Back to Journals » OncoTargets and Therapy » Volume 9
Pragmatic medicine in solid cancer: a translational alternative to precision medicine
Authors Brabek J, Rosel D, Fernandes M
Received 7 January 2016
Accepted for publication 18 February 2016
Published 5 April 2016 Volume 2016:9 Pages 1839—1855
DOI https://doi.org/10.2147/OTT.S103832
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr William C. Cho
Jan Brábek,1 Daniel Rosel,1 Michael Fernandes2
1Department of Cell Biology, Faculty of Science, Charles University in Prague, Prague 2, Czech Republic; 2Medbase, Chapel Hill, NC, USA
Abstract: The precision medicine (PM) initiative is a response to the dismal outlook in solid cancer. Despite heterogeneity, common mechanistic denominators may exist across the spectrum of solid cancer. A shift from conventional research and development (R&D) toward PM will require conceptual and structural change. As individuals and as a society, we welcome innovation, but question change. We ask: In solid cancer, does PM identify and address the causes of prior failures, and, if so, are the proposed solutions feasible? And, when may we expect safer, more effective and affordable drugs in the clinic? Considerations that prompt a pragmatic rethink include a failure analysis of translational R&D in solid cancer suggesting that trials and regulations need to be aligned with the natural history of the disease. In successful therapeutic interventions in chronic, complex disease, surrogate markers and endpoints should be consistent with the Prentice’s criteria. In solid cancer, drug induced tumor shrinkage, is a drug effect and not a disease response; tumor shrinkage does not reflect nor predict interruption of the disease. Overall, we support a pragmatic, multidisciplinary, and collaborative R&D, and suggest that direction be set by clinical need and utility, and by questions, not answers. PM will prove worthwhile if it could improve clinical outcomes. The lag in therapeutics relative to diagnostics is a cause for confusion. Overdiagnosis adds to fear and harm, especially in the absence of effective interventions. A revised initiative that prioritizes metastasis research could replicate the successful HIV/AIDS model in solid cancer. A pragmatic approach may further translational efforts toward meaningfully effective, generally available, and affordable solutions.
Keywords: precision medicine, pragmatism, solid cancer, translation, metastasis, RECIST, 21st Century Cures Act, Paul Ehrlich
Introduction
“What is the question?”
In scientific programs, especially one with health care and economic implications, questions usually precede answers.1 The precision medicine (PM) initiative lists several solutions, but the questions remain elusive. In the context of remarkable successes of the pharmaceutical industry in combating serious and widespread acute and chronic diseases, the question is: why are effective and affordable therapies still not available for solid cancer? In the absence of effective and affordable therapies, the value of national screening programs is questionable, and there is a concern that diagnostics may outpace therapeutics. In an earlier era, there were no medicines for serious diseases such as solid cancer. Today, curative medicines exist, but are unaffordable for the large majority of patients. In the treatment of solid cancer, we have progressed from the incurable to the unaffordable (Figure 1).
 | Figure 1 “You’ve got something unaffordable”. |
Here, we make the case for a shift in research and development (R&D) focus to relevant clinical outcomes, antimetastasis, and away from tumor shrinkage.2 Tumor shrinkage does not qualify as a surrogate marker of efficacy relative to the natural history of the disease. A focus on antimetastatic interventions may not only be appropriate to the natural history of solid cancer, but will also justify earlier diagnosis via meaningful population screening.
Precision medicine
The PM initiative is a major component of the 21st Century Cures Act, which, in part, is structured to accelerate the discovery, development, and delivery of useful drugs.3,4 PM is a modest proposal with two components: a focus on cancer and the development of precisely targeted drugs, and a longitudinal study of at least 1 million people to explore genetic and environmental determinants of health and disease.5,6 With the current and increasing concern on affordable medicines, especially in cancer, it is commendable that this initiative was prompted and levered by a significant reduction in the price of genome sequencing.7 If the lower cost of goods translates to lower drug prices, then the PM initiative could be an ideal “fit” with the Affordable Care Act.8
The response to the PM initiative has been mixed (Table 1).7,9–16 The bias toward genomics at the expense of the environment, and on the individual at the expense of the population needs to be clarified. It is difficult to target an intervention to both individuals and populations simultaneously. The role of the environment in disease and the therapeutic decision to target sick individuals or populations have been debated for decades.17,18 Today, we realize that genetic and environmental factors, within the context of disease, are distinct entities, and that, though connected, are inseparable.19 Will the focus on genomics underestimate the role of the environment in gene–environment interaction research?
 | Table 1 Joyner and Paneth pose seven questions for PM |
To strategize a rational response to disease, problems need to be precisely defined before precise solutions are presented. The history of medicine indicates that a better understanding of epidemiology and pathogenesis precedes diagnostic and therapeutic advances; following Kuhn,20 “failures, not successes have been the driving force behind innovation”. The earlier experience with antibiotics in combating infection was a huge success, but today, science is driven toward overcoming failure in combating drug resistance. The analogy to the treatment of solid cancer is evident – the challenge is toward the development of meaningfully effective drugs at affordable prices.2
Relative to solid cancer, we ask:
- Is our method for the clinical evaluation of targeted anticancer drugs (response evaluation criteria in solid tumors, RECIST) consistent with the natural history of solid cancer?
- Is intratumoral heterogeneity a conceptual or operational obstacle to a precision approach in medicinal chemistry?
- Why do precisely targeted drugs invite resistance?
- Why, commensurate to its relevance, is dedicated antimetastasis research neither recognized nor funded?
In contrast to Jameson and Longo,13 a careful and conceptual analysis of needs may be more relevant to the justification of the PM initiative than answers to unasked questions – why are we continuing to overpromise and underdeliver in the treatment of solid cancer?21
In the context of serial failures in drug development in solid cancer, nonmeaningful improvements in efficacy, the almost predictable development of drug resistance, and astronomical pricing,22–24 a novel rethink of the R&D and business models is appropriate and timely. High prices do not always reflect the costs of innovation; they are necessary to cover the loss from failures in late-phase trials and recalls following market authorization.24 High drug prices are not universal; they are especially evident in free-market economies where the optimal price is what the market can bear.
The immediate need is an expansion of cancer care and control, especially in low- and middle-income countries.25 A reduction in clinical trial failures may lower the price of goods and allow for effective, accessible, and affordable medicines for all.2 Complexity of cancer and the costs of drug development are weak excuses for failure. We succeeded with an equally if not more complex disease, which was an epidemic, communicable, characterized by rapid mutations, and a fatal course – HIV/AIDS.26–28 Edmondson29 explains that in business and public policy, learning from failures is a sound principle for strategy formulation. A failure analysis suggests that a directional rethink – trial end points and regulations – and an emphasis on antimetastasis drug development are high on the list of immediate needs.
A shift in approach toward a precision-based and personalized frame – from the phenotype to the genotype – will require major structural changes in medicine and therapeutics. However, a larger policy question lurks in the background: Will these initiatives presage a personalized and private medicine framework for all (oxymoron intended), and the imminent demise of public health?30,31 The premise of PM – prevention and treatment in the individual – may be at odds with public health strategy in that population-wide interventions are much more effective in reducing the incidence of disease than interventions in high-risk individuals.18,30,31
Clinical trials
Burton et al32 define genetic epidemiology as a discipline closely allied to traditional epidemiology that focuses on the familial, and in particular genetic, determinants of disease and the joint effects of genes and nongenetic determinants. Crucially, appropriate account is taken of the biology that underlies the action of genes and the known mechanisms of inheritance.32 Accordingly, novel elements of clinical trial design will likely include the development and validation of prognostic and predictive biomarkers and classifiers, novel trial designs,33 and a prospective plan of analysis.34 This is important because clinical trials are the foundation upon which PM will be evaluated and implemented in individuals and populations.
The transition from conventional practice to PM will be neither simple nor easy. Problems have been identified and solutions have been proposed.9,13,35–39 Specifically, the European Society of Medical Oncology has published a comprehensive position paper detailing the promises and challenges in the delivery of PM in oncology.39 In brief, PM requires specialized centers to provide guidance, precise and standardized genotyping, and appropriate management of patients in trials and in the clinic. In this context, Booth and Tannock40 propose that a few well-executed clinical trials performed at centers of excellence can complement population-based observational studies to expedite the evaluation of novel interventions. This proposal receives support from Kocher and Roberts41 who state that the most expensive step in creating a new drug is conducting clinical trials, and that high-frequency, material information on clinical efficacy and safety comes from the first few hundred patients studied in a trial. Accordingly, large trials that add unwanted noise into the system may not be necessary. In fact, N-of-1 clinical trials, for biomarker studies rather clinical end points, may represent an efficient format for PM.42–44
Regulations
Contrary to received wisdom, advances in science rarely extrapolate passively and painlessly to the approval of new drugs – regulations define and allow for the efficacy profile of products destined to the market. It is self-evident that translation to PMs requires precision regulations.
For too long, drug approval in solid cancer has been based on the RECIST convention, namely, tumor shrinkage45,46 (Figure 2). This is the case despite the near-universal recognition that the tumor shrinkage end point is a relic of the cytotoxic era and far from a precision end point.47 For targeted drugs, RECIST may not be fit for purpose.48–50 “Progressive disease”, as defined by RECIST, is an increase in size, not progression of the disease. The latter is defined by continuing local invasion and metastasis. And again, “response”, as defined by RECIST, is a decrease in tumor size, but this is unrelated to inhibition of invasion or of the metastatic process. A complete response, disappearance of all lesions, is rarely seen. We maintain that tumor shrinkage is a treatment effect, and not a disease response. In a commentary in J Natl Cancer Inst, Oxnard et al51 make our point, but in a confusing manner. Their title reads: When progressive disease does not mean treatment failure: reconsidering the criteria for progression. They state that the RECIST progression criteria were not developed as a surrogate for survival, and this makes interpretation of a “response” difficult. Their likely point is that a RECIST-based increase in tumor size does not imply treatment failure. We agree, and ask: in that case, why should a RECIST-based tumor shrinkage signify treatment success?
 | Figure 2 Precision science meets imprecise regulations. |
Krajewski et al52 at the Dana-Farber Cancer Institute report that a 10% or greater shrinkage in tumor diameter in the first follow-up scan predicts clinical outcome in patients with metastatic renal cell carcinoma treated with angiogenesis inhibitors (Figure 2). On the basis of this study, and a survey of the literature, Chen et al53 at the National Cancer Institute (NCI) support a 10% rule for declaring a response to noncytotoxic drugs. If confirmed, the implications to oncologists are substantial; the stable disease category will now be partitioned into a responsive subcategory (tumor size change from −10% to −19%, and merge with partial response) and a nonresponse subcategory (change from −9% to +29%, and merge with progressive disease). This modification will result in three categories: progressive disease, partial response, and complete response.
