Back to Journals » Diabetes, Metabolic Syndrome and Obesity » Volume 9
Practical combination therapy based on pathophysiology of type 2 diabetes
Authors Levin PA
Received 25 March 2016
Accepted for publication 30 April 2016
Published 31 October 2016 Volume 2016:9 Pages 355—369
DOI https://doi.org/10.2147/DMSO.S109216
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Ming-Hui Zou
Philip A Levin
MODEL Clinical Research, Endocrinology, Department of Medicine, Greater Baltimore Medical Center, Baltimore, MD, USA
Abstract: Type 2 diabetes is a complex, chronic, and progressive condition that often necessitates the use of multiple medications to achieve glycemic goals. Clinical guidelines generally recommend intensifying pharmacotherapy if glycemic goals are not achieved after 3 months of treatment. However, for many patients with type 2 diabetes, treatment intensification is delayed or does not occur. Initiating combination therapy early in the disease course has the potential to delay disease progression and improve patient outcomes. Guidelines generally provide a list of agents that may be used in combination regimens and emphasize individualization of treatment. The purpose of this review is to discuss the rationale for combination therapy, considering treatment effects on pathophysiologic aspects of type 2 diabetes and individual drug profiles. The combination of newer antidiabetes therapies with complementary mechanisms of action provides the opportunity to target multiple sites of tissue, organ, and cellular dysfunction.
Keywords: type 2 diabetes, combination therapy, oral antidiabetes drugs, insulin
Introduction
More than 415 million adults worldwide are estimated to have diabetes, with an increase to 642 million expected by 2040.1 Type 2 diabetes (T2D) is the most common type of diabetes in adults, and the prevalence is expected to increase largely because of a parallel increase in the prevalence of obesity.1 There have been significant advances in the understanding of glucose homeostasis and the pathophysiology of T2D over the past few decades.2 It is now understood that glucose concentrations are maintained by a complex network of finely tuned feedback mechanisms involving multiple organs and hormonal systems and that dysfunctions in this network contribute to the development and progression of insulin resistance and β-cell failure, hallmarks of T2D.2 Although there is still much to be learned about the disease process,2 current understanding suggests that early intervention with targeted pharmacotherapy to correct multiple sites of dysfunction might slow T2D progression and improve patient outcomes.
Treatment guidelines recommend metformin as initial oral pharmacotherapy for most adults,3,4 although study findings show that only 58% of patients are started on metformin in clinical practice.5 Guidelines consistently advise follow-up at 3 months and a general stepwise approach to pharmacotherapy with the addition of another antidiabetes agent if glycemic goals are not met3,4; however, many patients with T2D are subject to notable delays in treatment intensification, generally referred to as clinical inertia.6 A retrospective cohort study in patients with T2D and glycated hemoglobin (A1C) ≥7% or ≥8% taking one oral antidiabetes drug (OAD) found that the median time to treatment intensification with an additional OAD was 2.9 and 1.6 years, respectively, and more than 7.2 and 6.9 years in patients already taking two OADs.6 A recent retrospective database study found that in nearly 80% of patients with uncontrolled glycemia despite two OADs, a third OAD was added to the treatment regimen rather than insulin, which provided greater A1C reduction and lower health care costs, suggesting resistance to prescribing and taking insulin in current practice.7 These prolonged periods of inadequate glycemic control can have a detrimental effect on long-term outcomes.6,8
Guidelines from the American Association of Clinical Endocrinologists (AACE) suggest early use of combination pharmacotherapy for patients with high A1C at treatment initiation (Figure 1).3 AACE recommends initial treatment with dual therapy for patients with an A1C ≥7.5% and advancement to triple therapy if A1C goals are not met after 3 months. For patients with an A1C >9.0% and no hyperglycemia symptoms (eg, polyuria, polydipsia, polyphagia), initial pharmacotherapy with three antidiabetes agents may be appropriate.3 AACE guidelines further note that combination therapy should include medications with complementary mechanisms of action to maximize effectiveness3,9 to correct multiple sites of dysfunction. Targeting multiple sites of dysfunction using combination therapy early in the disease course may also produce more durable efficacy, particularly when using agents that preserve β-cell function.10 The purpose of this review is to discuss the rationale for combination therapy based on the pathophysiology of T2D, providing an overview of the underlying defects in T2D and the primary mechanisms of action of commonly used antidiabetes medications for a greater insight into their use in combination.
