Back to Journals » Clinical Pharmacology: Advances and Applications » Volume 10
Pramlintide, an antidiabetic, is antineoplastic in colorectal cancer and synergizes with conventional chemotherapy
Authors Al-Keilani MS ![]() , Alsmadi DH, Darweesh RS, Alzoubi KH
, Alsmadi DH, Darweesh RS, Alzoubi KH ![]()
Received 11 October 2017
Accepted for publication 27 December 2017
Published 5 March 2018 Volume 2018:10 Pages 23—29
DOI https://doi.org/10.2147/CPAA.S153780
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Arthur E. Frankel
Maha S Al-Keilani,1 Dua H Alsmadi,1 Ruba S Darweesh,2 Karem H Alzoubi1
1Department of Clinical Pharmacy, College of Pharmacy, Jordan University of Science and Technology, Irbid, Jordan; 2Department of Pharmaceutical Technology, College of Pharmacy, Jordan University of Science and Technology, Irbid, Jordan
Background: Approximately 90% of patients with metastatic colorectal cancer fail therapy mainly due to resistance. Taking advantage of currently approved agents for treatment of disease conditions other than cancer for the identification of new adjuvant anticancer therapies is highly encouraged. Pramlintide is a parenteral antidiabetic agent that is currently approved for treatment of types 1 and 2 diabetes mellitus.
Objectives: To address the antineoplastic potential of pramlintide in colorectal cancer and to evaluate the ability of pramlintide to enhance the cytotoxicity of 5-fluorouracil, oxaliplatin, and irinotecan against colorectal cancer cell lines expressing wild-type and mutant p53.
Materials and methods: The antiproliferative effect of pramlintide alone or in combination with 5-fluorouracil, oxaliplatin, or irinotecan in HCT-116 and HT-29 colorectal cancer cell lines was investigated using MTT cell proliferation assay. IC50 values were calculated using Compusyn software 1.0. Synergy values (R) were calculated using the ratio of IC50 of each primary drug alone divided by combination IC50s. For each two pairs of experiments, Student’s t-test was used for analysis. For combination studies, one-way analysis of variance and Tukey post hoc testing was performed using R 3.3.2 software. A p-value of <0.05 was considered significant.
Results: Pramlintide inhibited the growth of HCT-116 and HT-29 in a dose-dependent manner, with higher efficacy against the latter (IC50s; 48.67 and 9.10 µg/mL, respectively; p-value =0.013). Moreover, the addition of 5, 10, and 20 µg/mL of pramlintide to HCT-116 and HT-29 with 5-fluorouracil, oxaliplatin, or irinotecan induced the antiproliferative effect synergistically (R>1.6, p-value <0.05).
Conclusion: Pramlintide enhances the cytotoxicity of conventional chemotherapy against colorectal cancer cell lines harboring wild-type or mutant p53. Thus, pramlintide is a promising potential adjuvant chemotherapy in colorectal cancer.
Keywords: colorectal cancer, resistance, amylin analog, pramlintide, adjuvant chemotherapy, synergism
Introduction
Colorectal cancer is the third most common cancer in both sexes, and it is among the most common causes of cancer-related deaths in the United States.1 In Jordan, colorectal cancer accounts for 10.3% and 8.9% of cancer-related deaths in males and females, respectively.2 Surgical resection of the primary tumor and involved lymph nodes is the primary treatment for early-stage localized tumors. However, for advanced-stage metastatic tumors, adjuvant chemotherapy is the mainstay of therapy.3 Approximately 55% of colorectal cancer patients present with advanced stages at time of diagnosis, and half of the patients who undergo surgery ultimately develop a metastatic disease.4–6 Adjuvant chemotherapy consisting mainly of 5-fluorouracil (5-FU), oxaliplatin (OXA), and irinotecan (IRN) has been associated with improved response rates, reduced risk of tumor recurrence and mortality, and enhanced disease-free and median survival rates.4,7,8
Despite the advances in chemotherapy and targeted molecular agents, approximately 90% of patients with advanced metastatic colorectal cancer fail therapy, which is mainly attributed to resistance, either intrinsic or acquired.9 p53, is a tumor suppressor protein that works intracellularly to promote apoptosis or autophagy of the tumor cells.10,11 p53 is the most commonly mutated gene in cancer, with a mutation rate of approximately 50% in colorectal cancer.12,13 There is controversy regarding the effect of p53 status on the sensitivity of tumors to anticancer therapy. Taking into account the heterogeneity of cancer and the mutations affecting the p53 gene, it is now postulated that the p53-dependent response to anticancer therapy varies by cancer type, anticancer agent used, and the presence of mutations in other genes.14,15 Some studies proved that p53 is an important determinant of colorectal cancer cells’ response to 5-FU and OXA;7,14 cancer cell lines with mutation in p53 gene were more prone to resistance than wild-type cell lines, while other studies on various cancers revealed that p53-mutant cells were more sensitive to anticancer therapy such as OXA,14,16–18 thus indicating that other p53-independent pathways play a role in the response to chemotherapeutic agents. One suggestion is the coactivation of p53 homologues such as p73 that is independent of p53 status.19
The high resistance and toxicity profile associated with the current chemotherapeutic agents highlight the urgent need for the identification of new adjuvant therapies. Such agents will act synergistically to sensitize tumor cells to the currently approved anticancer agents, overcome therapy resistance, and reduce dose requirements and thus the drugs-induced adverse effects. Taking advantage of potential antineoplastic agents that are currently approved and used for treatment of disease conditions other than cancer is highly encouraged to evade the costly and exacting processes of development of new anticancer agents.