The RECIST scheme is related to changes in tumor size.45,46 It is a categorical classification based on the longest diameters of tumor images. The categories are arbitrary and wide. The stable disease category spans a change of −29% to +19%. Recently, a 10% or greater decrease in tumor size has been proposed as a cutoff limit to declare response for targeted drugs, mainly angiogenesis inhibitors.52,53
Returning to conventional RECIST, the appearance of a new lesion (metastasis) indicates progressive disease.45,46 But with protocol constraints to focus on selected target lesions, new metastases may be overlooked, especially if the primary tumor shows marked shrinkage; the desired and publicized end point is tumor shrinkage. Therefore, it may be advisable to programmatically examine organs and sites of metastatic predilection (nontargets) for new lesions. Clearly, the appearance of a new metastasis on treatment indicates that the drug is ineffective. Today, radiology is faced with the challenge of evaluating changes during therapy quantitatively and of visualizing therapeutic effects that are more discrete (eg, necrosis, altered tumor perfusion). Höink et al54 have recently addressed innovative developments and requirements for radiology across the spectrum of solid cancer.
In Greek mythology, Procrustes either stretched or cut his guests to make them fit the bed. Today, cancer clinical trials recapitulate a Procrustean scenario: precisely targeted drugs are being resized on a RECIST bed, and the dismal outcome is predestined.55 Novel biomarkers and advanced imaging that track drug response in the context of the natural history of solid cancer (not just tumor shrinkage) are essential to further the oncologic objectives of PM.56–61 Validity of a biomarker is established by authenticating its correlation with clinical, not regulatory outcomes based on tumor shrinkage.62 It stands to reason that existing and investigational biomarkers, identified by mining clinical trial data based on RECIST (tumor shrinkage) and “validated” by regulatory decisions, may not be useful. If the aim of PMs is to prevent progression of the disease, then we need biomarkers of local invasion and the propensity to metastasis. Following Aronson,63 the chain of events in a disease process linking pathogenesis to outcome is fragile, and the better we understand the nature of the path a disease takes and the pharmacology of a drug that affects it, the better biomarkers we will be able to develop in diagnosing, staging, and monitoring disease and its response to therapy. Accordingly, it may be best that drug and biomarker development and the selection of surrogate end points for clinical trials are aligned with the natural history of the disease64 (Figure 3).
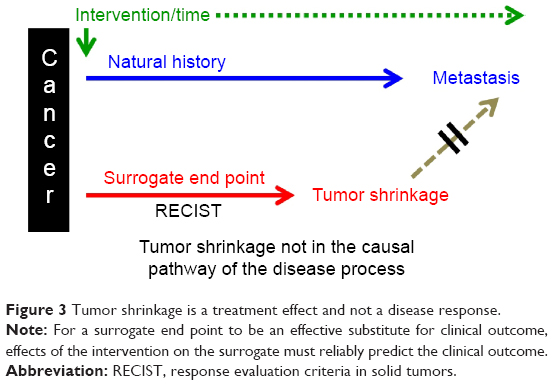 | Figure 3 Tumor shrinkage is a treatment effect and not a disease response. |
The Prentice criteria are a set of conditions that specify the conditional independence of the impact of treatment on the true end point, given the surrogate end point. In brief, the surrogate end point must be correlated with the clinical outcome and must fully capture the net effect of treatment on the clinical outcome.65 It is widely accepted that this set of criteria ensures the validity of a surrogate end point (Figure 3).
However, Berger66 disputes this claim and points out that these criteria alone ensure that an observed effect of the treatment on the true end point implies a treatment effect also on the surrogate end point, but contrary to popular belief, it does not ensure the converse, specifically, that the observation of a treatment effect on the surrogate end point can be used to infer a treatment effect on the true end point.
Failures in development may logically be attributed to a mismatch between precision science and imprecise regulations.47,67 The need for a regulatory rethink has been both anticipated and covered by Collins and Varmus:3 “achieving the goals of precision medicine will also require advancing the nation’s regulatory framework”. In solid cancer, we support a closer fit between related science (epidemiology, cell biology, pharmacology, and medicinal chemistry) and clinical trials, and also an agreement on novel translational end points that reflect a meaningful interruption in the progression of the disease, not just tumor shrinkage (Figure 4). Although translation is important, funding should not be at the expense of the related and contextual nonclinical and preclinical sciences. Otherwise, there may be little to translate!68
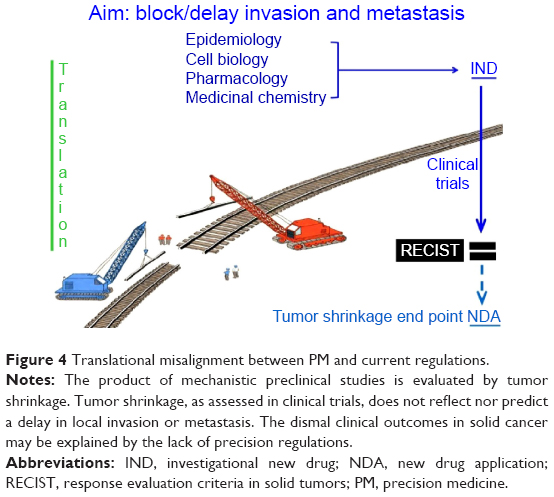 | Figure 4 Translational misalignment between PM and current regulations. |
Despite the dismal consequences of the RECIST regime, even today, authors continue to emphasize that end points need to reflect benefit to patients, but advise that changes in tumor size either in absolute terms (response and progression) or relative to control (progression) are clinically relevant.69
Metastasis
On average, approximately 5% of total cancer research funding is spent on investigating metastases in Europe, and most likely in the USA and Japan, yet metastatic disease is the direct or indirect cause of over 90% of all cancer deaths.70
In 2007, in a landmark publication, Eccles and Welch71 stated the obvious: “the prevention and/or elimination of spread of the disease, will represent the most important improvement in morbidity and mortality”. Today, patients fear the “M” word more than the “C” word,70 and it is surprising that the “M” word does not appear in PM manifesto.3 Oncologists ask: why does cancer therapy lack effective antimetastasis drugs?72 And pharma scientists bemoan the lack of regulatory guidances for antimetastatic drugs.73 This is the case despite the large and growing backlog of possibly antimetastatic drug candidates in the pipeline.74 In solid cancer, precision science, related to local invasion and metastasis, has been long practiced by cell biologists and medicinal chemists.75–86 Interestingly, the cell-migration-related signaling machinery is similar in both normal development as well as in invasion and metastasis.82,86 This could represent a problem, or an opportunity, for antimetastatic drug discovery. In our opinion, the shift in discovery focus from the cancer cell alone and toward the tumor stroma is both a promising and pragmatic one.47 Steeg et al,87 in an open letter to regulatory agencies, suggested that preclinical drug development must consider the impact on metastasis. This is good advice, but the message, perhaps, would be best delivered to clinical oncologists.
In 2016, a major and unanswered question related to metastasis is the timing of dissemination – early or late, and whether the mechanisms act in parallel or sequentially.88–90 Would precise treatment of the primary tumor cover that of the metastases? The parallel and independent evolution model proposed by Schmidt-Kittler et al88 suggests that therapies that target properties of advanced primary tumors may be ineffective against metastatic cells that evolved independently after early separation from the primary tumor89 (Figure 5). Gray90 expands on the possible consequences of sequential or parallel progression of metastasis (Figure 6).