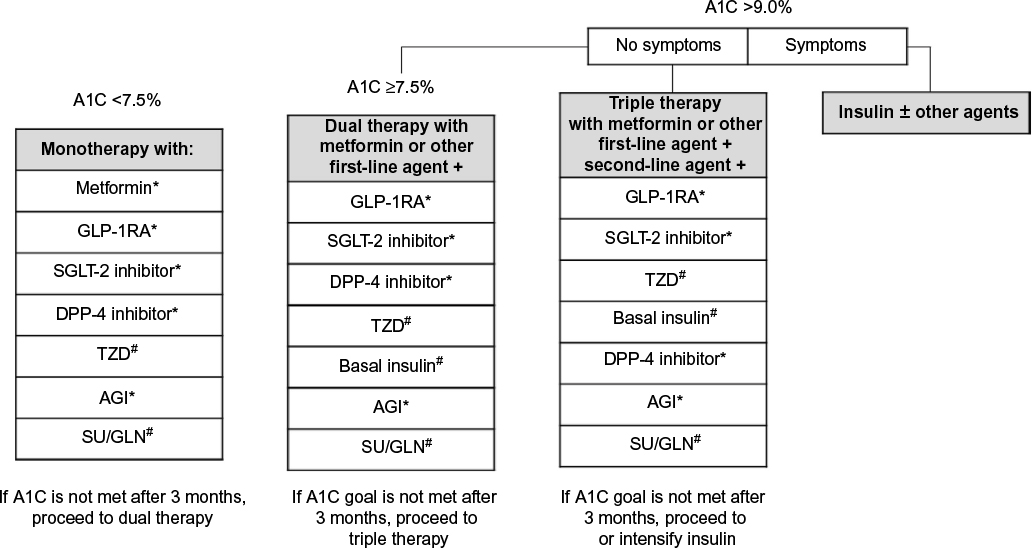 | Figure 1 Treatment algorithm based on A1C at entry. Notes: Order of medications suggests hierarchy of usage by the AACE. Adapted with permission from American Association of Clinical Endocrinologists © 2016 AACE. Garber AJ, Abrahamson MJ, Barzilay JI, et al. AACE/ACE comprehensive type 2 diabetesmanagement algorithm 2016. Endocr Pract. 2016;22:84–113.3 *Indicates few adverse events or possible benefits. #Use with caution. Abbreviations: A1C, glycated hemoglobin; AACE, American Association of Clinical Endocrinologists; ACE, American College of Endocrinology; AGI, α-glucosidase inhibitor; DPP-4, dipeptidyl peptidase-4; GLN, glinide; GLP-1RA, glucagon-like peptide-1 receptor agonist; SGLT-2, sodium-glucose cotransporter-2; SU, sulfonylurea; TZD, thiazolidinedione. |
Pathophysiology of T2D
Feedback regulation to maintain glucose homeostasis and the critical role of β-cells
In glucose-tolerant individuals, a feedback loop between β-cells and insulin-sensitive tissues maintains normal glucose homeostasis. In this cyclic process, insulin secreted in response to β-cell stimulation suppresses glucose production in the liver and facilitates uptake of glucose, amino acids, and fatty acids in adipose tissue and muscle. Plasma glucose concentrations are maintained within a narrow range (70–90 mg/dL) and are primarily regulated by the hormones insulin, glucagon, and incretins, with input from the central and peripheral nervous systems.11 Pancreatic glucagon plays an important role in maintaining glucose homeostasis by stimulating glycogenolysis and gluconeogenesis, metabolic processes in the liver and kidney that convert glycogen and other substrates (lactate, amino acids, glycerol) to free glucose for circulation in the fasted state.12,13 It is estimated that renal glucose release accounts for 20% of overall endogenous glucose release and ~40% of all gluconeogenesis.14 Glycogen is also stored in the skeletal muscle; however, muscle glycogen is not directly available to other tissues because it lacks the enzyme glucose-6-phosphatase needed to produce glucose.14 When exogenous glucose is available, insulin suppresses glycogenolysis and gluconeogenesis. In patients with T2D, however, hepatic insulin resistance results in the inability of normal concentrations of insulin to inhibit glycogenolysis and gluconeogenesis,15 resulting in increases in basal hepatic glucose production (HGP) and hyperglycemia in the fasted state.16
Normally functioning adipose tissue is one of the key regulatory tissues involved in maintaining lipid and glucose homeostasis.17 Thus, when adipose tissue is in a state of chronic low-level inflammation as occurs with obesity, a multitude of pathologies emerge, including peripheral insulin resistance.18,19 Preclinical studies have shown macrophage accumulation occurring at the onset of weight gain, and inflammation and macrophage-specific gene expression are upregulated in correlation with increased adiposity.20 In both animals and humans, macrophage accumulation is in direct proportion to adipocyte size and body mass index.21 This increased infiltration of macrophages into adipose tissue perpetuates the production of proinflammatory molecules, including free fatty acids that impair insulin sensitivity, which initiate additional activation and infiltration of peripheral monocytes and macrophages.20,21 Eventually, the continued state of inflammation and impaired insulin signaling in adipose tissue leads to lipolysis, necrosis, and the development of systemic insulin resistance.20