It was revealed by several studies that patients with diabetes mellitus were at higher risk of cancer such as colorectal cancer,20–23 breast, pancreatic, and endometrial cancers among others.24–26 This encouraged many researchers to study the potential anticancer activity of antidiabetic agents. For example, metformin exhibited antineoplastic activities in various types of cancers such as breast cancer, colorectal cancer, and prostate cancer.27–30
Pramlintide is a parenteral antidiabetic agent that is currently approved as adjunctive therapy to insulin in types 1 and 2 diabetes mellitus. Pramlintide is administered subcutaneously at doses ranging from 30 to 150 µg three to four times daily with a bioavailability of 30%–40%.31 It is a safe drug and is usually well tolerated with minimal adverse effects such as hypoglycemia, headache, and gastrointestinal upset.31,32 Pramlintide is a synthetic analog of amylin, a glucoregualtory hormone that is cosecreted with insulin from pancreatic β-cells. It acts on the three amylin receptors (AMY1, AMY2, and AMY3) that consist of a G protein-coupled receptor, calcitonin receptor, and three receptor activity-modifying protein receptors (1, 2, and 3).33
To the best of our knowledge, the anticancer effect of pramlintide was studied only in one previous study by Venkatanarayan et al.34 It was shown that the anticancer effect of pramlintide is related to p53 status and that it works through calcitonin receptors and receptor activity-modifying protein 3 receptors, which eventually resulted in the regression of p53-deficient thymic lymphomas.34 In the same study, it was found that different members of the p53 family, TAp63 and TAp73, can compensate for p53 knockdown through upregulating islet amyloid polypeptide, the gene encoding for amylin.34 Modulation of islet amyloid polypeptide or treatment with pramlintide resulted in regression of thymic lymphoma tumors with knocked-down p53,34 thus suggesting the beneficial antineoplastic role of pramlintide in p53-mutated tumors.
In this study, we investigated the antiproliferative effect of pramlintide in two different colorectal cancer cell lines; HCT-116 (wild-type p53) and HT-29 (mutant p53). After that, we investigated the synergistic effect of pramlintide combined with 5-FU, OXA, or IRN in the same cell lines. The results of our study demonstrate that pramlintide alone inhibits the growth of HCT-116 and HT-29, but with higher efficacy against the latter. Moreover, the addition of pramlintide to HCT-116 and HT-29 with 5-FU, OXA, or IRN induced growth inhibition synergistically.
Materials and methods
Drugs
IRN was purchased from Sigma-Aldrich (St. Louis, MO, USA) and dissolved in dimethyl sulfoxide (DMSO) at a concentration of 160 mM, and 5-FU and OXA were purchased from Combi-Blocks (San Diego, CA, USA) and dissolved in DMSO at concentrations of 385 mM and 378 mM, respectively. Pramlintide was obtained from R&D systems-Biotechne (Abingdon, UK) and dissolved in nuclease-free water at a concentration of 1 mg/mL. All drugs stocks were stored in a dark-colored bottles at –20°C as stock solutions.
Cell culture and drugs treatment
HCT-116 and HT-29 human colorectal cancer cell lines were purchased from the American Type Culture Collection (Manassas, VA, USA). Both cell lines were cultured in Dulbecco’s modified Eagle’s medium (Euroclone, Pero, Italy) supplemented with 10% fetal bovine serum (Euroclone) and 1% penicillin/streptomycin (100 IU/mL penicillin and 100 µg/mL streptomycin, Euroclone). Both cell lines were maintained in a humidified incubator with 95% air and 5% CO2 atmosphere at 37°C.
Drugs stocks were diluted to the required concentrations with culture media immediately before use. Before treatment with the drugs, the medium was removed when cells were adherent and approximately 80% confluent.