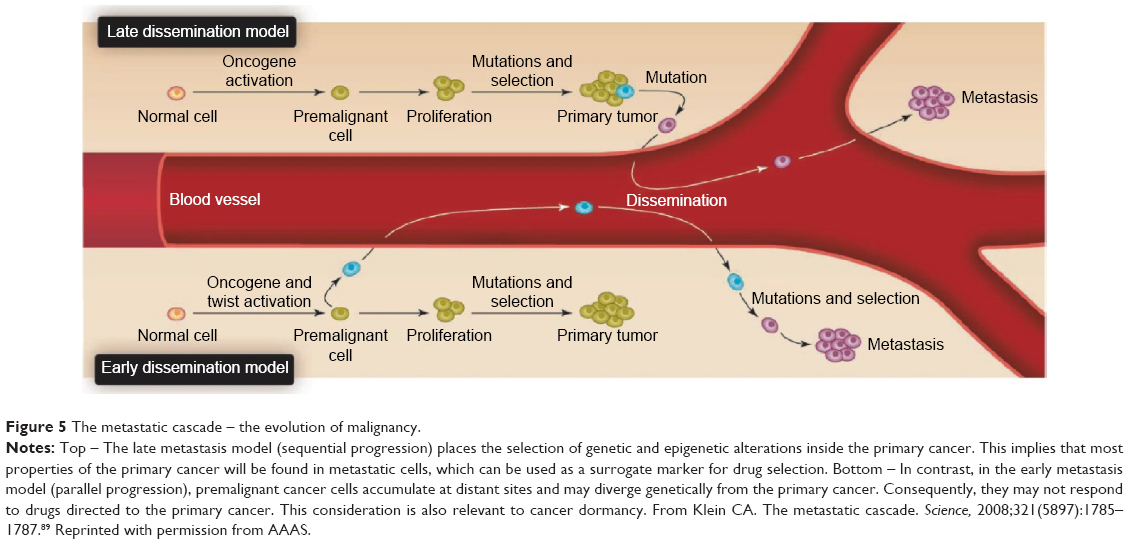 | Figure 5 The metastatic cascade – the evolution of malignancy. |
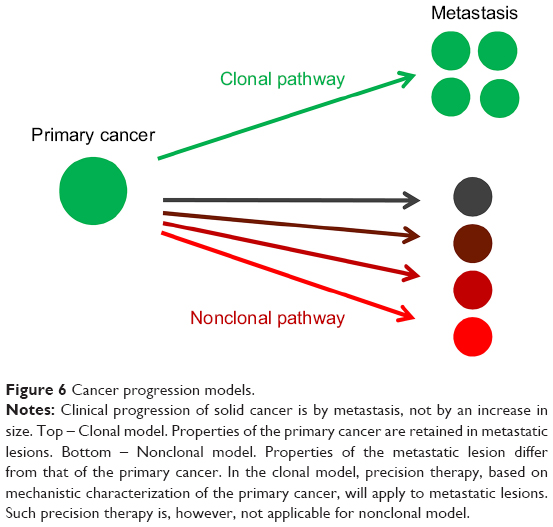 | Figure 6 Cancer progression models. |
Often, the first question addressed to academic scientists submitting a translational proposal is: What are the chances of clinical benefit? On the other hand, the first question posed to pharma scientists proposing an anticancer drug candidate directed to metastasis is: Is there a clear regulatory pathway? The usual and honest answer is: no, our drug inhibits invasion and may delay metastasis, but it does not cause tumor shrinkage. Accordingly, promising antimetastatic drug candidates are not advanced to the clinic and may explain, in part, the backlog noted earlier.91 This interaction reflects the dichotomous and tragic outcome in cancer R&D: cell biologists and medicinal chemists advance knowledge on the mechanisms of invasion and metastasis, while pharma, oncologists, and regulators are locked in the tumor-shrinkage box. The former suffer from a lack of funds, while the latter face clinical trial failures. But both camps, and especially patients and society, are the losers.
Issues
Before proceeding further, a summary may be in order. Mechanisms fundamental to solid cancer are clearly differentiated from that of hematologic cancer and benign tumors.92 Hematologic cancer is predominantly a disorder of clonal cell proliferation,93 while solid cancer is primarily a disease of cell migration, invasion across the basement membrane into normal adjacent tissue,92,94 and distant spread, termed metastasis.
In addition:
- an increase in tumor size bears no direct relationship to the timing or intensity of metastasis,
- treatment-induced tumor shrinkage is, by definition a treatment response, not a disease response, and
- metastasis is the direct or indirect cause of over 90% of morbidity and mortality.
The clinical challenge is earlier detection of curable disease, and this implies both enlightened screening and the availability of antimetastatic therapies.
Here, the central question is: which failures in the current practice medicine and public health will genomic medicine address? Based primarily on the teaching of Rose,18,95 we list seven concerns:
- Unifying molecular biology and medicine at the bench and at the bedside
- Genomics-driven oncology: framework for an emerging paradigm?
- PMs and drug resistance
- Earlier detection of disease and its consequences
- Genes and the environment
- 21st Century Cures Act
- Program costs.
Unifying molecular biology and medicine at the bench and at the bedside
Bridging the gap between the bench and the bedside is not easy, and Bates et al96 at the NCI explain that 1) not every altered protein or pathway is a valid anticancer target; 2) drugs must effectively engage the target; 3) the biology of the systems we use must be well understood; and 4) clinical trials must be designed to assess whether the drug reached and impaired the target.
Discoveries from genome-wide association studies may improve the prediction of common diseases, but the question is whether this improvement is sufficient to enable PM? Janssen and van Duijn15 argue that new gene discoveries may not improve the prediction of common diseases to a degree that it will change the management of individuals at increased risk. Improvements may only be expected if we manage to understand the complete causal mechanisms of common diseases to a similar extent as we understand those of monogenic disorders. Furthermore, although genomics research will make a major contribution to this understanding, the complexity of complex diseases may ultimately limit accurate prediction of disease in asymptomatic individuals, as unraveling their complete causal pathways may be impossible. Precision oncology is quite different from accurate oncology.
In disease, all risk factors are not created equal. The PM initiative, as presented, may give the impression that genomic considerations are more equal than traditional risk factors that have been epidemiologically validated. Horwitz et al97 wonder whether an overreliance on technology for solutions may undermine clinical and environmental sciences resulting in depersonalized medicine? Further, to maximize the potential of the PM initiative, a new taxonomy of solid cancer based on genomics will be needed. Clinicians need not fear a genomic taxonomy – yet; medicine is already personalized. It is the phenotype that walks into your clinic, is grateful for your care and kindness, and pays your bill.98
Genomics-driven oncology: framework for an emerging paradigm?
In an overview on precision oncology, Garraway et al99 present a comprehensive and systematic assessment of cancer genomic information and its accelerating clinical impact. This framework is based on the assumption that cancers are driven by genomic alterations that dysregulate oncogenic pathways, regulating cell growth and survival.100
In brief, implementation of this scheme requires:
- characterization of the tumor genome,
- filtration of genomic data through a knowledge base of US Food and Drug Administration (FDA) approved and investigational anticancer drugs, and
- presentation of an annotated drug list to the treating oncologist that can be incorporated into clinical decision making.
However, this framework fails to describe the genomic connection to the key clinical outcome in solid cancer – the development and spread of metastasis – and explain the strategy to overcome drug resistance secondary to intratumor heterogeneity. Unfortunately, none of the annotated targeted drugs have demonstrated clinical efficacy in inhibiting metastatic spread, or lack of resistance development.100 Tannock101 explains why intratumor heterogeneity may be a serious obstacle to precision treatments: “in the presence of heterogeneity, accurate genomic targeting is not possible, and resistant cells will be selected for survival”.
PMs and drug resistance
Despite outstanding advances in medicinal chemistry, the development of resistance is the major limiting factor of precisely targeted treatments.102 Vemurafenib (Zelboraf®), a B-Raf enzyme inhibitor, received marketing authorization in 2011 for the treatment of late-stage melanoma.103,104 Near-miraculous initial results105 were followed by the development of resistance and reappearance of the cancer within a year.106 Unfortunately, and in contrast to received wisdom, cancer mechanisms have not gone from a black box to a blueprint yet.21 The real chronicles of solid cancer R&D list unfulfilled promises and describe the triumph of hope and hyperbole over reality. This cycle of ecstasy and agony, as explained in the melanoma example, is mirrored in the experience of patients and their families, oncologists, and scientists.
In pharmaceuticals, imprecision has advantages, especially in multifactorial diseases. Multitargeted drugs and combinations have been successfully utilized for major chronic diseases such as hypertension, diabetes, and HIV/AIDS. Multikinase inhibitors in cancer generate much less resistance than selective ones.107 When drug resistance develops to a targeted drug, initial combination therapy has merit.108,109 In 2001, Goldie110 suggested that the “magic bullet” paradigm that had implicitly driven drug discovery until today needs to be changed to allow for the development of “magic volleys”. Fabbro,111 in his review on 25 years of small molecular weight kinase inhibitors, states that the various mechanisms of drug resistance reflect the plasticity of cancer cells and the many ways by which a tumor can evade precision therapies. Terminology is important; for a successful strategic outcome, accuracy precedes precision, not the other way round. In 2016, precisely targeted monotherapy in solid cancer is futile, and pragmatic medicine (empiric combinations, imprecise medicine) may have advantages over PM, especially in the context of drug resistance.
In solid cancer, the established dogma for managing recurrent or refractory disease is that an increase in tumor size following shrinkage should prompt a change in therapy. However, favorable responses have been reported on drug rechallenge and treatment beyond progression.112,113 These reports direct attention to the possibility of resensitization, or more analytically, a rethink of the operational definition of resistance. By “resistance” do we mean a new increase in tumor size, or a continuation of invasion and the metastatic process?47 If it implies the former, then the benefit of continuing therapy beyond “progression” (an increase in size) is not unexpected. However, if it implies a new mutationally induced driver, then, by definition, a rechallenge should not work. Intratumor heterogeneity poses a serious obstacle to PM relative to treatment response and resistance, disease progression, and relapse. Within the context of tumor evolution, Jamal-Hanjani et al114 have strategized translational approaches that involve combinatorial, adaptive, and tumor immune therapies.
Kang et al115 have reported improvement in progression-free survival in gastrointestinal stromal tumour (GIST) patients following rechallenge with imatinib, suggesting that residual bulk disease may contain clones with continuing sensitivity. Nishida and Doi116 extend this insight to targeted drugs in general and wonder whether radiographic disease progression (as assessed by RECIST criteria) always reflects disease progression and drug resistance?47 Recently, Auliac et al117 analyzed the impact of continuing first-line EGFR tyrosine kinase inhibitor (TKI) therapy beyond RECIST disease progression in patients with advanced EGFR-mutated non-small-cell lung cancer. There was no difference between the patients who did and did not continue TKI therapy with respect to progression-free survival (PFS1: 10.5 vs 9.5 months, P=0.4). Overall survival (OS) showed a nonsignificant trend in favor of continuing TKI therapy (33.0 vs 21.2 months, P=0.054). Accordingly, it is possible that with continuation of therapy, a delayed response, as with oncoimmunologics, may be noted. If prospectively confirmed, and in the context of targeted drugs, a new increase in tumor size following a response may not signify progression of the disease, nor the development of “resistance”. Accordingly, it is possible that with continuation of therapy, a delayed response, as with oncoimmunologics, may be noted. However, the central question remains: if RECIST cannot predict failure, how can it predict success?
