Technology, Inc.), Vimentin (cat. no. #5741, Cell Signaling Technology, Inc.), and β-actin (1:2000 dilution; cat. no. #ab107061, Abcam). Primary antibodies were incubated with the membranes overnight at 4°C. The diluted secondary antibodies were added to the membranes for 1 h. Finally, the protein was examined using an ECL reagent (EMD Millipore).
Immunofluorescence
The cells were washed 3 times with PBS, fixed with 4% paraformaldehyde for 10 min at room temperature, permeabilized with 0.1% Triton X-100, and blocked in PBS with 2% bovine serum albumin for 1 h. The staining was performed with a rabbit anti-human Prp8 antibody. Images were obtained using an Olympus IX81 microscope with an MT20/20 illumination system.
Short Hairpin RNA (shRNA) Method
The packaging construct (pHelper 1.0), the (vesicular stomatitis virus G, VSV-G) VSVG‐expressing construct (pHelper 2.0), pGCSIL-EGFP plasmid, pGCSIL-scramble vector and pGCSIL Prp8-shRNA construct were purchased from Genechem Biotech Co., Ltd. The shRNA-mediated knockdown was performed as previously described.22 HEK 293T cells (at 70–80% confluence) maintained in 6-well dishes were transfected with the aforementioned constructs using Lipofectamine (cat. no. 11668027; Thermo Fisher Scientific, Inc.), according to the manufacturer’s protocol. The viral stocks were concentrated via ultracentrifugation and dissolved in Hanks’ balanced salt solution. The HCC cells and hepatic astrocytes were transfected with the viral stocks at a multiplicity of infection of 200.
Cell Counting Kit-8 (CCK8) Assay
Transfected cells (4x103 cells/well) were seeded in RPMI-1640 with 10% FBS for 24, 48, 72 or 96 h. Next, the suspension of cells was incubated with 20 μL of CCK8 for 4 h. Then, 150μL DMSO was added into the medium. After 10 min, cell viability was assessed using a microplate reader (Olympus Corporation) to determine the optical density at 490 nm.
Colony Formation Assay
Briefly, stably transfected LX2 or Hep3B cells were plated in 6-well plates at 40 cells per well at 37°C for 10 days. the cell colonies were washed with PBS twice, and fixed with methanol for 30 min, and dyed with 0.1% crystal violet diluted in PBS for 15 min. The colonies which contained more than 120 cells were counted.
Transwell Chamber Assay
Transwell assay was used to assess cell invasion. Upper chambers were coated with Matrigel (BD Biosciences) to detect Hep3B or LX-2 cell invasion. The transfected cells (5x103 cells/well) were seeded in the upper chamber, and the lower chamber was filled with 10% FBS. The invasive cells were fixed and stained for 30 min. Finally, the invasive cells were fixed and stained for 30 min, and examined under a light microscope (Olympus Corporation).
Wound Healing Assay
A cell monolayer scratch assay was performed as described previously.23 Briefly, LX2 or Hep3B cells were seeded in 6 well plates at 3.5x105 cells per well, grown to ~100% confluence and pretreated with mitomycin C (10 μg/mL) for 2 h to inhibit cell proliferation before scratching. A scratch wound was created using a 20 μL pipette tip and was imaged at the same position at 0, 12 and 24 h.
Statistical Analysis
Data are presented as the mean ± SD, which were analyzed using SPSS 17.0 or Graphpad Prism 6. Chi-squared test, one-way ANOVA with Tukey’s post hoc test and Univariate Kaplan-Meier method with Log rank test were used to calculate differences between groups. P<0.05 was considered to indicate a statistically significant difference.
Results
The Expression of Prp8 Is Increased in HCC Tissues
The alternation of Prp8 and Prp31 expression was detected in HCC cells. RT-qPCR and Western blotting showed that the expression of Prp8 was increased in Hep3B, MHCC97H and Huh1 cells compared with LX-2 cells (P<0.01; Figure 1A–C), although there were no differences in Prp31 expression levels among LX-2, Hep3B, MHCC97H and Huh1 cells. Similarly, Prp8 expression was found to be higher in HCC tissues compared with normal hepatic tissues (P<0.01; Figure 1D and E). In addition, the correlation between abnormal Prp8 expression and clinical features in HCC patients was investigated. As shown in Table 1, the dysregulation of Prp8 was associated with tumor node and metastasis (TNM) stage (P<0.05) and distant metastasis (P<0.01). Furthermore, patients with HCC presenting high Prp8 expression showed a shorter overall survival time, indicating that upregulation of Prp8 predicted poor prognosis in patients with HCC (P<0.01, Figure 1F). The present results indicated that Prp8 may function as an important regulator of the pathogenesis of HCC.