MTT assay
The HCT-116 (wild-type p53) and HT-29 (mutant p53) cells were plated into the 96 well plates at a density of 5×103 in 200 µL of medium per well and the cells were incubated and allowed to attach overnight. The attached cells in the plates were treated with a series of drug concentrations: pramlintide (0–102.4 µg/mL), 5-FU (0–200 µM), OXA (0–300 µM), or IRN (0–160 µM) alone or in combination with three different concentrations of pramlintide (5, 10, and 20 µg/mL) that correspond to 0.5×IC50, IC50, and 2×IC50 in HT-29. Cells grown in medium alone (for treatment with pramlintide only) or containing an equivalent amount of DMSO served as control (for other treatment conditions).
Cells were incubated with the drugs at the indicated concentrations for 72 hours. All measurements were done in triplicate. After that, cell proliferation assay was performed per the manufacturer’s protocol. Briefly, MTT dye was added to the treated cells at a final concentration of 0.5 mg/mL in PBS. Then, the plates were incubated at 37°C for 3 hours and the MTT was discarded and the formazan product was dissolved by adding 100 µL of DMSO to each well, followed by shaking for 5 minutes. Then, the plates were read using an enzyme-linked immunosorbent assay plate reader at 570 nm with a reference wavelength of 690 nm. Cell viability was calculated as follows: absorbance of the experimental group/absorbance of the control group. The IC50 value was defined as the concentration needed for a 50% reduction in cell viability. Dose–effect analyses and IC50 calculations were performed using Compusyn software 1.0 (Combosyn Inc., Paramus, NJ, USA).
Analysis of the effects of drug combinations
The effects of different drug combinations were determined as previously described by Martinez-Marignac et al.35 Synergy values (R) were calculated using the ratio of IC50 of each primary drug alone (5-FU, OXA, or IRN) divided by combination IC50s. R value reflects the extent of synergy or antagonism for two drugs: R>1.6, synergy; R=1, additive effect; R<1, antagonism.
Statistical analyses
All results were expressed as the mean ± standard deviation. For each two pairs of experiments, Student’s t-test was used for analysis. For comparison of IC50 values of monotherapy versus combination also, Student’s t-test was used. For combination studies, one-way analysis of variance and Tukey post hoc testing was performed using R 3.3.2 software. Statistical significance was considered if p-value <0.05.