Earlier detection of disease and its consequences
The aim of early detection is to identify disease at an earlier (asymptomatic) and curable stage. To be effective, antimetastatic drugs are required. Detection screens are not simple; there is a trade-off. Sensitive screens are not specific and may lead to false-positives (termed overdiagnosis), while specific screens can only detect advanced disease, which may not be curable. Early detection, especially if addressed with effective treatment that could delay the progression of disease, is the broader objective of screening.
However, earlier detection inflates survival by:
- Lead time bias – the time of diagnosis is brought forward, to the left, ie, earlier118 (Figure 7).
- Overdiagnosis – finding cancers never destined to progress at all, or which progress so slowly that the person dies of other causes.119–121
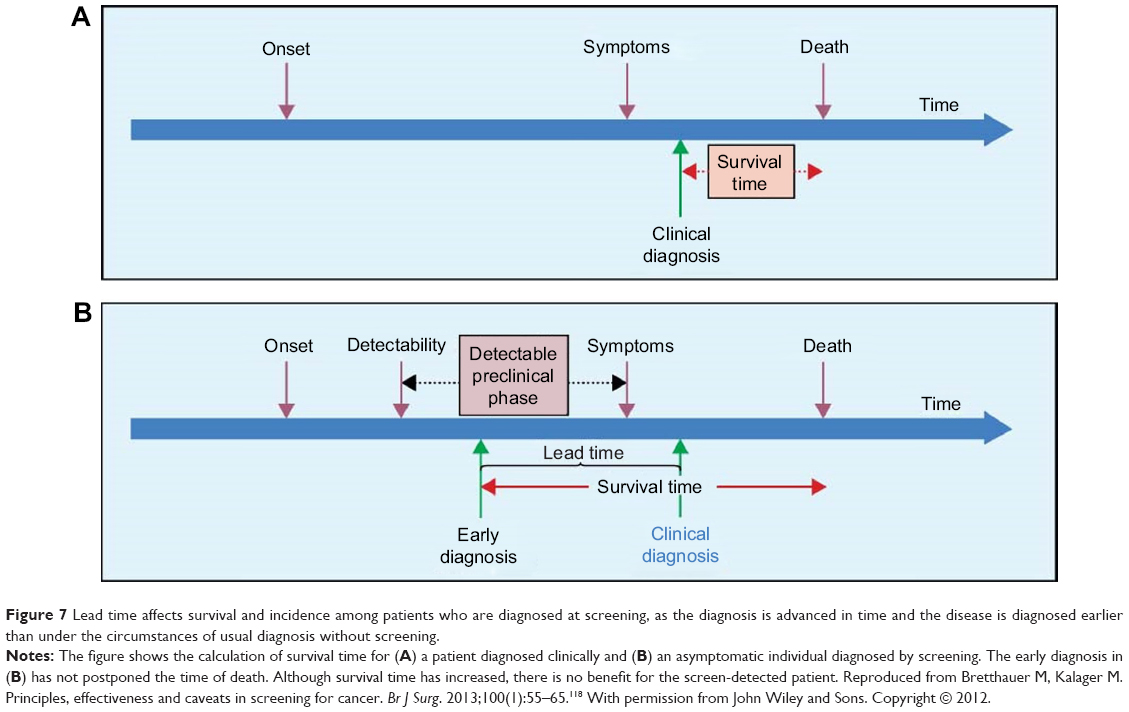 | Figure 7 Lead time affects survival and incidence among patients who are diagnosed at screening, as the diagnosis is advanced in time and the disease is diagnosed earlier than under the circumstances of usual diagnosis without screening. |
Clearly, intervention is not indicated for noncancers, but the consequences of false-positive results, namely, labeling, invasive testing to confirm a benign nature, and mental anguish, are serious considerations. This is demonstrated by the recent Korean Thyroid-Cancer “Epidemic”.122 Thyroid cancer is now the most common type of cancer diagnosed in South Korea. More than 40,000 people in the country were diagnosed with the disease in 2011 – a figure that is more than 100 times the number of people who die from thyroid cancer, which for the past decade has been between 300 and 400 each year. Despite the surge in incidence, mortality was unchanged. False-positive individuals are now regarded as “cancer survivors”. This epidemic was a consequence of the addition of thyroid screening with ultrasonography to government-funded cancer screening tests. A year later, in 2015, and in response to increased awareness of overdiagnosis, a 35% decrease in thyroid surgery was noted.123
Esserman et al term these minor, noncancer abnormalities detected on screening as “indolent lesions of epithelial origin” (IDLE) and suggest that screening guidelines need to be revised to lower the detection of IDLE’s with the same energy used to increase sensitivity.119 If not, and in contrast to the expectations of Dzau et al,11 genome diagnostics will likely increase the incidence of disease, and, exponentially, may qualify as the mother of all overdiagnoses and labeling. Here, Cho et al120 make a relevant and important suggestion – cancer registries do not routinely collect the mode of diagnosis (ie, symptomatic or screened). The addition of this data, and partitioning of analyses, would further interpretation. The more sensitive the screen, the higher the incidence of IDLE. In their 2000 perspective, Holtzman and Marteau124 explain that genotypes for common, complex diseases are incompletely penetrant, and correlations between the genotype and the phenotype are therefore weak. The operational consequences of incomplete genomic penetrance in common cancers are that there is a low magnitude of conferred risks, and therefore, a limited therapeutic potential for precision treatments.
It should be noted that the primary reason for screening is, with effective intervention, to extend meaningful life and delay mortality, not to reduce the incidence of disease. For primary prevention in populations, risk factor reduction makes more sense than genomic diagnoses, for example, smoking cessation and lung cancer. Genomic diagnostics may identify possible markers of early disease, but appropriate intervention is required to slow progression. To advocate a rush to develop genomic diagnostics before revising regulations governing useful drug approval, and refining screening guidelines and reporting, is to put the cart before the horse. In solid cancer, without effective medicines, an extension of useful life, via screening only, is an illusion.
A valuable insight into the perils of indiscriminate cancer screening has recently been publicized by Gilbert Welch and Peter Albertsen125 at The Dartmouth Institute and University of Connecticut Health Center, respectively: the heterogeneity in cancer can be described through the metaphor of birds, rabbits, and turtles. The goal of early detection is not to let any of the animals escape the barnyard and cause a cancer death.
- But the birds have already flown away. They are the most aggressive cancers, the ones that have already spread by the time they are detectable, the ones that are beyond cure.
- The rabbits, potentially lethal cancers that might benefit from treatment, are ready to hop out at any time. These are the cancers we hope to control with early detection.
- Then there are the turtles – these are nonlethal cancers that are not going anywhere. Screening is really good at finding these cancers.125
Following Hall et al,30 before genetic screening is launched on a national scale, the following criteria need to be satisfied.
- Genomic information will predict disease risk better than phenotypic information.
- Cost-effective interventions exist for those at increased genetic risk.
- These interventions will be more cost effective than population-level interventions.
- Genetic risk information will motivate desired behavior change.
Currently, and for the foreseeable future, there are no examples of genetic screening for disease risk that satisfy these criteria.30
Genes and the environment
Gene–environmental interactions are fundamental to the pathogenesis of disease, especially cancer; these influences cannot be separated. In some cancers, genetic determinants predominate, while in others, the environment is more relevant. Genetic factors usually dominate individual susceptibility but do not explain population differences in incidence. And genetic heterogeneity is much greater within, than between, populations. This is opposite to that seen for environmental factors. Several types of cancer vary in incidence by more than an order of magnitude between different populations. The convergence toward local cancer rates seen among immigrants excludes a genetic explanation of these differences.18 A high-penetrance gene defines the phenotype in individuals who carry it. A weakness of the PM logic in oncology is a consequence of the incomplete penetrance of genotypes in solid cancer, the limited ability to tailor treatment to genotypes, and the low magnitude of risks conferred by various genotypes for the population at large. The vast majority of solid cancers are associated with low penetrance genes that require an environmental contribution to allow for expression. It would be truly revolutionary if we could determine the genotypes of the majority of people who will get common cancers.124
In 1985, Geoffrey Rose18 demonstrated that the determinants of the population distribution of disease have less to do with the individual-level risk factors that promote disease within a population, and more to do with the macrolevel structural causes that shape entire distributions of disease across populations. As a young scientist, his grant application was rejected because the reviewers stated that even in national/international programs, clinical research should concentrate on individuals, not populations. The opposite opinion is now a fundamental principle in epidemiology and environmental medicine: the population carries a collective responsibility for its own health and well-being, including that of its deviants (those with disease).126
In 2015, Keyes et al127 at the Columbia University, New York, USA, analyzed the mathematical limits of genetic prediction for complex chronic disease, and confirmed Rose’s 1985 insight. When common germline genetic variants are insufficient to produce disease, the predictive capacity of the genetic variant alone will be determined by the prevalence of factors that interact with the variant and the rate of the disease in the population. Genetic variants will be strong predictors of disease only when the factors that interact with the disease are common and when the background rate of disease is rare.
21st Century Cures Act
In May 2015, the 21st Century Cures Act was introduced in the US House of Representatives, with the goal of promoting the development and speeding the approval of new drugs and devices.4,11 In committee, the Bill was approved unanimously (51-0). The main focus was funding for the National Institutes of Health/NCI, and reform at FDA toward simplifying and accelerating the new drug and diagnostics approval process, and incorporating PM-related elements,3 specifically the inclusion of genomic-based biomarkers and surrogate end points in clinical trials. Several “simplifying proposals”, namely, a reliance on clinical experience, observational studies, and registries, instead of randomized, controlled trials, and on biomarkers and other surrogate measures rather than actual clinical end points, are used to define clinical outcome. Gonsalves et al,128 leaders in regulatory science, have stated that these proposals could lead to the weakening of FDA’s process and controls, and to the approval of drugs that are less safe or effective, or trial failure of a truly innovative medicines. In their opinion, it is unlikely that better drugs would emerge via diluted regulations.