 |
Table 1 Expression of Prp8 and the Clinicopathological Characteristics in HCC Patients |
 |
Figure 1 The expression of Prp8 is increased in HCC cells and tissues. (A) Prp8 mRNA expression in HCC cells compared with hepatic astrocytes. **P<0.01, compared with the hepatic astrocytes. (B) Prp8 protein expression in HCC cells compared with hepatic astrocytes. (C) Relative protein expression of Prp8 in HCC cells versus hepatic astrocytes. **P<0.01, compared with the hepatic astrocytes. (D) Prp8 protein expression in HCC tissues was explored via IHC. (E) Prp8 protein expression in HCC tissues was examined via western-blotting. **P<0.01, compared with the hepatic tissues. (F) High Prp8 expression is associated with poor prognosis in HCC patients. |
Overexpression of Prp8 Promotes Cell Viability and Migration of Human Hepatic Astrocyte Line
Prp8 was overexpressed in human hepatic astrocyte line LX-2 to perform a gain-of-function experiment. Prp8 expression was upregulated after overexpression in LX-2 cells (P<0.01, Figure 2A and B). The effect of Prp8 on the PI3K/Akt pathway was investigated to further examine its role in HCC. Prp8 overexpression was found to promote the expression level of PI3K-p110α (P<0.01) and phosphorylated Akt (P<0.01) in LX-2 cells (Figure 2A and B). Besides, immunofluorescence data discovered that Prp8 was mainly expressed in the cell nucleus (Figure 2C). CCK8 and colony formation assay revealed that overexpression of Prp8 promoted cell proliferation and viability in LX-2 cells (P<0.01, Figure 2D and E). Transwell and wound healing assay showed that cell migration and invasion were increased following Prp8 overexpression in LX-2 cells (P<0.01, Figure 2F–H). Collectively, Prp8 promoted the proliferative, migratory and invasive abilities of hepatic astrocytes.
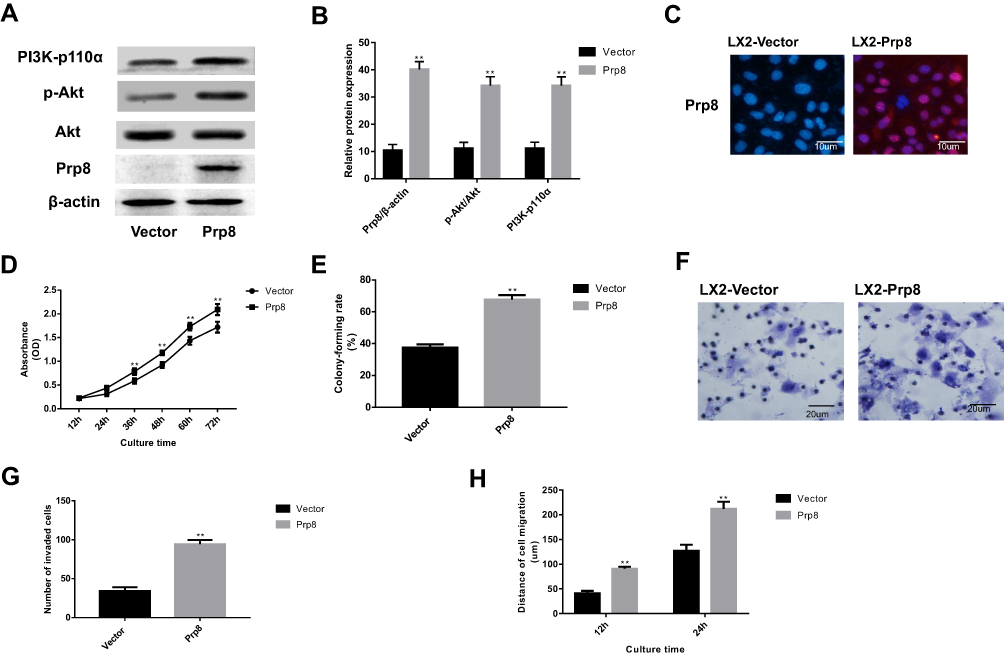 |
Figure 2 Upregulation of Prp8 promoted cell viability and migration in a human hepatic astrocyte line. (A) The effect of Prp8 on the PI3K/Akt pathway was investigated. (B) The relative protein expression of Prp8, PI3K-p110α and phosphorylated Akt. **P<0.01, compared with the empty vector groups. (C) The expression location of Prp8 was explored via immunofluorescence (D) CCK8 assay revealed that impact of Prp8 on cell proliferation. **P<0.01, compared with the empty vector groups. (E) Colony formation assay revealed that overexpression of Prp8 promoted cell viability. **P<0.01, compared with the empty vector groups. (F) Transwell assay was used to explore the impact of Prp8 on cell invasion. (G) The analyze of the invaded cell number. **P<0.01, compared with the empty vector groups. (H) The ability of cell migration was explored via wound healing assay. **P<0.01, compared with the empty vector groups. |
Loss of Prp8 Has an Inhibitory Effect on Cell Viability, Migration and Invasion in HCC Cells
Hep3B cells were selected for further functional assay due to the high expression level of Prp8 in this cell type. Prp8 was silenced in Hep3B cells to perform a loss-of-function experiment. Western-blot and immunofluorescence assay revealed that Prp8 expression was decreased following Prp8 knockdown in Hep3B cells (P<0.01, Figure 3A–C). Prp8 knockdown was found to inhibit the expression level of PI3K-p110α (P<0.01) and phosphorylated Akt (P<0.01) in Hep3B cells (Figure 3A and B). CCK8 and colony formation assay revealed that loss of Prp8 reduced cell proliferation and viability in Hep3B cells (P<0.01, Figure 3D and E). Similarly, knockdown of Prp8 reduced cell invasion cells (P<0.01, Figure 3F and G). Wound healing assay showed that cell migration was inhibited following Prp8 knockdown in Hep3B cells (P<0.01, Figure 3H). Taken together, Prp8 knockdown reduced the proliferative, migratory and invasive abilities of HCC cells.
