")
Back to Journals » Therapeutics and Clinical Risk Management » Volume 19
Practical Guidance for the Use of Patisiran in the Management of Polyneuropathy in Hereditary Transthyretin-Mediated Amyloidosis
Authors Dixon S, Kang X, Quan D
Received 5 August 2023
Accepted for publication 13 November 2023
Published 27 November 2023 Volume 2023:19 Pages 973—981
DOI https://doi.org/10.2147/TCRM.S361706
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Garry Walsh
Stacy Dixon, Xuan Kang, Dianna Quan
Department of Neurology, University of Colorado, Aurora, CO, USA
Correspondence: Dianna Quan, Department of Neurology - University of Colorado, 12631 East 17th Avenue, Mailstop B185, Aurora, CO, 80045, USA, Tel +1 (303) 724-2188, Fax +1 (303) 724-2202, Email [email protected]
Abstract: Variant transthyretin amyloidosis (ATTRv) is an autosomal dominant inherited genetic disorder that affects 5000– 10,000 people worldwide. It is caused by mutations in the transthyretin (TTR) gene and results in amyloid deposition in a variety of organs due to abnormal accumulation of TTR protein fibrils. Although this is a multisystem disorder, the heart and peripheral nerves are the preferentially affected organs. Over 150 TTR gene mutations have been associated with this disease and the clinical phenotype can vary significantly. Severe forms of the disorder can be fatal. Fortunately, the oligonucleotide-based therapy era has resulted in the development of several novel treatment options. Patisiran is a small interfering RNA (siRNA) encapsulated in a lipid nanoparticle that targets both mutant and wild-type TTR and results in significant reductions of the TTR protein in the serum and in tissue deposits. Patisiran has been approved for treatment of adults with polyneuropathy due to hereditary TTR-mediated amyloidosis in both the United States (US) and European Union (EU). In this review, we will discuss the development of patisiran, the clinical trials that lead to treatment approval, and provide guideline parameters for use in clinical practice.
Keywords: patisiran, transthyretin, TTR, neuropathy, siRNA
Introduction
Transthyretin (TTR) amyloidosis is a multisystemic disorder in which amyloid deposition disrupts function in multiple tissues throughout the body, most prominently in peripheral nerve and the heart. Transthyretin is a homotetrameric protein that normally serves to transport thyroxine and retinol binding protein. The majority of transthyretin is made in the liver with low levels of production within the retinal pigment epithelium and choroid plexus. Approximately 150 mutations in the TTR gene have been reported, most pathogenic and transmitted dominantly.1 Mutations alter protein fibril structure predisposing the tetramer to dissociate into monomeric fibrils that aggregate into insoluble amyloid deposits (Figure 1). Wild type transthyretin (ATTRwt) is also known to form amyloid. The prevalence and phenotype of variant TTR amyloidosis (ATTRv) varies depending on the specific genotype, and expression varies based on factors that are incompletely understood. The disorder is endemic in some parts of the world such as Portugal where prevalence of ATTRv is estimated to be 23/100,000.2 In other parts of the world such as the United States, symptomatic disease is more phenotypically variable due a more ethnically diverse population and a greater mix of variants is observed, most commonly V122I followed by T60A. An estimated 3% to 4% of African Americans carry the V122I variant, though not all carriers become symptomatic.3,4 The carrier frequency of other TTR variants among large populations is not as well studied.
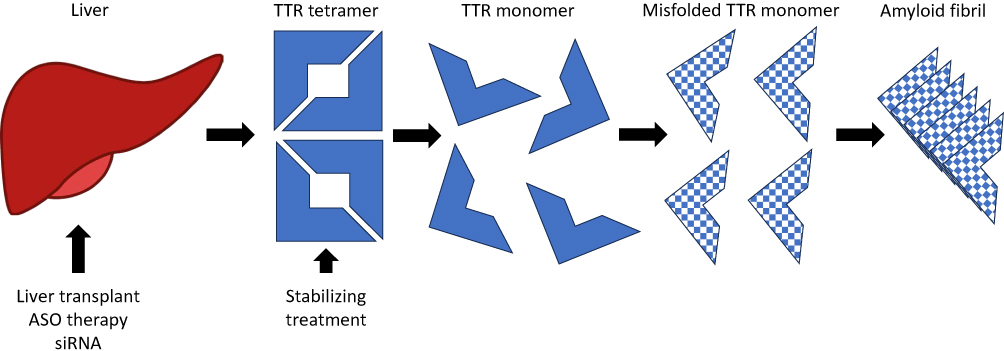 |
Figure 1 Mechanism of action of ATTRv treatments. Abbreviations: ASO, Antisense oligonucleotide; siRNA, Small interfering RNA. |
In 1952, Corino Andrade described the classic clinical disease features in a Portuguese population with familial amyloid polyneuropathy,5 now known to be due to a single amino acid substitution, V30M, in the TTR gene. Subsequent observations by other investigators revealed the same mutation in Swedish and some Japanese kindreds, with generally later onset polyneuropathy.6–8 Disease from ATTRv is now recognized in virtually all human populations though phenotype varies with different mutations and even among different populations with the same mutation. Disease penetrance and age of onset may vary within different family members carrying the same variant. Not all carriers become symptomatic, nor is the presence of a TTR variant a prerequisite to developing TTR amyloidosis. The phenotypes of more common variants are relatively well characterized and fall into 3 broad categories: predominantly neuropathic, predominantly cardiogenic, or mixed disease. Wild type transthyretin amyloidosis results in a predominantly cardiac phenotype with restrictive cardiomyopathy and heart failure. With mutations previously considered primarily neuropathic and those considered primarily cardiogenic, significant overlap is increasingly recognized.9