Program costs
Today, cancer drug pricing is dominating the news. At the 2015 American Society of Clinical Oncology meeting, Dr Leonard Salz of the Memorial Sloan Kettering Cancer Center, New York, stated: These drugs cost too much. PM emphasizes expected benefits to patients but is silent on costs. Recently, 100 oncologists presented an initiative to lower cancer drug prices,129 but the proposal did not define the effectiveness and value of drugs for solid cancer – is metastasis delayed and is there an extension of meaningful life?22 Efforts to lower the price of ineffective drugs may be to miss the point? Young130 notes that the costs of cancer drugs amount to only 5%–20% of the total costs of cancer care, depending on how many of the multiple cost components are included. Accordingly, and to be fair, attention may also be directed to the costs of care that are determined and driven by oncologists, hospitals, and health insurance.
Advances in sequencing have accelerated the identification of several new gene mutations or amplifications that may be appropriate for genomic-driven diagnostics and drug development. We assume that with the significant decline in the costs of goods – genomic sequencing – R&D costs will undergo a similar decline, and that pricing for PM-translated drugs will be affordable7 (Figure 8).
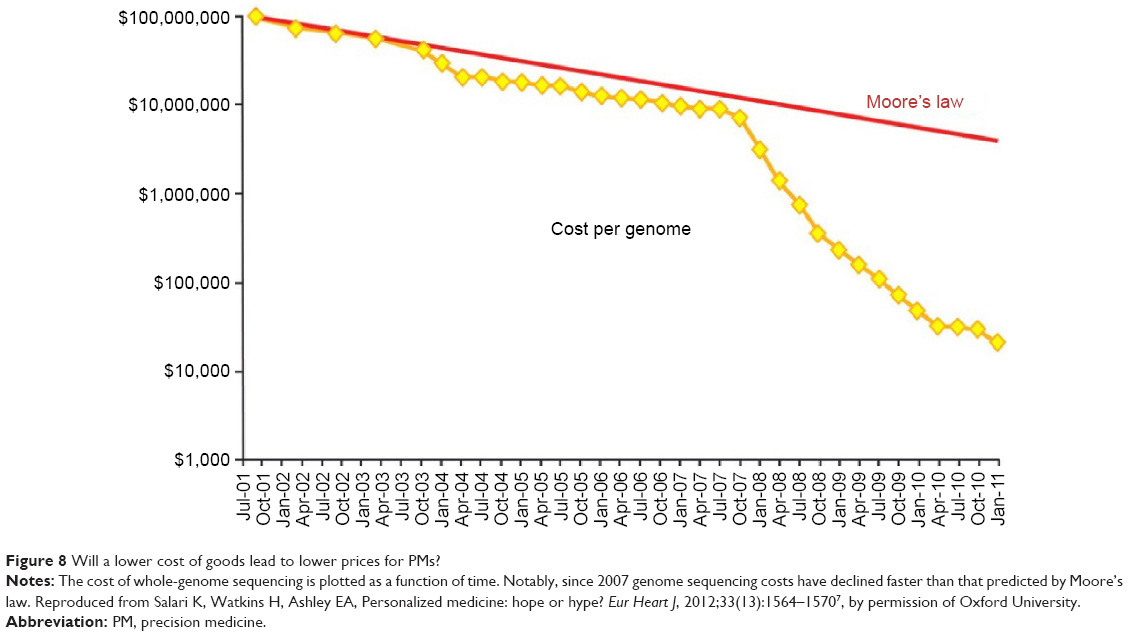 | Figure 8 Will a lower cost of goods lead to lower prices for PMs? |
The dramatic drop in costs of sequencing technologies in genomic testing has resulted in widespread consumer marketing. These tests provide individual risks for several complex diseases and phenotypic traits, and are targeted to healthy individuals as a form of genomic screening. Bunnik et al131 suggest that a rethink of medical evaluation and regulation, with a focus on the reduction of harm, may be necessary and timely.
In contrast to most research-intensive industries, consumer electronics for example, we have not seen improved medicines at affordable prices. Disruptive invention occurs in academic and pharma laboratories, but regulations both favor and force the development of commodity drugs – tumor shrinkage. As a consequence, pricing is unrelated to novel utility and meaningful benefit to patients, but based solely on what the market can bear.24 Clearly, the business model is broken, and can only be corrected by a regulatory and reimbursement rethink from a rational and public health standpoint. With strong and disciplined stakeholder efforts, we have succeeded with HIV/AIDS in both high- and low-income countries.26,27,132 Why not in solid cancer?
The cancer “moonshot” program – 2016
Overall, the PM initiative should be viewed as an encouragement to the furtherance of science in the service of medicine. It would be best for scientists and physicians to outline the program. Unfortunately, in this case, the government has identified genomics as the sole platform for diagnostics and therapeutics. Complex diseases require multidisciplinary effort, and the PM initiative with its emphasis on genomics may exclude related disciplines of epidemiology, pharmacology, biology, chemistry, and especially public health, from recognition and support. In 2015, universities have already created Genome Institutes, and we note a migration of students toward these institutes and away from traditional and proven disciplines. When asked for the reason, they quote the legendary bank robber Willie Sutton, who replied to the judge’s inquiry as to why he robbed banks by saying “because that is where the money is”. If prudence and caution are not exercised, Vice President Biden’s “moonshot” program, based on the promise of genomics, may suffer the same fate as a previous plan based on retrovirology.133
Conclusion – Paul Ehrlich: magister mundialis pragmaticae medicinae
The fundamental question is: whether conventional research in solid cancer therapeutics is on the right track, and if not, is PM the solution?1 The answer requires serious introspection.134 All things considered, we arrive at the impression that the PM initiative represents a technology push driven by the rapidly decreasing costs of genomic technology (solutions), rather than a demand pull (patient needs). Consistent with PM strategy, Dyson,135 at the Institute for Advanced Study, Princeton, in clear contrast to Kuhn,20 makes a case for the primacy of new tools, rather new ideas as a driver of innovation. However, the enthusiasm toward genomic technology, especially screening,11 in the absence of a novel disease/treatment paradigm, may be akin to the tail wagging the dog. In solid cancer, advances in technology may have outpaced an understanding of disease, and medical and public health issues may not have been addressed in a thoughtful, structured, and disciplined manner.15,30,31,64,124,136
PM is predicated on a complete understanding of disease mechanisms. According to the Centers of Disease Control and Prevention (USA), sickle-cell disease is a life-threatening disorder that affects millions of people throughout the world and is particularly common among those whose ancestors came from sub-Saharan Africa; Spanish-speaking regions in the Western hemisphere (South America, the Caribbean, and Central America); Saudi Arabia; India; and Mediterranean countries such as Turkey, Greece, and Italy. The disease affects nearly 100,000 individuals of African origin in the USA.137 A complete understanding of the mechanism of sickle-cell anemia has been known for over a half century, but we are still awaiting a PM. While we are still far from a precise understanding of mechanisms of hypertension, diabetes, and infectious diseases, especially HIV/AIDS, this has not prevented the development of medicines that are meaningfully effective, globally available, and affordable. Bentires-Alj et al138 explain that translational research leaves no one indifferent, and everyone expects a particular benefit. The collective objective is to identify measures to promote translational research without undermining basic exploratory research and academic freedom. Multidisciplinary basic research should be translated in a pragmatic manner.68,138
The American philosopher, James (1842–1910),139 explains that pragmatism is a new name for some old ways of thinking, namely, the test of expected utility. He states that pragmatism emphasizes practical consequences in determining the criterion of meaning, truth, or value. The relationship of pragmatism to translation is obvious and focuses on the development of products that demonstrate meaningful efficacy, here, and now. He advises that applicable knowledge is derived from verified facts and actions, rather than “logical proofs, or principles, or databases”. We consider Paul Ehrlich (1854–1915), as the father of pragmatic medicine.140,141 When the world, a century ago, faced an epidemic scourge, syphilis, Paul Ehrlich and Sahachiro Hata, with training in biology and chemistry, but scant resources, initiated an R&D program on organic arsenicals. In 1910, after 605 failures, they synthesized and marketed the first, relatively safe, and effective treatment for syphilis – Salvarsan or compound 606.
In 2010, Ledford142 reported that the cancer database would soon be flooded with genome sequences from 25,000 cancers, and interpretation would be the challenge. If asked, William James would tell us that building a bigger haystack is not the best way of finding the needle. And he would likely agree with Roberts et al143 at the Johns Hopkins, Baltimore, MD, USA, who states that it is going to take good old-fashioned biology to really determine what is going on. But today, the prospects of getting funded for good old-fashion biology are slim, if any. Again, it is self-evident that without multidisciplinary basic research in biology, there will be little to translate.68
The promise of huge clinical and monetary benefits of the PM initiative, as outlined by Collins and Varmus3 and Dzau et al,11 is based on the expected prediction of common multifactorial diseases using an economic simulation model. Roberts et al143 had earlier demonstrated that an analysis on whole-genome sequencing of monozygotic twin pairs (twins of a pair share the same genome type and therefore identical genetic risk factors) did not predict risk of most of 24 common diseases, including type 2 diabetes, cancers, stroke, and coronary heart diseases.144 On the one hand, Matuchansky,144 at the Diderot University, Paris, questions the prediction made by Dzau et al11 that PM would lead to substantial cumulative gains (expressed using US$100,000 per quality-adjusted life-year, with a $33 billion gain at a reduced disease incidence of 10% and up to a $607 billion gain at a 50% incidence reduction) in life expectancy and quality-adjusted life expectancy during the subsequent 50 years. He states that the proposed alignment of incentives to fulfill the potential of PM is only a promise drawn from a model, which is, itself, still a promise.144 On the other hand, Doble et al145 at the Garvan Institute, Australia, agree that genomics may have a role in population health. However, and in contrast to Dzau et al,11 the priority and benefits of PM will be limited to individuals at high risk of imminent, serious, preventable, penetrant disorders that have large health-care costs. These expert views have important implications for the valuation of new genomic initiatives by industry, public policy makers, university administrators, health insurance companies, patients, and of course, cancer-funding agencies. Following Welch,123,125 we hope that examples cited here will encourage physicians to find their voice when medical trends run counter to their patients’ interests.