Results
Pramlintide inhibited the growth of HCT-116 and HT-29
We utilized two different colorectal cancer cell lines; HCT-116 (wild-type p53) and HT-29 (mutant p53). Initially, we investigated the effects of pramlintide on the proliferation of the two cell lines using the MTT assay. As shown in Figure 1, pramlintide exerted antiproliferative effects in a dose-dependent manner against both cell lines, with a statistically significant higher efficacy against HT-29 (p-value =0.013).
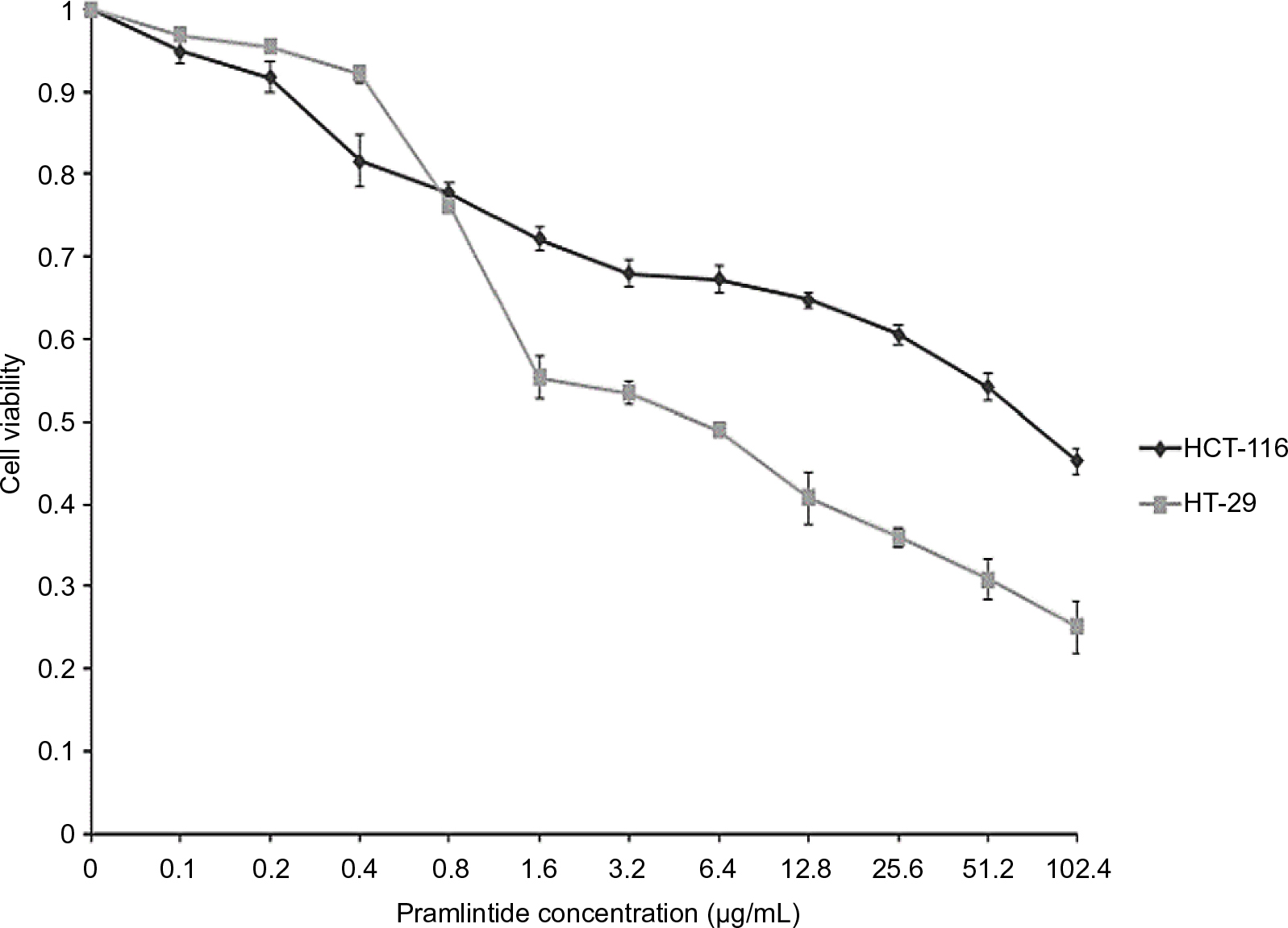 | Figure 1 Antiproliferative effect of pramlintide on HCT-116 and HT-29 colorectal cancer cell lines. Notes: Pramlintide concentrations ranged from 0.1 to 102.4 µg/mL. Attached cells were maintained in the drug-containing medium for 72 hours before being analyzed by MTT assay. Experiments were performed in triplicate. Pramlintide inhibited the growth of HCT-116 and HT-29 cell lines in a dose-dependent manner. HT29 and HCT116 differed significantly in their response to pramlintide (p-value =0.013). Student’s t-test was used for analysis, and statistical significance was considered if p-value <0.05. |
After 72 hours of treatment, 5 µg/mL pramlintide inhibited the growth of HT-29 and HCT-116 by 45.8% and 32.2%, respectively. The inhibitory effect was increased with 10 µg/mL (53.7% and 36.7%, respectively), and reached higher levels with 20 µg/mL (61.5% and 41.2%, respectively).
There was a difference between the IC50 values determined after treatment of HCT-116 and HT-29 with pramlintide. The IC50 value for pramlintide was approximately 5.4-fold higher in HCT-116 cells compared to HT-29 cells, 48.67 and 9.10 µg/mL, respectively, as shown in Table 1.
 | Table 1 IC50s for pramlintide and chemotherapy drugs in HCT-116 and HT-29 Abbreviations: 5-FU, 5-fluorouracil; IRN, irinotecan; OXA, oxaliplatin. |
Synergistic growth inhibition by pramlintide in combination with 5-FU, OXA, or IRN
The effect of 5, 10, and 20 µg/mL pramlintide on 5-FU-, OXA-, and IRN-induced growth inhibition effect was measured in HCT-116 and HT-29 cell lines by MTT assay (Figure 2).