In patients with a predominantly neurologic phenotype the disease most commonly presents as a painful length dependent peripheral neuropathy with numbness and later weakness. More localized manifestations of peripheral nerve disease, especially carpal tunnel syndrome, may occur years earlier. Another early feature may be autonomic dysfunction, with orthostasis, constipation, diarrhea, or both, urinary dysfunction with incontinence or urinary retention, sweating changes, and pupillary abnormalities. Patients with amyloid deposition in the heart develop restrictive cardiomyopathy with common signs of heart failure including reduced exercise capacity, dyspnea, and fluid overload. Other organ systems including the kidneys, musculoskeletal system, eyes, and brain may be affected. Untreated, average life expectancy is 5–15 years from symptom onset with death resulting from cardiac failure or inanition.10,11
The key to diagnosis is inclusion of ATTRv in the differential diagnosis of patients with painful peripheral neuropathy, cardiomyopathy, or both. Early carpal tunnel syndrome, dysautonomia, and family history of similar complaints are useful triggers for further genetic evaluation. However, the absence of family history is not exclusionary as disease manifestations may be present for many generations before they are accurately attributed to ATTRv. Genetic testing confirms carrier status, but it is important to exclude other hereditary and acquired causes of neuropathy, as well as other causes of amyloidosis such as light chain disease, non-TTR hereditary amyloidosis, or systemic inflammation leading to amyloid formation. Affected tissues may show biopsy evidence of amyloid deposition, and mass spectrometry allows typing of the amyloid material. A 99m-Tc-DPD or 99m-Tc-PYP bone scan or cardiac imaging with technetium pyrophosphate may also be helpful.12,13
Therapeutic strategies for symptomatic patients are directed at three main targets: TTR production, TTR stabilization, and removal of amyloid deposits. Modern treatment began in the 1990s with organ transplantation, liver transplantation to remove the primary source of aberrant TTR production, and cardiac transplantation for end-stage cardiomyopathy. The scarcity of available organs for transplant and the often-debilitated state of patients at the time of diagnosis limited this approach. Furthermore, ongoing production of wild type TTR by a transplanted liver remained a source of TTR that could continue to build upon existing amyloid deposits in tissue.14 Stabilizers of the TTR tetramer were developed and identified in later years. Diflunisal is a readily available nonsteroidal anti-inflammatory drug originally marketed in 1981 that has been shown to slow ATTRv neuropathy progression modestly.15 Tafamidis slows progression of ATTRv cardiomyopathy and may benefit neuropathy patients with mild disease.16,17 Neither stabilizer is sufficient to halt disease worsening. Research is ongoing for better treatments directed at all aspects of amyloid production, stabilization, and clearance.
Development and Pharmacologic Properties of Patisiran
The rise of oligonucleotide-based medical therapies has ushered in a new era in medical technology aimed at designing therapeutic agents to treat gene-specific disorders. The pioneer leading the charge into the land of antisense oligonucleotides was Paul C. Zamecnik.18 In 1978 he published work that demonstrated a synthetic oligonucleotide could bind Rous sarcoma virus RNA and block RNA translation, thereby inhibiting viral replication.19 Following his work, several advances in chemical synthesis and the understanding of RNAs in cellular biology paved the way for development of oligonucleotide medical therapies.20–22 With the advent of these technologies, several biotechnology companies were founded with the goal of developing gene-targeted therapeutics.