The β-cell is highly attuned to changes in insulin sensitivity and can maintain glucose homeostasis by increasing insulin secretion during the early stages of insulin resistance. When the β-cell can no longer produce enough insulin to compensate for a diminished insulin response, glucose concentrations rise.2 Exposure to chronically elevated glucose concentrations leads to production of reactive oxygen species that cause oxidative stress and increased production of free fatty acids, leading to glucolipotoxicity. This in turn interferes with β-cell compensatory mechanisms, collectively contributing to worsened β-cell function and apoptosis.2,22 The overall extent to which β-cell function is impaired determines the severity of hyperglycemia, and the progressive failure of β-cells accounts for the transition from impaired glucose tolerance to T2D.2 The rate at which β-cell impairment occurs varies among patients, although certain factors, most notably poor metabolic control and duration of T2D, contribute to progression.23
Multiple genetic and environmental factors also contribute to the development of insulin resistance and β-cell dysfunction.2 Insulin resistance and reduced β-cell function are largely heritable and present in individuals with normal glucose tolerance but at risk for hyperglycemia (eg, first-degree relatives of patients with T2D).24,25 Obesity, nutrient composition (particularly intake of saturated fats), and other factors, such as age-related declines in β-cell response, also contribute to glucose intolerance.2,26 A genetic propensity for obesity has also been demonstrated27; obesity and related insulin resistance are thought to arise when genes for abnormal body adiposity interact with certain environmental factors (eg, increased caloric intake, decreased energy expenditure).2 Although it is not yet known how genes interact with the environment to cause progressive β-cell dysfunction, and there is inherent variability among patients, current data suggest that both a genetic propensity for β-cell dysfunction and increased body adiposity may be necessary for the interaction with environmental factors that results in T2D.2
Dysfunction in other body systems
Gastrointestinal tract
In healthy subjects, more insulin is secreted after nutrient ingestion compared with a similar nutrient load given intravenously.28 The gastrointestinal-derived incretin hormones, glucagon-like peptide-1 (GLP-1) and glucose-dependent insulinotropic hormone (GIP), are largely responsible for this effect and primarily stimulate insulin secretion in a glucose-dependent manner by acting at their receptors on β-cells.28 GLP-1 has also been shown to directly inhibit glucagon secretion via GLP-1 receptors on pancreatic α cells or indirectly by stimulating insulin and somatostatin secretion.29 In vivo, GLP-1 and GIP facilitate β-cell proliferation and inhibit apoptosis, in addition to stimulating insulin secretion. GLP-1 also slows gastric emptying and decreases food intake, likely through multiple mechanisms.29,30 Under basal conditions, circulating GLP-1 and GIP concentrations are low but rise rapidly in the postprandial state and then decline as GIP and GLP-1 are rapidly degraded by enzymes, primarily dipeptidyl peptidase-4 (DPP-4).31 In patients with T2D, the insulinotropic response to GIP is notably reduced,32 which likely accounts for much of the decrease in incretin activity.33 In contrast, the insulinotropic response to GLP-1 remains largely intact in T2D,28 although certain patient factors, such as high body mass index or high glucagon concentrations, may reduce GLP-1 secretion.33