The challenge facing precision oncology is huge; solid cancer is not a single entity, and as noted may involve 25,000 cancer subtypes. Complexity aside, the cost of R&D for precision diagnostics and drugs, especially clinical trials, and regulatory requirements, would be enormous. Here, pragmatic medicine offers guidance and advises that we focus on “common denominators and not differences”. Cancer is conventionally classified as hematologic or solid. In hematologic cancer (eg, leukemias), clonal proliferation is the predominant mechanism,93 while solid cancer is mainly characterized by tissue invasion and metastasis, and the cancer tumor microenvironment (stroma) displays varying degrees of desmoplasia.47,146
Tissue invasion and metastasis relies primarily on motility and migration of the cancer cell,77,80–84,94,147 assisted by epithelial–mesenchymal plasticity,83,86,148–151 while desmoplasia is a consequence of fibroblastic activity. Economy of effort suggest that attention be best directed to inhibiting cell motility and factors promoting cell aggressiveness, namely hypoxia83,151 and desmoplastic containment of the cancer stroma.146,152 Whatcott et al152 assessed desmoplasia in metastatic lesions of panceatic ductal adenocarcinoma and compared it with that of primary tumors. Both primary tumors and metastases had a highly fibrotic stroma. A negative correlation was noted between patient survival and extracellular matrix (collagen I or hyaluronan) deposition. The authors suggest that stromal-targeting agents have the potential to benefit patients with solid cancer, even those with metastatic disease.152 We infer that a focus on the initial steps in the metastatic cascade may be more rewarding than exploring downstream mechanisms complicated by dense signaling cross-talk.
In this context, we draw attention to lines of inquiry that illustrate the pragmatic approach in solid cancer: that of Cox and Erler153 at the Biotech Research and Innovation Centre, Copenhagen, on their work on connecting tissue stiffness, fibrosis, and solid tumor metastasis; and Olson et al at the Cancer Research UK Beatson Institute, Glasgow; the late Marshall et al at the Institute of Cancer Research, London; and Yun et al at the Penn State Hershey College of Medicine, USA, on the demonstration of Rho kinase/MRCK inhibitors in blocking cancer cell migration and metastasis.154–156
In conclusion, we hope that a consideration of the points raised in this essay may be relevant to the ongoing debate on PM and the 21st Century Cures Act. In particular, the successful HIV/AIDS program serves as a global model of Pragmatic Medicine in operation – tangible utility, here, and now. We should recall with both pride and humility that this program was a pragmatic, community-initiated, translation-driven, regulatory-supported, public health initiative, and a direct response to expressed need of patients and their families. It can be repeated for solid cancer!
Acknowledgment
We thank Roberto Fiorentini, MD, Washington, DC, for his comments on pharma and regulatory strategy.
Author contributions
All authors contributed equally, made substantial contributions to the conception, design, and drafting this essay, and have approved the final version.
Disclosure
JB and DR are supported by grants from the Kellner Family Foundation Principal Investigator Grant, the project BIOCEV–Biotechnology and Biomedicine Center of the Academy of Sciences and Charles University (CZ.1.05/1.1.00/02.0109), and from the European Regional Development Fund. MF is at Medbase, an independent think tank focused on pragmatic innovation in pharmaceutical products and medical devices. The authors report no other conflicts of interest in this work.
References
Leek, JT, Peng, RD. What is the question? Science. 2015;347(6228):1314–1315. | ||
Brábek J, Fernandes M. Affordable cancer care. Lancet Oncol. 2012;13(1):2–3. | ||
Collins FS, Varmus H. A new initiative on precision medicine. N Engl J Med. 2015;372(9):793–795. | ||
Avorn J, Kesselheim AS. The 21st Century Cures Act – will it take us back in time? N Engl J Med. 2015;372(26):2473–2475. | ||
Wild CP, Bucher JR, de Jong BW, et al. Translational cancer research: balancing prevention and treatment to combat cancer globally. J Natl Cancer Inst. 2015;107:dju353. | ||
Khoury MJ, Evans JP. A public health perspective on a national precision medicine cohort: balancing long-term knowledge generation with early health benefit. JAMA. 2015;313(21):2117–2118. | ||
Salari K, Watkins H, Ashley EA. Personalized medicine: hope or hype? Eur Heart J. 2012;33(13):1564–1570. | ||
Blumenthal D, Abrams M, Nuzum R. The Affordable Care Act at 5 years. N Engl J Med. 2015;372(25):2451–2458. | ||
Topol EJ. Individualized medicine from prewomb to tomb. Cell. 2014;157(1):241–253. | ||
Rubin R. Precision medicine: the future or simply politics? JAMA. 2015;313(11):1089–1091. | ||
Dzau VJ, Ginsburg GS, Van Nuys K, Agus D, Goldman D. Aligning incentives to fulfill the promise of personalised medicine. Lancet. 2015;385(9982):2118–2119. | ||
Aronson SJ, Rehm HL. Building the foundation for genomics in precision medicine. Nature. 2015;526(7573):336–342. | ||
Jameson JL, Longo DL. Precision medicine – personalized, problematic, and promising. N Engl J Med. 2015;372(23):2229–2234. | ||
Doshi P. 21st century cures: is US medicines bill a colossal mistake? BMJ. 2015;351. | ||
Janssens AC, van Duijn CM. Genome-based prediction of common diseases: advances and prospects. Hum Mol Genet. 2008;17(R2):R166–R173. | ||
Joyner MJ, Paneth N. Seven questions for personalized medicine. JAMA. 2015;314(10):999–1000. | ||
Bradford Hill A. The environment and disease: association or causation. Proc R Soc Med. 1965;58:295–300. | ||
Rose G. Sick individuals and sick populations. Int J Epidemiol. 2001;30(3):427–432. | ||
Vineis P. A self-fulfilling prophecy: are we underestimating the role of the environment in gene–environment interaction research? Int J Epidemiol. 2004;33(5):945–946. | ||
Kuhn TS. The Structure of Scientific Revolutions. 50th Anniversary edition. Chicago, IL: University of Chicago Press; 2012. | ||
DeVita VT, Rosenberg SA. Two hundred years of cancer research. N Engl J Med. 2012;366(23):2207–2214. | ||
Hoag H. Cancer drugs should add months, not weeks, say experts. Nature Med. 2011;17(1):7. | ||
Ward, A. Healthcare: counting the cost of cancer. Financial Times, January 15, 2015. | ||
Mailankody S, Prasad V. Five years of cancer drug approvals innovation, efficacy, and costs. JAMA Oncol. 2015;1(4):539–540. | ||
Farmer P, Frenk J, Knaul FM, et al. Expansion of cancer care and control in countries of low and middle income: a call to action. Lancet. 2010;376(9747):1186–1193. | ||
Gallo RC, Montagnier L. The discovery of HIV as the cause of AIDS. N Engl J Med. 2003;349(24):2283–2285. | ||
Volberding P. The impact of HIV research on health outcome and healthcare policy. Ann Oncol. 2011;22(Suppl 7):50–53. | ||
Piot P, Quinn, TC. Response to the AIDS pandemic – a global health model. N Engl J Med. 2013;368(23):2210–2218. | ||
Edmondson AC. Strategies for learning from failure. Harvard Bus Rev. 2011;89(4):48–55. | ||
Hall WD, Mathews R, Morley KI. Being more realistic about the public health impact of genomic medicine. PLoS Med. 2010;7(10):e1000347. | ||
Bayer R, Galea S. Public health in the precision-medicine era. N Engl J Med. 2015;373(6):499–501. | ||
Burton PR, Tobin MD, Hopper JL. Key concepts in genetic epidemiology. Lancet. 2005;366(9489):941–951. | ||
Ananthakrishnan R, Menon S. Design of oncology clinical trials: a review. Crit Rev Oncol Hematol. 2013;88(1):144–153. | ||
Simon R, Roychowdhury S. Implementing personalized cancer genomics in clinical trials. Nat Rev Drug Discov. 2013;12(5):358–369. | ||
Marzuillo C, De Vito C, D’Andrea E, Rosso A, Villari P. Predictive genetic testing for complex diseases: a public health perspective. QJM. 2014;107(2):93–97. | ||
Arnedos M, Soria JC, Andre F, Tursz T. Personalized treatments of cancer patients: a reality in daily practice, a costly dream or a shared vision of the future from the oncology community? Cancer Treat Rev. 2014;40(10):1192–1198. | ||
Meric-Bernstam F, Mills GB. Overcoming implementation challenges of personalized cancer therapy. Nat Rev Clin Oncol. 2012;9(9):542–548. | ||
Stahel R, Bogaerts J, Ciardiello F, et al. Optimising translational oncology in clinical practice: strategies to accelerate progress in drug development. Cancer Treat Rev. 2015;41(2):129–135. | ||