 |
Figure 3 Loss of Prp8 has an inhibitory effect on cell viability and metastasis in HCC cells. (A) Prp8 expression and the PI3K/Akt pathway were inhibited following Prp8 knockdown in Hep3B cells. (B) The relative protein expression of Prp8, PI3K-p110α and phosphorylated Akt. **P<0.01, compared with the scramble group. (C) The expression location of Prp8 was explored via immunofluorescence (D) CCK8 assay revealed that loss of Prp8 reduced cell proliferation. **P<0.01, compared with the scramble group. (E) Colony formation assay revealed that the loss of Prp8 inhibited the ability of colony formation. **P<0.01, compared with the scramble group. (F) Transwell assay showed that cell invasion was inhibited by Prp8 knockdown in Hep3B cells. (G) The analyze of the invaded cell number. **P<0.01, compared with the scramble group. (H) The ability of cell migration was explored via wound healing assay. **P<0.01, compared with the scramble group. |
Prp8 Activates Epithelial-Mesenchymal Transition (EMT) in HCC
The effect of Prp8 on the EMT process was investigated to further examine its role in HCC. Prp8 overexpression was found to promote the expression level of N-cadherin and Vimentin, and to inhibit the expression of E-cadherin in LX-2 cells, suggesting that Prp8 may be involved in EMT in hepatic astrocytes (Figure 4A and B). Conversely, Prp8 knockdown reduced the expression levels of N-cadherin and Vimentin, and increased the expression of E-cadherin in HCC Hep3B cells (Figure 4C and D).
 |
Figure 4 Prp8 activates EMT in HCC. (A) Prp8 regulated the expression levels of E-cadherin, N-cadherin and Vimentin in LX-2 cells. (B) The relative protein expression of E-cadherin, N-cadherin and Vimentin in LX-2 cells. **P<0.01, compared with the empty vector groups. (C) Prp8 regulated the expression levels of E-cadherin, N-cadherin and Vimentin in Hep3B cells. (D) The relative protein expression of E-cadherin, N-cadherin and Vimentin in Hep3B cells. **P<0.01, compared with the scramble group. |
Overexpression of Prp8 Has a Positive Effect on Cell Viability, Migration and Invasion in HCC Cells
Prp8 was overexpressed in human HCC cell line Huh1 to perform a gain-of-function experiment. Prp8 expression was upregulated after overexpression in Huh1 cells (P<0.01, Figure 5A and B). The effects of Prp8 on the PI3K/Akt pathway and EMT process were investigated to further examine its role in HCC. Prp8 overexpression was found to promote the expression level of PI3K-p110α, phosphorylated Akt, N-cadherin and Vimentin, but inhibit the expression of E-cadherin in Huh1 cells (P<0.01; Figure 5A and B). CCK8 and colony formation assay revealed that overexpression of Prp8 promoted cell proliferation and viability in Huh1 cells (P<0.01, Figure 5C and D). Transwell and wound healing assay showed that cell migration and invasion were increased following Prp8 overexpression in Huh1 cells (P<0.01, Figure 5E–G). Collectively, Prp8 promoted the proliferative, migratory and invasive abilities of HCC cells.