Two commonly utilized technologies for medical therapeutics are single-stranded antisense oligonucleotides (ASOs) and double-stranded small interfering RNAs (siRNAs). Both types of oligonucleotide constructs are chemically synthesized (often 12–30 nucleotides in length) to modulate RNA function by binding specific RNA sequences following Watson-Crick base pairing.21 ASOs can either sterically block or occupy specific complementary RNA sequences, thereby modulating protein expression, or promote RNA cleavage and degradation.21,23 In contrast, double-stranded siRNAs result in gene silencing through incorporation into the cellular RNA-induced silencing complex (RISC) leading to RNA interference (RNAi) degradation of messenger RNAs (mRNAs) that are complementary to the guide sequence.23 Unintended off-target effects are possible with both constructs if there is partial sequence homology to other regions. Additionally, unmodified oligonucleotides are unstable and can be rapidly degraded. Therefore, chemical modifications to the constructs are necessary to enhance the mechanism of action, increase stability, and affect pharmacokinetic properties including tissue targeting.21,23
Patisiran is a double-stranded siRNA oligonucleotide that was developed to treat ATTRv. The siRNA construct is 21 bases in length on each strand and is chemically modified with eleven 2’-O-methoxy-modified sugar residues and four 2’-deoxythymidine residues.24,25 The siRNA construct is encapsulated into a lipid nanoparticle allowing targeted delivery to hepatocytes where most TTR protein is synthesized.24,25 Patisiran was one of the first oligonucleotide therapeutics to utilize a lipid nanoparticle to protect the siRNA from nuclease digestion and allow for delivery into the target cell population of hepatocytes. Details of the lipid nanoparticle and the mechanism for cellular delivery are well summarized in several review articles.24,26,27
Patisiran binds to a genetically conserved sequence in the 3’ untranslated region (3’UTR) of both wild-type and mutant TTR mRNA.25 Using RISC and RNAi mechanisms, TTR mRNA is degraded leading to reduction of serum and tissue levels of TTR protein.25,28 Patisiran demonstrates linear and dose-proportional increases in serum concentration after single intravenous dose range of 0.01 to 0.5mg/kg.25 More than 95% of patisiran in circulation is associated with the lipid complex and there was minimal protein binding observed during in vitro studies, indicating plasma protein binding of patisiran is low.25 Patisiran is metabolized by nucleases to variable length nucleotides and a minimal amount (less than 1%) is excreted unchanged into the urine.25 The terminal elimination half-life has been reported at 3.2 ±1.8 days (mean ± SD).25 Dose adjustments are not needed for mild to moderate renal or mild hepatic impairment but patisiran has not been studied in patients with severe renal disease or moderate to severe hepatic impairment (see manufacturer prescribing information for complete details).25 Patisiran is not an inhibitor, inducer, or substrate of cytochrome P450 enzymes.25 Drug–drug interactions are not expected, although formal clinical drug interaction studies have not been performed.25
Serum TTR protein is a carrier of retinol-binding protein involved in the transport of vitamin A. Persistent dosing of patisiran for more than 18 months resulted in a 45% reduction in serum retinol-binding protein and a 62% reduction in serum vitamin A.25 However, Phase 1 data showed no significant change in thyroid, renal, liver, or hematologic functions.29 The LNP contains polyethylene glycol (PEG)-lipid as one of its components. Biological agents modified with PEG have been shown to produce anti-drug antibodies (ADAs).30,31 In Phase 3 testing, ADAs were seen in 3.4% of Patisiran treated versus 1.3% of placebo treated subjects; no significant effect on safety, efficacy, pharmacodynamics, or pharmacokinetics was observed and ADA positivity was transient.32
Clinical Trial Experience
Both preclinical and clinical studies have supported the efficacy and safety of patisiran for the treatment of neuropathy associated with TTR amyloidosis. Clinical efficacy and safety data are discussed below and summarized in Table 1.
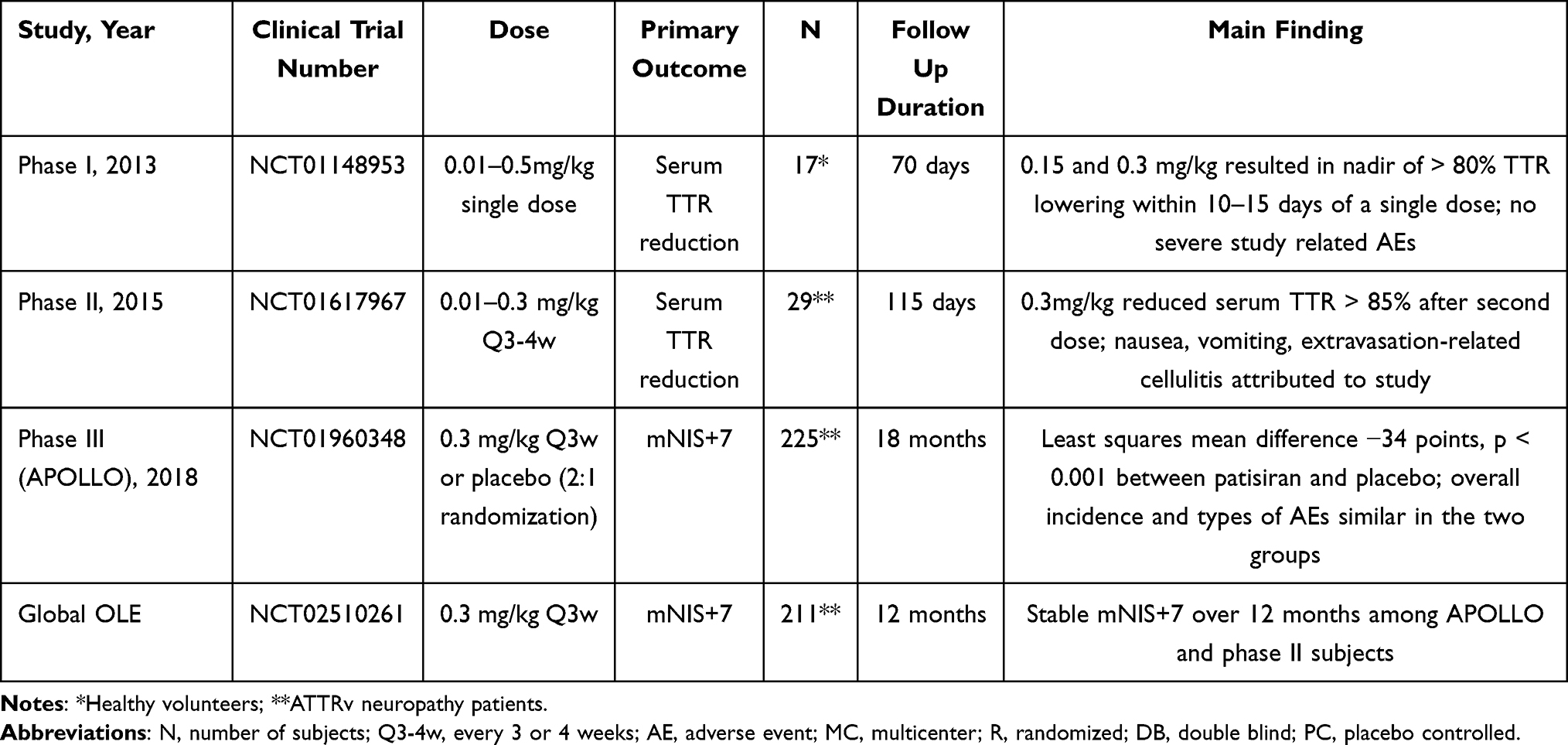 |
Table 1 Summary of Clinical Trials of Patisiran for Treatment of ATTRv |
Phase I
During phase 1 testing, a single-dose of two forms of intravenously (IV) infused siRNA encapsulated in lipid nanoparticle targeting TTR mRNA were tested against placebo.29 The first, ALN-TTR01 (0.01 to 1 mg/kg) was administered to 8 groups of 4 patients each, all with biopsy proven transthyretin amyloidosis and mild to moderate neuropathy. The second, ALN-TTR02, later named patisiran, was administered to 4 groups of 4 healthy volunteers each at doses of 0.01, 0.5, 0.15, 0.3 mg/kg. An additional subject received a dose of 0.5 mg/kg, with subsequent enrollment halted due to minimal incremental dose effect. In each four-subject dose group, volunteers were randomized to active medication or placebo in a 3:1 ratio.