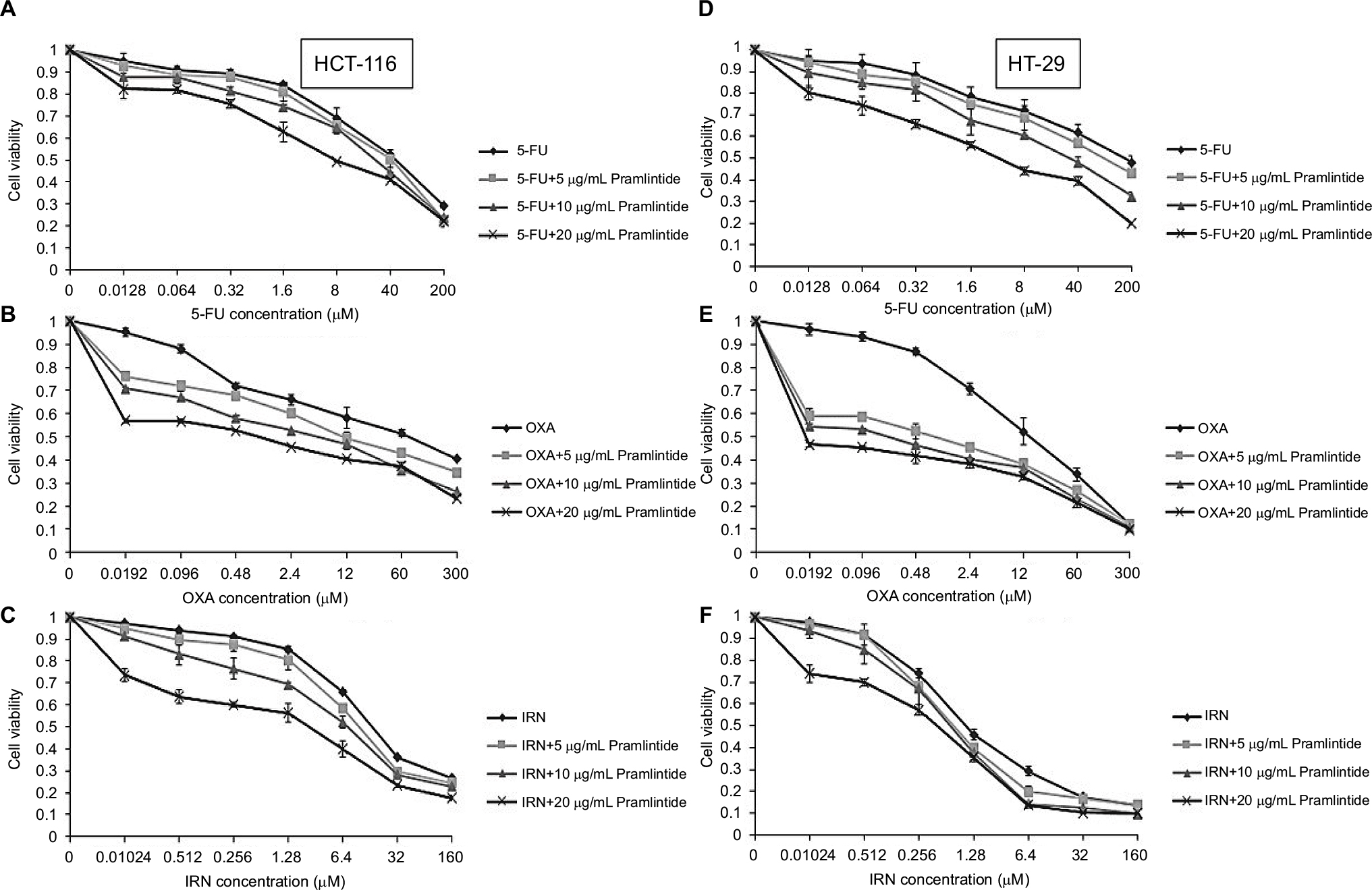 | Figure 2 Effect of treatment with 5-FU, OXA, orIRN, alone or combined with pramlintide on cell proliferation of HCT-116 (A, B, and C) and HT-29 (D, E, and F) colorectal cancer cell lines. Notes: Cell viability was assessed using MTT assay. Cell viability was measured after treatment with a series of drug concentrations: 5-FU (0–200 µM) (A, D), OXA (0–300 µM) (B, E), or IRN (0–160 µM) (C, F) alone or combined with three different concentrations of pramlintide (5, 10, and 20 µg/mL) for 72 hours. Cells grown in medium containing an equivalent amount of DMSO served as control. Each treatment condition was performed in triplicate. Data are expressed as mean±standard deviation. One-way ANOVA and Tukey post hoc was used for analysis and statistical significance was considered if p-value <0.05. Abbreviations: 5-FU, 5-fluorouracil; ANOVA, analysis of variance; DMSO, dimethyl sulfoxide; IRN, irinotecan; OXA, oxaliplatin. |
Our results revealed that at clinically achievable and nontoxic concentrations, pramlintide enhanced the antiproliferative of the three tested anticancer agents in a dose-dependent manner.
Synergy values (R) were quantified by using the ratio of IC50 of each primary drug alone (5-FU, OXA, or IRN) divided by combination IC50s.
Pramlintide showed a statistically significant synergistic effect against both cell lines when combined with 5-FU, OXA, and IRN, as indicated by (R) values (p-value <0.05) (Table 2).