 |
Figure 5 Upregulation of Prp8 promoted cell viability and migration in HCC cells. (A) The effects of Prp8 on the PI3K/Akt pathway and EMT process were investigated. (B) The relative protein expression of Prp8, PI3K-p110α, phosphorylated Akt, E-cadherin, N-cadherin and Vimentin. **P<0.01, compared with the empty vector groups. (C) CCK8 assay revealed that impact of Prp8 on cell proliferation. **P<0.01, compared with the empty vector groups. (D) Colony formation assay revealed that overexpression of Prp8 promoted cell viability. **P<0.01, compared with the empty vector groups. (E) Transwell assay was used to explore the impact of Prp8 on cell invasion. (F) The analyze of the invaded cell number. **P<0.01, compared with the empty vector groups. (G) The ability of cell migration was explored via wound healing assay. **P<0.01, compared with the empty vector groups. |
LY294002 Treatment Reversed the Carcinogenesis Effect of Prp8 in Hepatic Astrocytes
Functionally, the effect of Prp8 on PI3K/Akt pathway in hepatic astrocytes was abolished following treatment with 20 nM PI3K tyrosinase inhibitor LY294002 for 24 h. In addition, the effect of LY294002 on the PI3K/Akt pathway was also identified in LX-2 cells (Figure 6A and B). LY294002 treatment was found to inhibit the expression level of N-cadherin and Vimentin, and to promote the expression of E-cadherin in LX-2 cells with Prp8 overexpressing (P<0.01, Figure 6A and B). The LY294002 treatment suppressed the effects of Prp8 on cell proliferation and viability in LX-2 cells (Figure 6C and D). Similarly, the effect of Prp8 on LX-2 cell invasion (P<0.01, Figure 6E and F) and migration (P<0.01, Figure 6G) were also inhibited following treatment with LY294002. Collectively, LY294002 treatment reversed the carcinogenic effects of Prp8 in hepatic astrocytes. In summary, Prp8 was identified to serve a carcinogenic role via PI3K/Akt pathway in HCC progression.
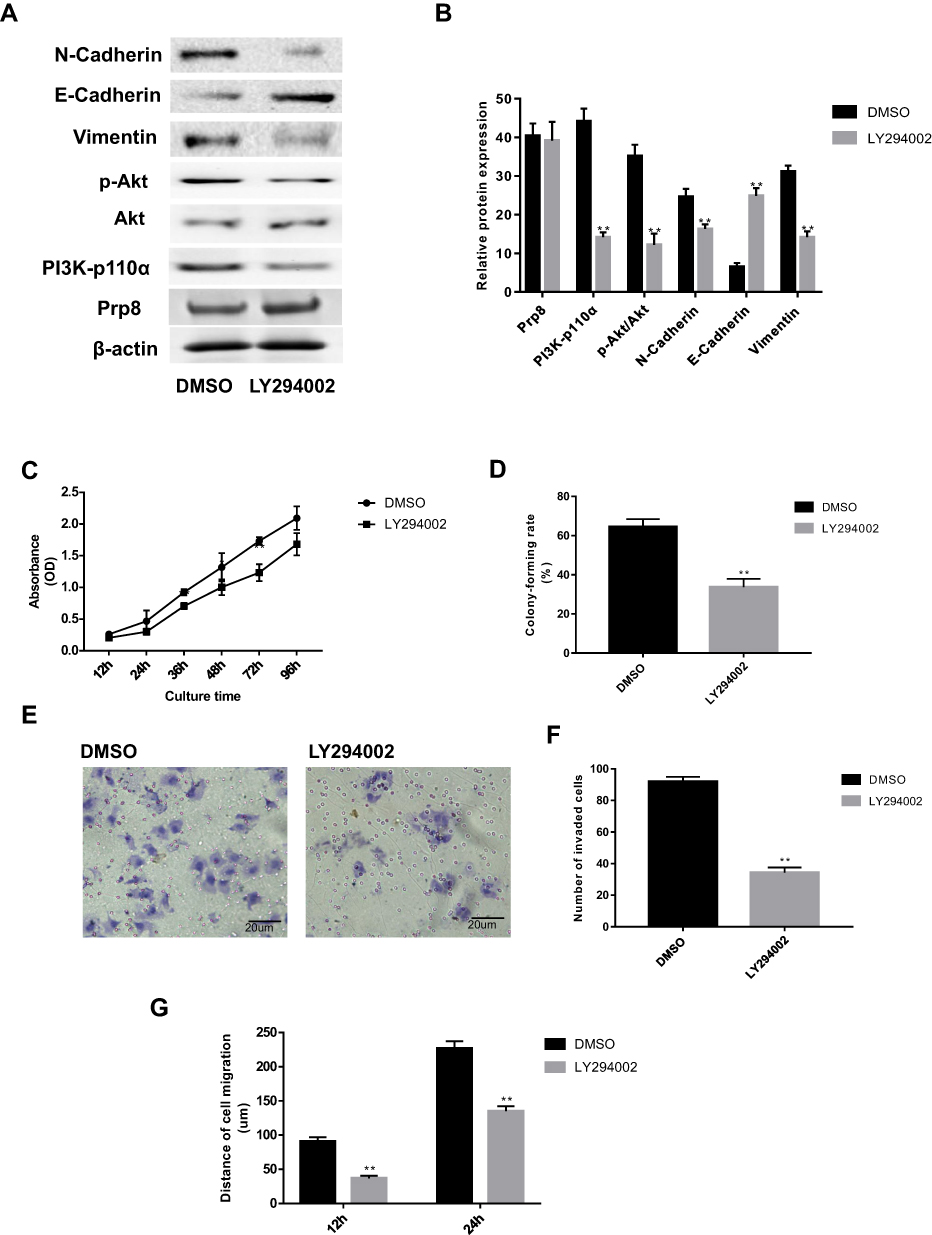 |