Patisiran demonstrated a more significant and consistent mean reduction in TTR level compared to ALN-TTR01. ALN-TTR01 1 mg/kg reduced TTR levels a mean of 38% at day 7. In comparison, patisiran 0.3 mg/kg achieved more than 50% TTR lowering by day 3 and patisiran 0.15 and 0.3 mg/kg respectively resulted in nadirs of 82.3% and 86.8% TTR lowering between days 10 to 15. Fewer infusion-related reactions were observed with patisiran (7.7% subjects) compared to ALN-TTR01 (20.8% subjects). Overall, phase 1 testing demonstrated patisiran 0.15 to 0.3 mg/kg provided the optimal TTR lowering and safety profile.29 Based on these Phase I results, development of patisiran in Phase II and Phase III trials was continued.
Phase II
In Phase II, adults with biopsy proven ATTRv and mild to moderate polyneuropathy were studied.33 Patients with prior liver transplants, unstable angina or myocardial infarct within 6 months of the study, New York heart associated (NYHA) class III or IV heart failure, pregnancy, or other systemic medical conditions were excluded. Nine groups of 3 patients each were planned with all patients to receive 2 infusions of patisiran. The first 3 groups received patisiran at 0.01, 0.05, and 0.15 mg/kg, 2 doses separated by 4 weeks. The fourth and fifth groups received 0.3 mg/kg, 2 doses separated by 4 weeks. Patients in groups 6 to 9 received 0.3 mg/kg, 2 doses separated by 3 weeks. Twenty-six of 29 enrolled patients completed the study.
The largest and most sustained reduction of TTR was observed with the 0.3 mg/kg 3 week interval cohorts, totaling 12 subjects. Mean reduction of TTR levels after the first and second doses reached a nadir of 83.8% and 86.7%, respectively. Infusion-related reactions seen in 10% of patients were the most common adverse event, but none led to treatment discontinuation. Urinary tract infection with associated sepsis, nausea, and vomiting was reported in one patient treated with 0.3mg/kg at the 3 week interval; this subject discontinued the study due to nausea and vomiting attributed to the treatment. Another patient in the 0.3 mg/kg 4 week interval cohort experienced extravasation-related cellulitis attributed to the study. All patients who completed phase II were invited to participate in an open label extension (OLE) study of the 0.3 mg/kg every 3 week regimen.34
Phase III APOLLO Trial
The phase III APOLLO trial was an 18-month, international multicenter, double blind, randomized, placebo-controlled study of ATTRv patients with polyneuropathy. A total of 225 patients were randomized in a 2:1 ratio to either receive patisiran 0.3mg/kg (148 patients) or placebo every 3 weeks. The primary outcome measure was the change in the modified Neuropathy Impairment Score +7 (mNIS+7) from baseline. The mNIS+7 is a 0 to 304 point validated measure developed to quantitate the spectrum of sensory, motor, and autonomic neuropathy in ATTRv.35 Secondary endpoints included measures of quality of life, motor strength, disability, gait speed, nutritional status, and patient reported autonomic symptoms.
At 18 months, patients in the patisiran group experienced significantly less worsening in the mNIS + 7 scores from baseline compared to the placebo group (least squares mean difference −34 points, p < 0.001), with difference between the groups becoming apparent by 9 months. At 18 months, 56% of patisiran patients had improved mNIS+7 scores compared to 4% of placebo patients. Additionally, patients in the patisiran group had less decline in quality of life at 18 months as measured by the Norfolk Quality of Life-Diabetic Neuropathy questionnaire (possible range −4 to 136) with least squares mean difference between groups of −21.1 points, p < 0.001. Other secondary measures of strength, disability, gait speed, nutritional status, and autonomic symptoms were all statistically better in the patisiran group compared to placebo at 18 months. As reflected in primary and secondary endpoints, patisiran halted or reversed neuropathy progression in ATTRv.