Kidney
In glucose-tolerant subjects, nearly all glucose filtered by the renal glomeruli is reabsorbed by the proximal tubule for recirculation.34 Glucose is excreted into the urine to help maintain glucose homeostasis only if the maximum glucose transport capacity is exceeded.34 Glucose reabsorption from the glomerular filtrate is primarily mediated by sodium-glucose cotransporter-2 (SGLT-2), a low-affinity, high-capacity transporter located in the proximal convoluted tubule, in a process independent of insulin secretion or action.34 Compared with subjects with normal glucose tolerance, patients with T2D have increased renal glucose uptake and release in the postprandial state.35 As the threshold for glucose reabsorption increases, uptake continues despite elevated plasma glucose concentrations, minimizing glucosuria and exacerbating the hyperglycemic environment.36 In addition, the kidneys also contribute to glucose release through increased gluconeogenic activity.14
Brain
The nervous system also plays an important role in metabolic regulation. Through direct and indirect pathways, the nervous system controls glucose metabolism, affecting insulin and glucagon secretion and HGP.2 The hypothalamus, in particular, is an important mediator of β-cell function and plasma insulin concentrations.2,37 Insulin action at the hypothalamus also plays a critical role in regulating appetite and body weight,2,35 and high-fat diets have been shown to lead to inflammatory signaling in the hypothalamus and subsequent insulin and leptin resistance.38 Decreased activity at this site is associated with the development of obesity, which is consistent with imaging studies showing structural changes (increased gliosis) in the hypothalamus of obese subjects.2,39 In preclinical studies, disruptions in circadian rhythms were associated with the development of insulin resistance and obesity,40 as well as an increase in β-cell dysfunction and apoptosis.41
It is apparent that continuous interactions between β-cells and insulin-sensitive tissues and input from other essential organs, such as the brain, kidney, and gastrointestinal tract, collectively ensure an adequate supply of glucose to tissues and organs while keeping plasma glucose concentrations within a specific range to prevent hypoglycemia or hyperglycemia. However, disruptions in these interactions by genetic or environmental triggers, such as inherited insulin resistance, β-cell dysfunction, or obesity, lead to a series of interconnected disturbances that contribute to the development and progression of T2D.
Actions of commonly used antidiabetes medications
New approaches to targeted pharmacotherapy have been developed in response to a better understanding of the underlying pathophysiology of T2D. Knowledge about the mechanisms of action of the more traditional agents has also increased in recent years. Table 1 summarizes the primary actions and key clinical effects of each class of commonly used antidiabetes medications.