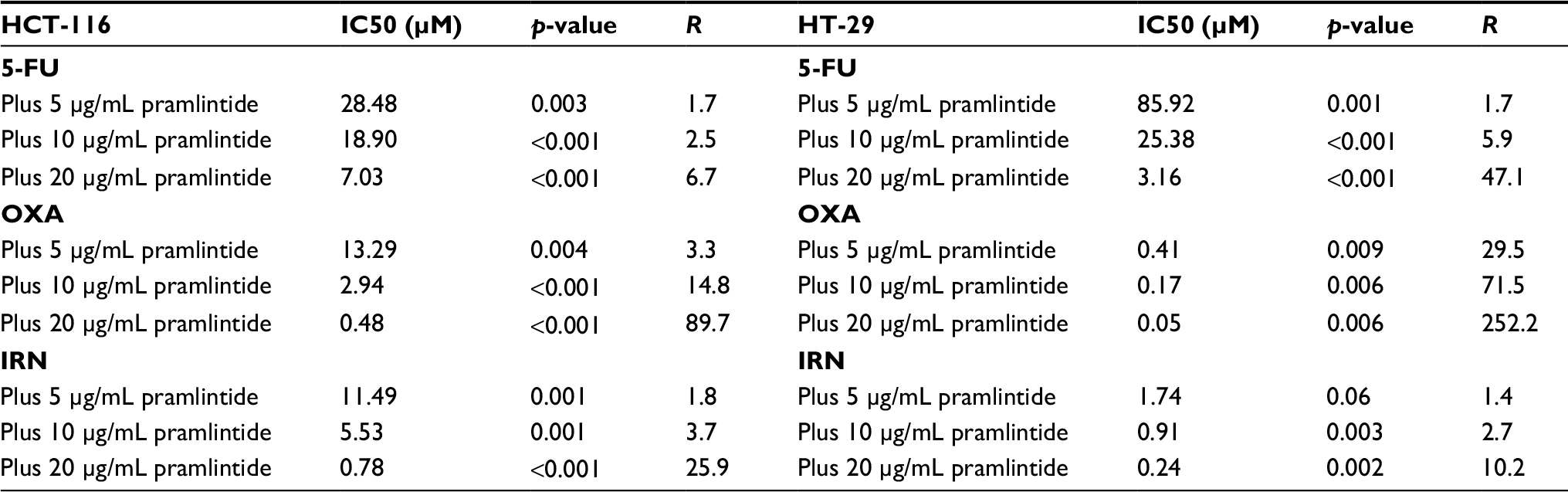 | Table 2 Combination IC50s and synergy values (R) in HCT-116 and HT-29 Note: IC50 values of monotherapy and combination were compared using the Student’s t-test. Abbreviations: 5-FU, 5-fluorouracil; IRN, irinotecan; OXA, oxaliplatin. |
Discussion
The current study was performed in an attempt to identify the antineoplastic potential of pramlintide colorectal cancer. For this, we utilized two colorectal cancer cell lines HCT-116 and HT-29. Our current study showed that this amylin analog inhibits the growth of colorectal tumor cell lines and synergizes with 5-FU, OXA, and IRN.
We found that pramlintide inhibited the growth of HCT-116 and HT-29 cell lines in a dose-dependent manner. However, HCT-116 was shown to be more resistant to pramlintide than HT-29, with approximately five-fold difference in IC50s. The difference in response to pramlintide may indicate that the anticancer effect of this agent is dependent on the p53 status; HCT-116 cell lines express p53 in wild-type conformation, while in HT-29 there is a G to A mutation in codon 273 of the gene that results in an arginine to histidine substitution (R273H). This missense mutation is associated with loss of wild-type activity and overexpression of a mutant p53 with oncogenic functions, ie, “gain of functions” phenotypes.36 Differential antineoplastic activity of pramlintide based on p53 status was previously shown in thymic lymphoma in mice.34 The function of p53 could have been restored or reactivated by pramlintide in HT-29, which is one mechanism for targeting p53 suggested by several studies.37,38 Further investigations are required to identify the mechanism of action and the mutant p53-dependent antiproliferative effect of pramlintide.
Despite the controversy regarding the effect of p53 status on the sensitivity of tumors to anticancer therapy, many studies revealed that p53 overexpression, a surrogate marker of p53 mutation, is associated with increased resistance to 5-FU, OXA, and IRN.7,39 Our results indicate that pramlintide may have activated the transcriptional activity of p53 in HT-29 cell line, which as a consequence resulted in enhanced apoptosis. This suggests that pramlintide is of high benefit to overcome p53-mutated tumors’ resistance toward chemotherapeutic agents. It is worth noting that metformin-induced anticancer activity was more pronounced in HCT-116 cell lines with p53 knockout.28
To investigate the synergistic potential of pramlintide with chemotherapy agents in colorectal cancer cell lines, we sought to test three different concentrations of pramlintide that correspond to 0.5×IC50, IC50, and 2×IC50 in each cell line. Nevertheless, due to high concentration requirements in HCT-116 cell lines, the limited amount of the drug available, and to be consistent with the investigated comparisons, we utilized pramlintide at concentrations of 5, 10, and 20 µg/mL, which correspond to 0.5×IC50, IC50, and 2×IC50 in HT-29. We demonstrated for the first time that at low and clinically achievable concentrations, pramlintide could synergistically inhibit colorectal cancer cell proliferation in HCT-116 and HT-29 cell lines when combined with 5-FU, OXA, and IRN in a concentration-dependent manner. These results suggest that pramlintide is a novel potential adjuvant anticancer agent with beneficial role in overcoming resistance to 5-FU, OXA, and IRN. Further in vivo and clinical studies are required to establish pramlintide as a valid chemopreventive and chemotherapeutic agent in colorectal cancer.