While adverse events were reported in 97% of patients, the majority were mild to moderate in severity. The frequency of serious and severe adverse events was similar between the patisiran-treated and placebo-treated groups with more adverse events leading to discontinuation of the trial regimen in the placebo-treated group (14%) compared to the patisiran-treated group (5%). Peripheral edema and IRRs were more common in the patisiran group. 19% of patisiran-treated versus 9% of placebo-treated patients experienced IRRs, the majority of which occurred within the first two infusions; IRR frequency decreased with time.25,28 Infusion interruptions occurred in 5% of patients with IRRs and less than 1% of patients permanently discontinued treatment due to IRRs.25,28 Other adverse events that occurred in at least 5% of patisiran-treated patients and occurred at least 3% more frequently compared to placebo-treated patients included upper respiratory tract infections, dyspepsia, dyspnea, muscle spasms, arthralgia, erythema, bronchitis, and vertigo.25 Death occurred in 5% of patisiran and 8% of placebo patients, respectively.28 Causes of deaths were primarily cardiovascular in nature, consistent with expected disease progression in this patient population.
Pharmacodynamic analysis from APOLLO demonstrated mean serum TTR reduction of 84.3% at 18 months with mean maximum reduction of 87.8%.32 There were no statistically significant differences in the level of TTR reduction based on age, sex, race, genotype, weight, renal or hepatic function, prior stabilizer use, or antidrug antibody status. Dosing above an absolute dose of 30mg was predicted to fall in the plateau portion of the concentration effect curve, indicating 30 mg every 3 weeks compared to higher actual weight-based dosing results in similar TTR reduction in patients over 100 kg.32 Onset of serum TTR reduction was relatively rapid, falling by approximately 80% within 10 to 14 days after a single intravenous dose of 0.3mg/kg of patisiran.25,28 At the recommended dosing, steady state was achieved within 24 weeks of treatment.25
Global Open Label Extension Study
Study participants from phase II and phase III trials were invited to continue on or enter a multicenter OLE study. Of 187 eligible patients from APOLLO, 186 patients enrolled, joining all 25 patients from the phase II OLE study. Patients received 0.3mg/kg dosing by IV infusion every 3 weeks and continued treatment for 5 years.
There was sustained reduction of TTR concentration (mean 78.7%) through all groups compared to pre-patisiran levels. At a 12-month interim analysis, patients continued to demonstrate improvement of mNIS+7 scores compared to baseline at OLE enrollment (APOLLO-Patisiran group −4, phase II OLE −4.7, and APOLLO-placebo group −1.4).36 Continued improvement compared to APOLLO baseline was observed in Norfolk QOL-DN (−3.9 mean change), as well as autonomic function based on the Composite Autonomic Symptom Score or COMPASS-31 (−4 mean change) in the APOLLO-patisiran group. Additionally, for patients in the APOLLO-placebo group, the progression of neuropathy seen in APOLLO based on mNIS+7, QOL-DN, polyneuropathy disability (PND) and COMPASS-31 scores either stabilized or reversed once patisiran was initiated in the OLE. Positive effects on cardiac function were also observed in the global OLE. N-terminal prohormone of brain natriuretic peptide (NT-proBNP) remained stable in patients previously treated with patisiran, and improved in the APOLLO-placebo group after initiating patisiran.36 None of the neuropathy measures returned to APOLLO baseline, emphasizing the importance of timely disease recognition and treatment.
In a safety analysis, 97% of patients reported adverse events, which were mostly mild to moderate IRRs. 39% of patients reported serious adverse events and 11% of patients passed away. The APOLLO-placebo group reported more severe adverse events (47% compared to 26% in APOLLO-patisiran and 12% in phase II OLE patients). The death rate was higher in APOLLO-placebo patients (27% compared to 7% in APOLLO-patisiran patients). No deaths were reported in phase II OLE patients who received the longest duration of patisiran treatment.
Post-Hoc Analysis of Patisiran Studies
A post-hoc analysis of the phase II OLE and phase III trials compared patisiran alone and with concomitant use of a TTR stabilizer, either tafamidis or diflunisal.37 During the phase II OLE, 26% of patients received patisiran alone, 48% of patients received tafamidis with patisiran, and 26% of patients received diflunisal with patisiran. Patients in the phase III APOLLO trial were on patisiran alone, but 33% had previously used tafamidis and 20% had previously used diflunisal. Among phase II OLE patients, TTR reductions were comparable across groups through 24 months of patisiran treatment, regardless of concomitant stabilizer treatment (TTR reduction: -88% patisiran alone, −79.9% with tafamidis, –84% with diflunisal). Prior stabilizer use among APOLLO patients had no significant impact on the degree of patisiran benefit on mNIS+7 or Norfolk QOL score. In both phase II OLE and APOLLO, safety profiles were similar regardless of concomitant or past stabilizer use.