Figure 6 LY294002 treatment reverses the carcinogenic-inducing effects of Prp8 in hepatic astrocytes. (A) LY294002 treatment reversed the effects of Prp8 on PI3K/Akt pathway and EMT process in hepatic astrocytes. (B) The relative protein expression of Prp8, PI3K-p110α, phosphorylated Akt, E-cadherin, N-cadherin and Vimentin. **P<0.01, compared with the DMSO groups. (C) The LY294002 treatment suppressed the promoted effects of Prp8 on cell proliferation. **P<0.01, compared with the DMSO groups. (D) Colony formation assay revealed that the LY294002 treatment inhibited the ability of colony formation. **P<0.01, compared with the DMSO groups. (E) Promoted effect of Prp8 on cell invasion were inhibited by treatment with LY294002. (F) The analyze of the invaded cell number. **P<0.01, compared with the DMSO groups. (G) The ability of cell migration was explored via wound healing assay. **P<0.01, compared with the DMSO groups. |
Loss of Prp8 Has No Obvious Impact on Cell Viability in Hepatic Astrocytes
Prp8 was silenced in LX-2 cells to perform a loss-of-function experiment. Prp8 expression was reduced following Prp8 knockdown in LX-2 cells (P>0.05, Figure 7A and B). The loss of Prp8 has no obvious effect on the expression level of N-cadherin, Vimentin, E-cadherin, PI3K-p110α, and phosphorylated Akt in LX-2 cells (P>0.05, Figure 7A and B). CCK8 assay revealed that loss of Prp8 had no obvious impact on cell proliferation in LX-2 cells (P>0.05, Figure 7C). Colony formation assay revealed that the Prp8 knockdown have no obvious impact on the ability of colony formation in LX-2 cells (P>0.05, Figure 7D). Transwell assay (P>0.05, Figure 7E and F) and wound healing assay (P>0.05, Figure 7G) showed that cell migration and invasion were not altered fo llowing Prp8 knockdown in LX-2 cells. Collectively, Prp8 knockdown had no obvious impact on the proliferative, migratory and invasive abilities of hepatic astrocytes.