Ciardiello F, Arnold D, Casali PG, et al. Delivering precision medicine in oncology today and in future – the promise and challenges of personalised cancer medicine: a position paper by the European Society for Medical Oncology (ESMO). Ann Oncol. 2014;25(9):1673–1678. | ||
Booth CM, Tannock IF. Randomised controlled trials and population-based observational research: partners in the evolution of medical evidence. Br J Cancer. 2014;110:551–555. | ||
Kocher R, Roberts B. The calculus of cures. N Engl J Med. 2014;370:1473–1475. | ||
Kravitz RL, Duan N, Niedzinski EJ, Hay MC, Subramanian SK, Weisner TS. Whatever happened to N-of-1 trials? Insiders’ perspectives and a look to the future. Milbank Q. 2008;86(4):533–555. | ||
Schork, NJ. Personalized medicine: time for one-person trials. Nature. 2015;520:609–611. | ||
Collette L, Tombal B. N-of-1 trials in oncology. Lancet Oncol. 2015;16:885–886. | ||
Eisenhauer EA, Therasse P, Bogaerts J, et al. New response evaluation criteria in solid tumors: revised RECIST guideline version 1.1. Eur J Cancer. 2009;45(2):228–247. | ||
Ganten MK, Ganten TM, Schlemmer HP. Radiological monitoring of the treatment of solid tumors in practice. Fortschr Röntgenstr. 2014;186(5):466–473. | ||
Fernandes M, Rosel D, Brábek J. Translation in solid cancer: are size-based response criteria an anachronism? Clin Transl Oncol. 2015;17:1–10. | ||
Coche E. Recist and beyond. JBR-BTR. 2013;96:167–171. | ||
Diederich S. Imaging beyond RECIST: CT and MRI in molecular therapies. Cancer Imaging. 2012;12:347–350. | ||
Husband JE, Schwartz LH, Spencer J, et al. Evaluation of the response to treatment of solid tumours – a consensus statement of the International Cancer Imaging Society. Br J Cancer. 2004;90(12):2256–2260. | ||
Oxnard GR, Morris MJ, Hodi FS, et al. When progressive disease does not mean treatment failure: reconsidering the criteria for progression. J Natl Cancer Inst. 2012;104(20):1534–1541. | ||
Krajewski KM, Franchetti Y, Nishino M, et al. 10% Tumor diameter shrinkage on the first follow-up computed tomography predicts clinical outcome in patients with advanced renal cell carcinoma treated with angiogenesis inhibitors: a follow-up validation study. Oncologist. 2014;19(5):507–514. | ||
Chen HX, Rubinstein LV, Shankar LK, Abrams JS. Are we ready for the 10% solution? Oncologist. 2014;19(5):439–440. | ||
Höink AJ, Heindel W, Buerke B. Radiological evaluation of the therapeutic response of malignant diseases: status quo, innovative developments and requirements for radiology. Fortschr Röntgenstr. 2014;186(10):927–936. | ||
Fernandes M. Judging a cancer drug: Avastin’s story [letter]. New York Times. 2011;160(55423):section A. | ||
Gwyther SJ, Schwartz LH. How to assess anti-tumour efficacy by imaging techniques. Eur J Cancer. 2008;44(1):39–45. | ||
Bidard F-C, Pierga J-Y, Soria JC, Thiery JP. Translating metastasis-related biomarkers to the clinic – progress and pitfalls. Nat Rev Clin Oncol. 2013;10(3):169–179. | ||
Fein MR, Egeblad M. Caught in the act: revealing the metastatic process by live imaging. Dis Model Mech. 2013;6(3):580–593. | ||
Mankoff DA, Farwell MD, Clark AS, Pryma DA. How imaging can impact clinical trial design: molecular imaging as a biomarker for targeted cancer therapy. Cancer J. 2015;21(3):218–224. | ||
Li C. A targeted approach to cancer imaging and therapy. Nat Mater. 2014;13(2):110–115. | ||
Jung KH, Lee KH. Molecular imaging in the era of personalized medicine. J Pathol Transl Med. 2015;49(1):5–12. | ||
Ensor JE. Biomarker validation: common data analysis concerns. Oncologist. 2014;19(8):886–891. | ||
Aronson JK. Research priorities in biomarkers and surrogate end-points. Br J Clin Pharmacol. 2012;73(6):900–907. | ||
Fleming TR, DeMets DL. Surrogate end points in clinical trials: are we being misled. Ann Intern Med. 1996;125(7):605–613. | ||
Prentice RL. Surrogate endpoints in clinical trials: definition and operational criteria. Stat Med. 1989;8(4):431–440. | ||
Berger VW. Does the Prentice criterion validate surrogate endpoints? Statist Med. 2004;23(10):1571–1578. | ||
Huber P, Howard P. What failed, the new cancer treatment or regulators? The Wall Street Journal. February 3, 2015. | ||
Bertuzzi S, Cleveland DW. The curious incident of the translational dog that didn’t bark. Trends Cell Biol. 2015;25(4):187–189. | ||
Wilson MK, Collyar D, Chingos DT, et al. Outcomes and endpoints in cancer trials: bridging the divide. Lancet Oncol. 2015;16(1):e43–e52. | ||
Sleeman J, Steeg PS. Cancer metastasis as a therapeutic target. Eur J Cancer. 2010;46(7):1177–1180. | ||
Eccles SA, Welch DR. Metastasis: recent discoveries and novel treatment strategies. Lancet. 2007;369(9574):1742–1757. | ||
Weber GF. Why does cancer therapy lack effective anti-metastasis drugs? Cancer Lett. 2013;328(2):207–211. | ||
Elvin P, Garner AP. Tumour invasion and metastasis: challenges facing drug discovery. Curr Opin Pharmacol. 2005;5(4):374–381. | ||
Fabbro D, Cowan-Jacob, SW, Moebitz H. Ten things you should know about protein kinases: IUPHAR Review 14. Br J Pharmacol. 2015;172(11):2675–2700. | ||
Ordóñez-Morán P, Huelsken J. Complex metastatic niches: already a target for therapy? Curr Opin Cell Biol. 2014;31:29–38. | ||
Pein M, Oskarsson T. Microenvironment in metastasis: roadblocks and supportive niches. Am J Physiol Cell Physiol. 2015;309(10):C627–C638. | ||
Brabek J, Mierke CT, Rosel D, Vesely P, Fabry B. The role of the tissue microenvironment in the regulation of cancer cell motility and invasion. Cell Commun Signal. 2010;8:22. | ||
Sleeman JP, Christofori G, Fodde R, et al. Concepts of metastasis in flux: the stromal progression model. Semin Cancer Biol. 2012;22(3):174–186. | ||
Eckhardt BL, Francis PA, Parker BS, Anderson RL. Strategies for the discovery and development of therapies for metastatic breast cancer. Nat Rev Drug Discov. 2012;11(6):479–497. | ||
Wells A, Grahovac J, Wheeler S, Ma B, Lauffenburger D. Targeting tumor cell motility as a strategy against invasion and metastasis. Trends Pharmacol Sci. 2013;34(5):283–289. | ||
Kosla J, Paňková D, Plachý J, et al. Metastasis of aggressive amoeboid sarcoma cells is dependent on Rho/ROCK/MLC signaling. Mol Cancer Res. 2013;11(10):1235–1247. | ||
Bordeleau F, Alcoser TA, Reinhart-King CA. Physical biology in cancer. 5. The rocky road of metastasis: the role of cytoskeletal mechanics in cell migratory response to 3D matrix topography. Am J Physiol Cell Physiol. 2014;306(2):C110–C120. | ||
Gilkes DM, Semenza GL, Wirtz D. Hypoxia and the extracellular matrix: drivers of tumour metastasis. Nat Rev Cancer. 2014;14(6):430–439. | ||
Fife CM, McCarroll JA, Kavallaris M. Movers and shakers: cell cytoskeleton in cancer metastasis. Br J Pharmacol. 2014;171(24):5507–5523. | ||
Matsuoka T, Yashiro M. Rho/ROCK signaling in motility and metastasis of gastric cancer. World J Gastroenterol. 2014;20(38):13756–13766. | ||
Tsai JH, Yang J. Epithelial-mesenchymal plasticity in carcinoma metastasis. Genes Dev. 2013;27(20):2192–2206. | ||
Steeg PS, Anderson RL, Bar-Eli M, et al. An open letter to the FDA and other regulatory agencies: Preclinical drug development must consider the impact on metastasis. Clin Cancer Res. 2009;15:4529–4530. | ||
Schmidt-Kittler O, Ragg T, Daskalakis A, et al. From latent disseminated cells to overt metastasis: genetic analysis of systemic breast cancer progression. PNAS. 2003;100(13):7737–7742. | ||
Klein CA. The metastatic cascade. Science. 2008;321(5897):1785–1787. | ||
Gray JW. Evidence emerges for early metastasis and parallel evolution of primary and metastatic tumors. Cancer Cell. 2003;4(1):4–6. | ||
Brábek J, Fernandes M. Reasons for the R&D crisis. The Scientist, September 23, 2013. Available from: http://www.the-scientist.com/?articles.view/articleNo/37522/title/Opinion--Reasons-for-the-R-D-Crisis/. Accessed March 4, 2016. | ||
Lazebnik Y. What are the hallmarks of cancer? Nat Rev Cancer. 2010;10(4):232–233. | ||
Nowell PC. The clonal evolution of tumor cell populations. Science. 1976;194(4260):23–28. | ||
Tolde O, Rösel D, Veselý P, Folk P, Brábek J. The structure of invadopodia in a complex 3D environment. Eur J Cell Biol. 2010;89(9):674–680. | ||
Burton H, Sagoo GS, Pharoah P, Zimmern RL. Time to revisit Geoffrey Rose: strategies for prevention in the genomic era? Italian J Public Health. 2012;9(4):e8665, 1–9. | ||
Bates SE, Amiri-Kordestani L, Giaccone G. Drug development: portals of discovery. Clin Cancer Res. 2012;18(1):23–32. | ||