Despite the promising results we obtained from this study, there were some limitations. First, the antineoplastic potential of pramlintide was tested only using the short-term MTT assay. Second, we utilized only two representative cell lines to investigate the differential effect of pramlintide based on the p53 status; thus, the difference in the response to pramlintide between HT-29 and HCT-116 could be due to factors other than p53.
Conclusion
This study shows for the first time that pramlintide has anticancer activity against colorectal cancer and has a synergistic effect with 5-FU, OXA, and IRN. Future work will more fully explore the antiproliferative mechanisms of pramlintide and the underlying molecular mechanisms of synergism. Moreover, the antineoplastic potential of pramlintide will be analyzed using other long-term assays, such as colony-forming assay, and in vivo models of colorectal cancer.
Acknowledgment
This research was funded by the deanship of research at Jordan University of Science and Technology, Grant Number 20150046.
Author contributions
All authors contributed toward data analysis, drafting and critically revising the paper and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
Siegel RL, Miller KD, Jemal A. Cancer statistics, 2017. CA Cancer J Clin. 2017;67(1):7–30. | ||
Abdel-Razeq H, Attiga F, Mansour A. Cancer care in jordan. Hematol Oncol Stem Cell Ther. 2015;8(2):64–70. | ||
Midgley R, Kerr D. Colorectal cancer. Lancet. 1999;353(9150):391–399. | ||
Benson AB 3rd. Adjuvant chemotherapy of stage III colon cancer. Semin Oncol. 2005;32(6 Suppl 9):S74–S77. | ||
Kanwar SS, Poolla A, Majumdar AP. Regulation of colon cancer recurrence and development of therapeutic strategies. World J Gastrointest Pathophysiol. 2012;3(1):1–9. | ||
de Mestier L, Manceau G, Neuzillet C, et al. Primary tumor resection in colorectal cancer with unresectable synchronous metastases: a review. World J Gastrointest Oncol. 2014;6(6):156–169. | ||
Boyer J, McLean EG, Aroori S, et al. Characterization of p53 wild-type and null isogenic colorectal cancer cell lines resistant to 5-fluorouracil, oxaliplatin, and irinotecan. Clin Cancer Res. 2004;10(6):2158–2167. | ||
Carethers JM. Systemic treatment of advanced colorectal cancer: tailoring therapy to the tumor. Therap Adv Gastroenterol. 2008;1(1):33–42. | ||
Hammond WA, Swaika A, Mody K. Pharmacologic resistance in colorectal cancer: a review. Ther Adv Med Oncol. 2016;8(1):57–84. | ||
Flores ER, Tsai KY, Crowley D, et al. p63 and p73 are required for p53-dependent apoptosis in response to DNA damage. Nature. 2002;416(6880):560–564. | ||
Kenzelmann Broz D, Spano Mello S, Bieging KT, et al. Global genomic profiling reveals an extensive p53-regulated autophagy program contributing to key p53 responses. Genes Dev. 2013;27(9):1016–1031. | ||
Olivier M, Hollstein M, Hainaut P. TP53 mutations in human cancers: origins, consequences, and clinical use. Cold Spring Harb Perspect Biol. 2010;2(1):a001008. | ||
Kandoth C, McLellan MD, Vandin F, et al. Mutational landscape and significance across 12 major cancer types. Nature. 2013;502(7471):333–339. | ||
Brown JM, Wouters BG. Apoptosis, p53, and tumor cell sensitivity to anticancer agents. Cancer Res. 1999;59(7):1391–1399. | ||
Soussi T. p53 mutations and resistance to chemotherapy: a stab in the back for p73. Cancer Cell. 2003;3(4):303–305. | ||
Fan S, Smith ML, Rivet DJ 2nd, et al. Disruption of p53 function sensitizes breast cancer MCF-7 cells to cisplatin and pentoxifylline. Cancer Res. 1995;55(8):1649–1654. | ||
Gupta M, Fan S, Zhan Q, Kohn KW, O’Connor PM, Pommier Y. Inactivation of p53 increases the cytotoxicity of camptothecin in human colon HCT116 and breast MCF-7 cancer cells. Clin Cancer Res. 1997;3(9):1653–1660. | ||