 |
Figure 7 Loss of Prp8 has no obvious impact on cell viability in hepatic astrocytes. (A) The loss of Prp8 has no obvious effect on the expression level of N-cadherin, Vimentin, E-cadherin, PI3K-p110α, and phosphorylated Akt in LX-2 cells. (B) The relative expression of Prp8, PI3K-p110α, phosphorylated Akt, E-cadherin, N-cadherin and Vimentin in LX-2 cells. **P<0.01, compared with the scramble group. (C) CCK8 assay revealed that loss of Prp8 had no obvious impact on cell viability in LX-2 cells. (D) The impact on the ability of colony formation was explored via colony formation assay. (E) Transwell assay showed that cell invasion was not obviously affected by Prp8 knockdown in LX-2 cells. (F) The analyze of the invaded cell number. (G) Wound healing assay showed that cell migration was altered by Prp8 knockdown in LX-2 cells. |
Discussion
Inhibition of basal spliceosomal activity may have limited effect on splicing in all cells, but normal cells may tolerate the slightly lowered spliceosomal activity.24 However, cancer cells may be much more susceptible to a reduction in spliceosomal activity due to the rapid proliferation and high metabolic demand of cancer cells. This effect was identified for proteasome inhibitors, such as velcade, which have been successfully used for cancer therapy.25 In this present study, it was hypothesized that inhibition of multiple spliceosomal components in addition to SF3b can selectively inhibit cancer cell growth and survival with limited side effects on normal cells.26 The present results on Prp8 knockdown, a component of the tri-snRNP, in HCC Hep3B cells support the hypothesis of the present study. The present results revealed that the loss of Prp8 had no significant impact on cell viability of hepatic astrocytes, but significantly inhibited the viability and metastatic potential of HCC cells. To date, suppression of Prp8 function has never been clinically targeted in HCC. However, the expression patterns of Prps in patients with HCC requires further study. The present observations suggested that Prp8 was upregulated in HCC tissues and was associated with distant metastasis. In addition, the influence of Prp8 on the PI3K/Akt signaling pathway in hepatic astrocytes and HCC cells were examined, and the present results suggested that Prp8 promoted the viability and metastatic potential of hepatic astrocytes and HCC cells via the PI3K/Akt signaling pathway. Moreover, the present observations suggested that the inhibition of PI3K/Akt signaling pathway induced a reduction of the metastatic potential of HCC cells that stably expressed Prp8. Furthermore, knockdown of Prp8 in an HCC cell line (Hep3B) leads to a suppression of the PI3K/Akt signaling pathway and reduced the metastatic potential of Hep3B cells via suppressing EMT process. Nevertheless, the detailed molecular mechanisms of the signal transduced from nuclear Prp8 to PI3K require further investigation.
Recent studies also demonstrated that the splicing apparatus is a limiting factor and various pre-mRNAs may compete with each other when the availability of the splicing apparatus is limited. Inhibition of the basal spliceosomal components may lead to increased competition for a limited amount of functional spliceosome and selectively affect alternative splicing events that contain sub-optimal splicing sites. Therefore, inhibition of the basal splicing machinery can adversely affect cancer cells via changes in alternative splicing events that are critical to cancer cells. A number of natural products isolated from the fermentation broths of Pseudomonas spp. and Streptomyces spp. that have potent anti-tumor properties support this hypothesis.27 The spliceosome contains multiple enzymes, including eight RNA helicases, one GTPase, and various prolylisomerases and kinases.28 These enzymes and many protein interactions in the spliceosome may be inhibited by small molecules, and targeting these spliceosomal components may represent a unique approach for cancer therapy. The potential side effect of these splicing-targeted therapies on photoreceptors is not a significant concern due to the existence of blood-retinal barrier, which has a similar structure as the blood-brain barrier and can prevent most small molecules from penetrating the barrier.29 These compounds were first identified due to their potent cytotoxic and cell cycle arresting effect in multiple tumor cell lines, as these molecules exhibit an in vitro IC50 in the low nM range, and significant anti-tumor activity in animal models.30 Recent mechanistic studies found that these compounds bind most tightly to the SF3b complex, which contains five protein components, of the spliceosome in cellular extracts.7 Although the exact binding partner of these compounds in SF3b remains to be determined, accumulating evidence suggested that they bind to the interface between the subunits of SF3b proteins.31 These compounds may have more potent growth arresting and cytotoxic effects on cancer cells, with no apparent general toxic effects due to extensive inhibition of general splicing and gene expression. One analog of these compounds (E7107) is currently in Phase I clinical trial for treating solid tumors.32
Conclusion
In summary, the present study identified an upregulation in Prp8 in HCC, which was associated with poor prognosis in HCC patients. Functionally, the loss of Prp8 may specifically inhibit cell viability and metastasis and activated EMT and PI3K/Akt in HCC cells. Although the present study has preliminarily investigated the regulatory mechanism of Prp8, further studies on Prp8 in HCC are required.