Clinical Treatment Parameters for Patisiran
Patisiran was approved for treatment of polyneuropathy due to ATTRv in 2018 in both the United States (US) and European Union. Approval in Europe is limited to stage 1 (patient can walk unaided) and stage 2 (patient can still walk but needs help) polyneuropathy.38 Although patisiran can reduce both wild-type and mutant TTR protein, current approval is only for treatment of amyloidosis in patients with ATTRv neuropathy, as studies have not been completed in patients with amyloidosis from wild-type TTR. A pre-specified cardiac subgroup analysis of the APOLLO study population did show that patisiran decreased mean left ventricular wall thickness, global longitudinal strain, and NT-proBNT, but current patisiran approval is not for cardiomyopathy alone in ATTRv (ie without polyneuropathy).39 APOLLO-B is an ongoing clinical trial to further assess patisiran in relation to cardiomyopathy from both wild type and variant transthyretin amyloidosis.40
When the authors evaluate a patient with a known pathogenic TTR mutation, we assess for polyneuropathy both clinically (based on symptoms and neurological examination) and electrophysiologically with nerve conduction studies (NCS) and electromyography (EMG). If there is concern for polyneuropathy clinically, but with normal NCS/EMG, then skin biopsy to assess for reduced intraepidermal nerve fiber density (IENFD) and amyloid deposition can be considered. More invasive testing such as nerve biopsy or fat pad biopsy to demonstrate evidence of amyloidosis is rarely done in the setting of a known pathogenic TTR mutation and clinical symptoms of polyneuropathy without other explanation. Carpal tunnel syndrome (CTS), especially if bilateral, may be an early sign of polyneuropathy in patients with pathogenic TTR mutations. If the CTS is severe and warrants surgical release, the authors would typically request tissue sample from the surgery be analyzed for presence of amyloid protein. Some TTR mutations are more likely to be associated with polyneuropathy than others, so the type of TTR mutation is also considered. Once a clinical diagnosis of polyneuropathy in the setting of a pathogenic TTR mutation is made, we assess the severity of the polyneuropathy based on the Polyneuropathy Disability (PND) Score and the Familial Amyloid Polyneuropathy (FAP) Stage.41,42 Other clinical assessment scales, such as the modified neuropathy impairment score (mNIS) +7, six-minute walk test (6-MWT), or other measures as suggested in the guideline article by Ando et al, 2022 can be considered. Time limitations of clinical appointments and appropriate training for each scale may limit the utility of performing these in clinical practice.1
Once an appropriate patient has been identified for treatment with patisiran, we initiate the recommended dosing of 0.3 mg/kg intravenously once every 3 weeks for patients weighing <100 kg or 30mg intravenously once every 3 weeks for patients weighing ≥100 kg. It is recommended that the infusions be administered over approximately 80 minutes.25 Patients are appropriately pre-medicated with oral acetaminophen, intravenous corticosteroid, and intravenous or oral H1 blocker and H2 blocker (form differs based on availability and tolerability) to reduce infusion related reactions (IRRs).25 Infusion related reactions can also be mitigated by slowing the rate of infusion. Patisiran should be discontinued if there is a severe or life-threatening infusion related reaction. The severity of IRRs tends to decrease with longer treatment duration.28
Every patient started on patisiran is instructed to take daily vitamin A supplements at the recommended daily allowance of vitamin A as per the prescribing guidelines.25 Based on US National Institutes of Health (NIH) guidelines, the recommended daily dose of vitamin A for adult men is 900 mcg RAE (retinol activity equivalents) or approximately 3000 IU (international units). For adult women, the recommended dose is 700 mcg RAE or approximately 2333 IU.43 Often, the amount of vitamin A in commercial brand multi-vitamins is similar to these recommended daily values. As per prescribing guidelines, serum levels of vitamin A are not routinely checked nor are higher doses of vitamin A recommended as serum vitamin A levels do not reflect total body vitamin A levels.25 Patients are clinically monitored for signs of vitamin A deficiency (such as night blindness) and would be referred for evaluation by ophthalmology if suspected.
There are no studies of patisiran in pregnant women. Given vitamin A is essential for normal embryo fetal development but excessive levels of vitamin A (a fat-soluble vitamin) can cause adverse developmental events, patisiran use is not recommended in pregnancy unless the clinical treatment of the woman outweighs the risks to the fetus. Animal studies showed administration of patisiran lipid complex (patisiran-LC) compared to a rodent-specific (pharmacologically active) surrogate did not have adverse developmental effects in pregnant rats.25 However, in pregnant rabbits treated with doses of patisiran-LC that led to maternal toxicity, developmental fetal toxicity was also seen.25 There is a patisiran pregnancy exposure registry that should be utilized with contact information listed on the FDA website.44 In lactating rats, the lipid components of patisiran were detected in milk but not patisiran itself.25 No further studies have been completed in lactating women or breastfed infants. Patisiran has not been studied in pediatric patients.
Conclusions
The success of patisiran clinical trials is a major milestone for disease modifying treatment in TTR amyloidosis. These studies have demonstrated the efficacy of patisiran on reducing TTR serum concentration and preserving or improving neurologic and cardiac function in patients with ATTRv. Mild to moderate infusion related side effects were the main adverse events during clinical trials, but patisiran has demonstrated an otherwise favorable side effect profile. These studies of patisiran and ATTRv have led to a fundamental paradigm shift in the management of ATTRv and development of new treatments for many other genetic diseases using similar RNAi technology.
Disclosure
Dr Dianna Quan reports grants, personal fees, non-financial support from Alnylam; grants from Ionis, Pfizer, Cytokinetics, Momenta, VielaBio, and Avidity Biosciences, outside the submitted work. Dr Stacy Dixon is a sub-investigator for APOLLO study and HELIOS-A study for Alnylam, outside the submitted work. The authors report no other conflicts of interest in this work.