Varna M, Bousquet G, Plassa LF, Bertheau P, Janin A. TP53 status and response to treatment in breast cancers. J Biomed Biotechnol. 2011;2011:284584. | ||
Toscano F, Parmentier B, Fajoui ZE, et al. P53 dependent and independent sensitivity to oxaliplatin of colon cancer cells. Biochem Pharmacol. 2007;74(3):392–406. | ||
Hu FB, Manson JE, Liu S, et al. Prospective study of adult onset diabetes mellitus (type 2) and risk of colorectal cancer in women. J Natl Cancer Inst. 1999;91(6):542–547. | ||
Yang YX, Hennessy S, Lewis JD. Insulin therapy and colorectal cancer risk among type 2 diabetes mellitus patients. Gastroenterology. 2004;127(4):1044–1050. | ||
Larsson SC, Orsini N, Wolk A. Diabetes mellitus and risk of colorectal cancer: a meta-analysis. J Natl Cancer Inst. 2005;97(22):1679–1687. | ||
Kabat GC, Kim MY, Strickler HD, et al. A longitudinal study of serum insulin and glucose levels in relation to colorectal cancer risk among postmenopausal women. Br J Cancer. 2012;106(1):227–232. | ||
Inoue M, Iwasaki M, Otani T, Sasazuki S, Noda M, Tsugane S. Diabetes mellitus and the risk of cancer: results from a large-scale population-based cohort study in Japan. Arch Intern Med. 2006;166(17):1871–1877. | ||
Pandey A, Forte V, Abdallah M, et al. Diabetes mellitus and the risk of cancer. Minerva Endocrinol. 2011;36(3):187–209. | ||
Noto H, Tsujimoto T, Noda M. Significantly increased risk of cancer in diabetes mellitus patients: a meta-analysis of epidemiological evidence in Asians and non-Asians. J Diabetes Investig. 2012;3(1):24–33. | ||
Dowling RJ, Zakikhani M, Fantus IG, Pollak M, Sonenberg N. Metformin inhibits mammalian target of rapamycin-dependent translation initiation in breast cancer cells. Cancer Res. 2007;67(22):10804–10812. | ||
Buzzai M, Jones RG, Amaravadi RK, et al. Systemic treatment with the antidiabetic drug metformin selectively impairs p53-deficient tumor cell growth. Cancer Res. 2007;67(14):6745–6752. | ||
Ben Sahra I, Laurent K, Loubat A, et al. The antidiabetic drug metformin exerts an antitumoral effect in vitro and in vivo through a decrease of cyclin D1 level. Oncogene. 2008;27(25):3576–3586. | ||
Wang Y, Liu G, Tong D, et al. Metformin represses androgen-dependent and androgen-independent prostate cancers by targeting androgen receptor. Prostate. 2015;75(11):1187–1196. | ||
McQueen J. Pramlintide acetate. Am J Health Syst Pharm. 2005;62(22):2363–2372. | ||
Chase HP, Lutz K, Pencek R, Zhang B, Porter L. Pramlintide lowered glucose excursions and was well-tolerated in adolescents with type 1 diabetes: results from a randomized, single-blind, placebo-controlled, crossover study. J Pediatr. 2009;155(3):369–373. | ||
Gingell JJ, Burns ER, Hay DL. Activity of pramlintide, rat and human amylin but not Abeta1-42 at human amylin receptors. Endocrinology. 2014;155(1):21–26. | ||
Venkatanarayan A, Raulji P, Norton W, et al. IAPP-driven metabolic reprogramming induces regression of p53-deficient tumours in vivo. Nature. 2015;517(7536):626–630. | ||
Martinez-Marignac V, Shawi M, Pinedo-Carpio E, et al. Pharmacological targeting of eIF4E in primary CLL lymphocytes. Blood Cancer J. 2013;3:e146. | ||
Candeias MM, Hagiwara M, Matsuda M. Cancer-specific mutations in p53 induce the translation of Delta160p53 promoting tumorigenesis. EMBO Rep. 2016;17(11):1542–1551. | ||
Bykov VJ, Issaeva N, Selivanova G, Wiman KG. Mutant p53-dependent growth suppression distinguishes PRIMA-1 from known anticancer drugs: a statistical analysis of information in the national cancer institute database. Carcinogenesis. 2002;23(12):2011–2018. | ||
Wang Z, Sun Y. Targeting p53 for novel anticancer therapy. Transl Oncol. 2010;3(1):1–12. | ||
Magrini R, Bhonde MR, Hanski ML, et al. Cellular effects of CPT-11 on colon carcinoma cells: dependence on p53 and hMLH1 status. Int J Cancer. 2002;101(1):23–31. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.