Abbreviations
Prp8, pre-mRNA processing factor 8; IHC, immunohistochemical analysis; CCK-8, Cell Counting Kit-8; Akt, protein kinase B; HCC, hepatocellular carcinoma.
Data Sharing Statement
The datasets used and/or analyzed during the present study are available from the corresponding author on reasonable request.
Acknowledgments
We would like to thank American Journal Experts (AJE) and Spandidos Publications English Language Editing Service for help with this manuscript.
Disclosure
The authors declare that they have no competing interests.
References
1. Li C, Xu X. Biological functions and clinical applications of exosomal non-coding RNAs in hepatocellular carcinoma. Cell Mol Life Sci. 2019;76(21):4203–4219. doi:10.1007/s00018-019-03215-0
2. Naftelberg S, Schor IE, Ast G, Kornblihtt AR. Regulation of alternative splicing through coupling with transcription and chromatin structure. Annu Rev Biochem. 2015;84:165–198. doi:10.1146/annurev-biochem-060614-034242
3. Yoshimoto R, Kaida D, Furuno M, et al. Global analysis of pre-mRNA subcellular localization following splicing inhibition by spliceostatin A. Rna. 2017;23(1):47–57. doi:10.1261/rna.058065.116
4. Park E, Pan Z, Zhang Z, Lin L, Xing Y. The expanding landscape of alternative splicing variation in human populations. Am J Hum Genet. 2018;102(1):11–26. doi:10.1016/j.ajhg.2017.11.002
5. Shi Y. Mechanistic insights into precursor messenger RNA splicing by the spliceosome. Nat Rev Mol Cell Biol. 2017;18(11):655–670. doi:10.1038/nrm.2017.86
6. Finci LI, Zhang X, Huang X, et al. The cryo-EM structure of the SF3b spliceosome complex bound to a splicing modulator reveals a pre-mRNA substrate competitive mechanism of action. Genes Dev. 2018;32(3–4):309–320. doi:10.1101/gad.311043.117
7. Lee SC, Abdel-Wahab O. Therapeutic targeting of splicing in cancer. Nat Med. 2016;22(9):976–986. doi:10.1038/nm.4165
8. Effenberger KA, Urabe VK, Jurica MS. Modulating splicing with small molecular inhibitors of the spliceosome. Wiley Interdiscip Rev RNA. 2017;8(2):e1381. doi:10.1002/wrna.1381
9. Yan C, Hang J, Wan R, Huang M, Wong CC, Shi Y. Structure of a yeast spliceosome at 3.6-angstrom resolution. Science. 2015;349(6253):1182–1191. doi:10.1126/science.aac7629
10. Chen Z, Gui B, Zhang Y, et al. Identification of a 35S U4/U6.U5 tri-small nuclear ribonucleoprotein (tri-snRNP) complex intermediate in spliceosome assembly. J Biol Chem. 2017;292(44):18113–18128. doi:10.1074/jbc.M117.797357
11. Bao P, Boon KL, Will CL, Hartmuth K, Luhrmann R. Multiple RNA-RNA tertiary interactions are dispensable for formation of a functional U2/U6 RNA catalytic core in the spliceosome. Nucleic Acids Res. 2018;46(22):12126–12138.
12. Wilkinson ME, Lin PC, Plaschka C, Nagai K. Cryo-EM studies of Pre-mRNA splicing: from sample preparation to model visualization. Annu Rev Biophys. 2018;47:175–199. doi:10.1146/annurev-biophys-070317-033410
13. Scotti MM, Swanson MS. RNA mis-splicing in disease. Nat Rev Genet. 2016;17(1):19–32.
14. Diakatou M, Manes G, Bocquet B, Meunier I, Kalatzis V. Genome editing as a treatment for the most prevalent causative genes of autosomal dominant retinitis pigmentosa. Int J Mol Sci. 2019;20(10).