References
1. Ando Y, Adams D, Benson MD, et al. Guidelines and new directions in the therapy and monitoring of ATTRv amyloidosis. Amyloid. 2022;29(3):143–155. doi:10.1080/13506129.2022.2052838
2. Ines M, Coelho T, Conceicao I, Duarte-Ramos F, de Carvalho M, Costa J. Epidemiology of transthyretin familial amyloid polyneuropathy in Portugal: a nationwide study. Neuroepidemiology. 2018;51(3–4):177–182. doi:10.1159/000490553
3. Agbor-Etang BB, Okafor HE, Farber-Eger EH, Wells QS. Low prevalence of clinically apparent cardiac amyloidosis among carriers of transthyretin V122I variant in a large electronic medical record. Am J Med. 2021;134(2):e98–e100. doi:10.1016/j.amjmed.2020.06.031
4. Kozlitina J, Garg S, Drazner MH, et al. Clinical implications of the amyloidogenic V122I transthyretin variant in the general population. J Card Fail. 2022;28(3):403–414. doi:10.1016/j.cardfail.2021.09.015
5. Andrade C. A peculiar form of peripheral neuropathy; familiar atypical generalized amyloidosis with special involvement of the peripheral nerves. Brain. 1952;75(3):408–427. doi:10.1093/brain/75.3.408
6. Holmgren G, Costa PM, Andersson C, et al. Geographical distribution of TTR met30 carriers in northern Sweden: discrepancy between carrier frequency and prevalence rate. J Med Genet. 1994;31(5):351–354. doi:10.1136/jmg.31.5.351
7. Coelho T, Maurer MS, Suhr OB. THAOS - The Transthyretin Amyloidosis Outcomes Survey: initial report on clinical manifestations in patients with hereditary and wild-type transthyretin amyloidosis. Curr Med Res Opin. 2013;29(1):63–76. doi:10.1185/03007995.2012.754348
8. Ueda M, Yamashita T, Misumi Y, Masuda T, Ando Y. Origin of sporadic late-onset hereditary ATTR Val30Met amyloidosis in Japan. Amyloid. 2018;25(3):143–147. doi:10.1080/13506129.2018.1531842
9. Dispenzieri A, Coelho T, Conceicao I, et al. Clinical and genetic profile of patients enrolled in the Transthyretin Amyloidosis Outcomes Survey (THAOS): 14-year update. Orphanet J Rare Dis. 2022;17(1):236. doi:10.1186/s13023-022-02359-w
10. Benson MD, Teague SD, Kovacs R, Feigenbaum H, Jung J, Kincaid JC. Rate of progression of transthyretin amyloidosis. Am J Cardiol. 2011;108(2):285–289. doi:10.1016/j.amjcard.2011.03.040
11. Adams D, Coelho T, Obici L, et al. Rapid progression of familial amyloidotic polyneuropathy: a multinational natural history study. Neurology. 2015;85(8):675–682. doi:10.1212/WNL.0000000000001870
12. Carroll A, Dyck PJ, de Carvalho M, et al. Novel approaches to diagnosis and management of hereditary transthyretin amyloidosis. J Neurol Neurosurg Psychiatry. 2022;93(6):668–678. doi:10.1136/jnnp-2021-327909
13. Madhani A, Sabogal N, Massillon D, et al. Clinical penetrance of the transthyretin V122I variant in older black patients with heart failure: the SCAN-MP (screening for cardiac amyloidosis with nuclear imaging in minority populations) study. J Am Heart Assoc. 2023;12(15):e028973. doi:10.1161/JAHA.122.028973
14. Stangou AJ, Hawkins PN, Heaton ND, et al. Progressive cardiac amyloidosis following liver transplantation for familial amyloid polyneuropathy: implications for amyloid fibrillogenesis. Transplantation. 1998;66(2):229–233. doi:10.1097/00007890-199807270-00016
15. Berk JL, Suhr OB, Obici L, et al. Repurposing diflunisal for familial amyloid polyneuropathy: a randomized clinical trial. JAMA. 2013;310(24):2658–2667. doi:10.1001/jama.2013.283815
16. Coelho T, Maia LF, Martins da Silva A, et al. Tafamidis for transthyretin familial amyloid polyneuropathy: a randomized, controlled trial. Neurology. 2012;79(8):785–792. doi:10.1212/WNL.0b013e3182661eb1
17. Maurer MS, Schwartz JH, Gundapaneni B, et al. Tafamidis treatment for patients with transthyretin amyloid cardiomyopathy. N Engl J Med. 2018;379(11):1007–1016. doi:10.1056/NEJMoa1805689
18. Pederson T. Obituary: paul C. Zamecnik (1912–2009). Nature. 2009;462(7272):423. doi:10.1038/462423a
19. Stephenson ML, Zamecnik PC. Inhibition of Rous sarcoma viral RNA translation by a specific oligodeoxyribonucleotide. Proc Natl Acad Sci U S A. 1978;75(1):285–288. doi:10.1073/pnas.75.1.285
20. Caruthers MH. Gene synthesis machines: DNA chemistry and its uses. Science. 1985;230(4723):281–285. doi:10.1126/science.3863253
21. Bennett CF. Therapeutic antisense oligonucleotides are coming of age. Annu Rev Med. 2019;70:307–321. doi:10.1146/annurev-med-041217-010829
22. Crooke ST, Baker BF, Crooke RM, Liang XH. Antisense technology: an overview and prospectus. Nat Rev Drug Discov. 2021;20(6):427–453. doi:10.1038/s41573-021-00162-z
23. Chi X, Gatti P, Papoian T. Safety of antisense oligonucleotide and siRNA-based therapeutics. Drug Discov Today. 2017;22(5):823–833. doi:10.1016/j.drudis.2017.01.013
24. Urits I, Swanson D, Swett MC, et al. A review of patisiran (ONPATTRO(R)) for the treatment of polyneuropathy in people with hereditary transthyretin amyloidosis. Neurol Ther. 2020;9(2):301–315. doi:10.1007/s40120-020-00208-1
25. Food and Drug Administration 2018. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/label/2018/210922s000lbl.pdf. ONPATTRO-Prescribing Information.