Horwitz RI, Cullen MR, Abell J, Christian JB. (De)personalized medicine. Science. 2013;339(6124):1155–1156. | ||
Murray JF. Personalized medicine: been there, done that, always needs work! Am J Respir Crit Care Med. 2012;185(12):1251–1252. | ||
Garraway LA, Verweij J, Ballman KV. Precision oncology: an overview. J Clin Oncol. 2013;31(15):1803–1805. | ||
Garaway LA. Genomics-driven oncology: framework for an emerging paradigm. J Clin Oncol. 2013;31(15):1806–1814. | ||
Tannock, I. Words of wisdom. Re: Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. Eur Urol. 2014;65(4):846–847. | ||
Izar B, Rotow J, Gainor J, Clark J, Chabner B. Pharmacokinetics, clinical indications, and resistance mechanisms in molecular targeted therapies in cancer. Pharmacol Rev. 2013;65(4):1351–1395. | ||
Tsai J, Lee JT, Wang W, et al. Discovery of a selective inhibitor of oncogenic B-Raf kinase with potent antimelanoma activity. PNAS. 2008;105(8):3041–3046. | ||
Bollag G, Tsai J, Zhang J, et al. Vemurafenib: the first drug approved for BRAF-mutant cancer. Nat Rev Drug Discov. 2012;11(11):873–886. | ||
Chapman PB, Hauschild A, Robert C, et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med. 2011;364(26):2507–2516. | ||
Solit DB, Rosen N. Resistance to BRAF inhibition in melanomas. N Engl J Med. 2011;364(8):772–774. | ||
Faivre S, Djelloul S, Raymond E. New paradigms in anticancer therapy: targeting multiple signaling pathways with kinase inhibitors. Semin Oncol. 2006;33(4):407–420. | ||
Sawyers CL. Combined forces. Nature. 2013;498(7455):S7. | ||
Komarova NL, Boland CR. Cancer: calculated treatment. Nature. 2013;499(7458):291–292. | ||
Goldie JH. Drug resistance in cancer: a perspective. Cancer Metastasis Rev. 2001;20(1–2):63–68. | ||
Fabbro D. 25 Years of small molecular weight kinase inhibitors: potentials and limitations. Mol Pharmacol. 2015;87(5):766–775. | ||
Kuczynski EA, Sargent DJ, Grothey A, Kerbel RS. Drug rechallenge and treatment beyond progression – implications for drug resistance. Nat Rev Clin Oncol. 2013;10(10):571–587. | ||
Tonini G, Imperatori M, Vincenzi B, Frezza AM, Santini D. Rechallenge therapy and treatment holiday: different strategies in management of metastatic colorectal cancer. J Exp Clin Cancer Res. 2013;32:92. | ||
Jamal-Hanjani M, Quezada SA, Larkin J, Swanton C. Translational implications of tumor heterogeneity. Clin Cancer Res. 2015;21(6):1258–1266. | ||
Kang YK, Ryu MH, Yoo C, et al. Resumption of imatinib to control metastatic or unresectable gastrointestinal stromal tumours after failure of imatinib and sunitinib (RIGHT): a randomised, placebo-controlled, phase 3 trial. Lancet Oncol. 2013;14(12):1175–1182. | ||
Nishida T, Doi T. Rechallenge of drugs in the era of targeted therapy. Lancet Oncol. 2013;14(12):1143–1145. | ||
Auliac JB, Fournier C, Audigier Valette C, et al. Impact of continuing first-line EGFR tyrosine kinase inhibitor therapy beyond RECIST disease progression in patients with advanced EGFR-mutated non-small-cell lung cancer (NSCLC): retrospective GFPC 04-13 study. Target Oncol. Epub 2015 Aug 29. | ||
Bretthauer M, Kalager M. Principles, effectiveness and caveats in screening for cancer. Br J Surg. 2013;100(1):55–65. | ||
Esserman LJ, Thompson IM, Reid B, et al. Addressing overdiagnosis and overtreatment in cancer: a prescription for change. Lancet Oncol. 2014;15(6):234–242. | ||
Cho H, Mariotto AB, Schwartz LM, Luo J, Woloshin S. When do changes in cancer survival mean progress? The insight from population incidence and mortality. J Natl Cancer Inst Monogr. 2014;49:187–197. | ||
Mariotto AB, Noone AM, Howlader N, et al. Cancer survival: an overview of measures, uses, and interpretation. J Natl Cancer Inst Monogr. 2014;49:145–186. | ||
Ahn HS, Kim HJ, Welch HG. Korea’s thyroid-cancer “epidemic” – screening and overdiagnosis. N Engl J Med. 2014;371(19):1765–1767. | ||
Ahn HS, Welch HG. South Korea’s thyroid-cancer “epidemic” – turning the tide. N Engl J Med. 2015;373(24):2389–2390. | ||
Holtzman NA, Marteau TM. Will genetics revolutionize medicine? N Engl J Med. 2000;343(2):141–144. | ||
Welch HG, Albertsen PC. Why doctors shouldn’t be punished for giving prostate tests. The New York Times, January 7, 2016. | ||
Rose G, Day S. The population mean predicts the number of deviant individuals. BMJ. 1990;301(6759):1031–1034. | ||
Keyes KM, Davey Smith G, Koenen KC, Galea S. The mathematical limits of genetic prediction for complex chronic disease. J Epidemiol Commun Health. 2015;69(6):574–579. | ||
Gonsalves G, Harrington M, Kessler DA. Don’t weaken the FDA’s drug approval process. The New York Times, June 11, 2015. | ||
Tefferi A, Kantarjian H, Rajkumar SV, et al. In support of a patient-driven initiative and petition to lower the high price of cancer drugs. Mayo Clinic Proc. 2015;90(8):996–1000. | ||
Young RC. Value-based cancer care. N Engl J Med. 2015;373(27):2593–2595. | ||
Bunnik EM, Schermer MH, Janssens AC. Naming and framing in genomic testing. Trends Mol Med. 2014;20(2):63–65. | ||
Brandt AM. How AIDS invented global health. N Engl J Med. 2013;368(23):2149–2152. | ||
Joyner MJ. “Moonshot” medicine will let us down. New York Times, January 29, 2015. | ||
Jalali R, Mittra I, Badwe R. Cancer research: in need of introspection. Lancet Oncol. 2016;17:140–141. | ||
Dyson FJ. Is science mostly driven by ideas or by tools? Science. 2012;338(6113):1426–1427. | ||
Rothman KJ, Greenland S. Causation and causal inference in epidemiology. Am J Public Health. 2005;95(S1):S144–S150. | ||
Yawn BP, Buchanan GR, Afenyi-Annan AN, et al. Management of sickle cell diseasesummary of the 2014 evidence-based report by expert panel members. JAMA. 2014;312(10):1033–1048. | ||
Bentires-Alj M, Rajan A, van Harten W, et al. Stimulating translational research: several European life science institutions put their heads together. Trends Mol Med. 2015;21(9):525–527. | ||
James W. Pragmatism: A New Name for Some Old Ways of Thinking. London, UK: Longman, Green & Co.; 1907. | ||
Rösel D, Brábek J, Veselý P, Fernandes M. Drugs for solid cancer: the productivity crisis prompts a rethink. Onco Targets Ther. 2013;6:767–777. | ||
Valent P, Groner B, Schumacher U, et al. Paul Ehrlich (1854–1915) and his contributions to the foundation and birth of translational medicine. J Innate Immun. Epub 2016 Feb 5. | ||
Ledford H. Big Science: the cancer genome challenge. Nature. 2010;464(7291):972–974. | ||
Roberts NJ, Vogelstein JT, Parmigiani G, Kinzler KW, Vogelstein B, Velculescu VE. The predictive capacity of personal genome sequencing. Sci Transl Med. 2012;4(133):133ra58. | ||
Matuchansky C. The promise of personalised medicine. Lancet. 2015;386(9995):742. | ||
Doble B, Schofield DJ, Roscioli T, Mattick JS. The promise of personalised medicine. Lancet. 2016;387(10017):433–434. | ||
Olive KP. Stroma, stroma everywhere (far more than you think). Clin Cancer Res. 2015;21(15):3366–3368. | ||
Glentis A, Gurchenkov V, Matic Vignjevic D. Assembly, heterogeneity, and breaching of the basement membranes. Cell Adh Migr. 2014;8(3):236–245. | ||
Thiery JP, Acloque H, Huang RY, Nieto MA. Epithelial-mesenchymal transitions in development and disease. Cell. 2009;139(5):871–890. | ||
Panková K, Rösel D, Novotný M, Brábek J. The molecular mechanisms of transition between mesenchymal and amoeboid invasiveness in tumor cells. Cell Mol Life Sci. 2010;67(1):63–71. | ||
Somarelli JA, Schaeffer D, Marengo MS, et al. Distinct routes to metastasis: plasticity-dependent and plasticity-independent pathways. Oncogene. Epub January 11, 2016. | ||
Sada M, Ohuchida K, Horioka K, et al. Hypoxic stellate cells of pancreatic cancer stroma regulate extracellular matrix fiber organization and cancer cell motility. Cancer Lett. 2016;372(2):210–218. | ||
Whatcott CJ, Diep CH, Jiang P, et al. Desmoplasia in primary tumors and metastatic lesions of pancreatic cancer. Clin Cancer Res. 2015;21(15):3561–3568. | ||
Cox TR, Erler JT. Molecular pathways: connecting fibrosis and solid tumor metastasis. Clin Cancer Res. 2014;20(14):3637–3643. | ||
Unbekandt M, Croft DR, Crighton D, et al. A novel small-molecule MRCK inhibitor blocks cancer cell invasion. Cell Commun Signal. 2014;12:54. | ||
Sadok A, McCarthy A, Caldwell J, et al. Rho kinase inhibitors block melanoma cell migration and inhibit metastasis. Cancer Res. 2015;75(11):2272–2284. | ||
Kale VP, Hengst JA, Desai DH, et al. A novel selective multikinase inhibitor of ROCK and MRCK effectively blocks cancer cell migration and invasion. Cancer Lett. 2015;361(2):185–196. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.