15. Krausova M, Stanek D. snRNP proteins in health and disease. Semin Cell Dev Biol. 2018;79:92–102. doi:10.1016/j.semcdb.2017.10.011
16. Carey KT, Wickramasinghe VO. Regulatory potential of the RNA processing machinery: implications for human disease. Trends Genet. 2018;34(4):279–290. doi:10.1016/j.tig.2017.12.012
17. Stegeman R, Hall H, Escobedo SE, Chang HC, Weake VM. Proper splicing contributes to visual function in the aging Drosophila eye. Aging Cell. 2018;17(5):e12817. doi:10.1111/acel.12817
18. Sveen A, Kilpinen S, Ruusulehto A, Lothe RA, Skotheim RI. Aberrant RNA splicing in cancer; expression changes and driver mutations of splicing factor genes. Oncogene. 2016;35(19):2413–2427. doi:10.1038/onc.2015.318
19. Colloca G, Venturino A. Trial-level analysis of progression-free survival and response rate as end points of trials of first-line chemotherapy in advanced ovarian cancer. Med Oncol. 2017;34(5):87. doi:10.1007/s12032-017-0939-9
20. Zhang X, Ruan Y, Li Y, Lin D, Quan C. Tight junction protein claudin-6 inhibits growth and induces the apoptosis of cervical carcinoma cells in vitro and in vivo. Med Oncol. 2015;32(5):148. doi:10.1007/s12032-015-0600-4
21. Zhang X, Wang H, Li Q, Li T. CLDN2 inhibits the metastasis of osteosarcoma cells via down-regulating the afadin/ERK signaling pathway. Cancer Cell Int. 2018;18:160. doi:10.1186/s12935-018-0662-4
22. Zhang X, Wang X, Wang A, Li Q, Zhou M, Li T. CLDN10 promotes a malignant phenotype of osteosarcoma cells via JAK1/Stat1 signaling. J Cell Commun Signal. 2019;13(3):395–405. doi:10.1007/s12079-019-00509-7
23. Niu G, Ye T, Qin L, et al. Orphan nuclear receptor TR3/Nur77 improves wound healing by upregulating the expression of integrin beta4. FASEB J. 2015;29(1):131–140. doi:10.1096/fj.14-257550
24. Taylor J, Lee SC. Mutations in spliceosome genes and therapeutic opportunities in myeloid malignancies. Genes Chromosomes Cancer. 2019;58(12):889–902. doi:10.1002/gcc.22784
25. Li T, Ho L, Piperdi B, et al. Phase II study of the proteasome inhibitor bortezomib (PS-341, Velcade) in chemotherapy-naive patients with advanced stage non-small cell lung cancer (NSCLC). Lung Cancer. 2010;68(1):89–93. doi:10.1016/j.lungcan.2009.05.009
26. Brierley CK, Steensma DP. Targeting splicing in the treatment of myelodysplastic syndromes and other myeloid neoplasms. Curr Hematol Malig Rep. 2016;11(6):408–415. doi:10.1007/s11899-016-0344-z
27. Harvey AL, Edrada-Ebel R, Quinn RJ. The re-emergence of natural products for drug discovery in the genomics era. Nat Rev Drug Discov. 2015;14(2):111–129. doi:10.1038/nrd4510
28. Kastner B, Will CL, Stark H, Luhrmann R. Structural insights into nuclear pre-mRNA splicing in higher eukaryotes. Cold Spring Harb Perspect Biol. 2019;11(11). doi:10.1101/cshperspect.a032417
29. Diaz-Coranguez M, Ramos C, Antonetti DA. The inner blood-retinal barrier: cellular basis and development. Vision Res. 2017;139:123–137. doi:10.1016/j.visres.2017.05.009
30. Agrawal AA, Yu L, Smith PG, Buonamici S. Targeting splicing abnormalities in cancer. Curr Opin Genet Dev. 2018;48:67–74. doi:10.1016/j.gde.2017.10.010
31. Lin JC. Therapeutic applications of targeted alternative splicing to cancer treatment. Int J Mol Sci. 2017;19(1). doi:10.3390/ijms19010075
32. Kim YJ, Abdel-Wahab O. Therapeutic targeting of RNA splicing in myelodysplasia. Semin Hematol. 2017;54(3):167–173. doi:10.1053/j.seminhematol.2017.06.007
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.