26. Akinc A, Maier MA, Manoharan M, et al. The Onpattro story and the clinical translation of nanomedicines containing nucleic acid-based drugs. Nat Nanotechnol. 2019;14(12):1084–1087. doi:10.1038/s41565-019-0591-y
27. Suzuki Y, Ishihara H. Difference in the lipid nanoparticle technology employed in three approved siRNA (Patisiran) and mRNA (COVID-19 vaccine) drugs. Drug Metab Pharmacokinet. 2021;41:100424. doi:10.1016/j.dmpk.2021.100424
28. Adams D, Gonzalez-Duarte A, O’Riordan WD, et al. Patisiran, an RNAi Therapeutic, for hereditary transthyretin amyloidosis. N Engl J Med. 2018;379(1):11–21. doi:10.1056/NEJMoa1716153
29. Coelho T, Adams D, Silva A, et al. Safety and efficacy of RNAi therapy for transthyretin amyloidosis. N Engl J Med. 2013;369(9):819–829. doi:10.1056/NEJMoa1208760
30. Kozma GT, Shimizu T, Ishida T, Szebeni J. Anti-PEG antibodies: properties, formation, testing and role in adverse immune reactions to PEGylated nano-biopharmaceuticals. Adv Drug Deliv Rev. 2020;154–155:163–175. doi:10.1016/j.addr.2020.07.024
31. Suzuki T, Suzuki Y, Hihara T, et al. PEG shedding-rate-dependent blood clearance of PEGylated lipid nanoparticles in mice: faster PEG shedding attenuates anti-PEG IgM production. Int J Pharm. 2020;588:119792. doi:10.1016/j.ijpharm.2020.119792
32. Zhang X, Goel V, Attarwala H, Sweetser MT, Clausen VA, Robbie GJ. Patisiran pharmacokinetics, pharmacodynamics, and exposure-response analyses in the phase 3 APOLLO trial in patients with hereditary transthyretin-mediated (hATTR) amyloidosis. J Clin Pharmacol. 2020;60(1):37–49. doi:10.1002/jcph.1480
33. Suhr OB, Coelho T, Buades J, et al. Efficacy and safety of patisiran for familial amyloidotic polyneuropathy: a phase II multi-dose study. Orphanet J Rare Dis. 2015;10:109. doi:10.1186/s13023-015-0326-6
34. Coelho T, Adams D, Conceicao I, et al. A phase II, open-label, extension study of long-term patisiran treatment in patients with hereditary transthyretin-mediated (hATTR) amyloidosis. Orphanet J Rare Dis. 2020;15(1):179. doi:10.1186/s13023-020-01399-4
35. Dyck PJB, Gonzalez-Duarte A, Obici L, et al. Development of measures of polyneuropathy impairment in hATTR amyloidosis: from NIS to mNIS + 7. J Neurol Sci. 2019;405:116424. doi:10.1016/j.jns.2019.116424
36. Adams D, Polydefkis M, Gonzalez-Duarte A, et al. Long-term safety and efficacy of patisiran for hereditary transthyretin-mediated amyloidosis with polyneuropathy: 12-month results of an open-label extension study. Lancet Neurol. 2021;20(1):49–59. doi:10.1016/S1474-4422(20)30368-9
37. Lin H, Merkel M, Hale C, Marantz JL. Experience of patisiran with transthyretin stabilizers in patients with hereditary transthyretin-mediated amyloidosis. Neurodegener Dis Manag. 2020;10(5):289–300. doi:10.2217/nmt-2020-0020
38. European Medicine Agency 2018. Available from: https://www.ema.europa.eu/en/medicines/human/EPAR/onpattro. Onpattro-European public assessment report.
39. Solomon SD, Adams D, Kristen A, et al. Effects of Patisiran, an RNA interference therapeutic, on cardiac parameters in patients with hereditary transthyretin-mediated amyloidosis. Circulation. 2019;139(4):431–443. doi:10.1161/CIRCULATIONAHA.118.035831
40. National Library of Medicine. APOLLO-B: a study to evaluate patisiran in participants with transthyretin amyloidosis with cardiomyopathy (Attr amyloidosis with cardiomyopathy) 2019. Available from: https://clinicaltrials.gov/study/NCT03997383.
41. Yamamoto S, Wilczek HE, Nowak G, et al. Liver transplantation for familial amyloidotic polyneuropathy (FAP): a single-center experience over 16 years. Am J Transplant. 2007;7(11):2597–2604. doi:10.1111/j.1600-6143.2007.01969.x
42. Coutinho P. Amyloid and Amyloidosis. Excerpta Medica. Amsterdam. 1980;1980:1.
43. National Institutes of Health (NIH) Office of Dietary Supplements (ODS). Vitamin A and Carotenoids 2022. Available from: https://ods.od.nih.gov/factsheets/VitaminA-Consumer/.
44. Food and Drug Administration. List of pregnancy exposure registries; 2023. Available from: https://www.fda.gov/science-research/womens-health-research/list-pregnancy-exposure-registries.
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.