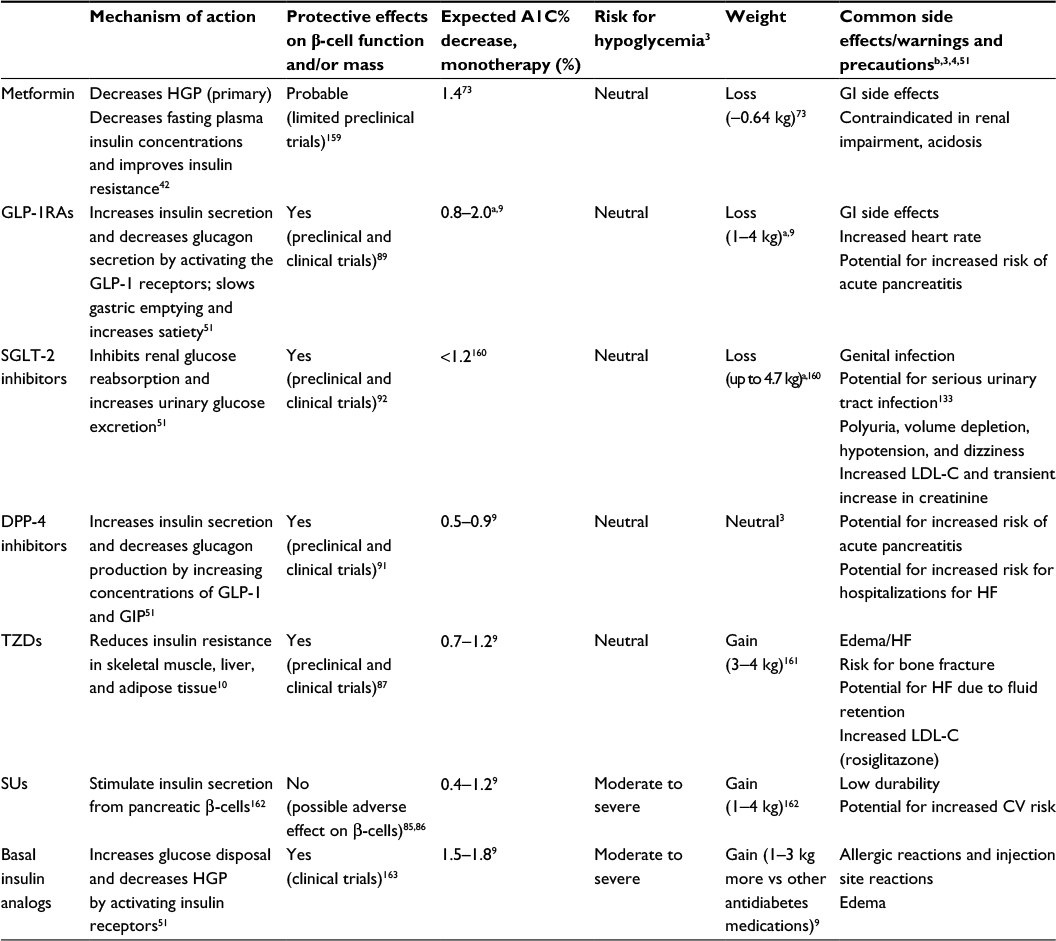 | Table 1 Overview of antidiabetes medications Notes: aIncludes monotherapy and add-on therapy evaluations. bNot inclusive of all treatment-related adverse effects or warnings. Abbreviations: A1C, glycated hemoglobin; CV, cardiovascular; DPP-4, dipeptidyl peptidase-4; GI, gastrointestinal; GIP, glucose-dependent insulinotropic hormone; GLP-1, glucagon-like peptide-1; GLP-1RA, glucagon-like peptide-1 receptor agonist; HF, heart failure; HGP, hepatic glucose production; LDL-C, low-density lipoprotein cholesterol; SGLT-2, sodium-glucose cotransporter-2; SU, sulfonylurea; TZD, thiazolidinedione. |
As recognized by the AACE, many patients will not reach glycemic goals with monotherapy and will require a combination of antidiabetes medications.3 However, association recommendations are generally not prescriptive with regard to which treatments to use in combination; rather, individualized treatment is emphasized. Current knowledge provides a sensible rationale for choosing medications that act in a synergistic manner (eg, improved insulin sensitivity and increased insulin secretion) to control glycemia, minimize adverse effects, including hypoglycemia, and correct underlying pathologies.
Suppression of HGP
The primary action of metformin is as an agonist of 5-adenosine monophosphate-activated protein kinase, which reduces HGP by inhibiting gluconeogenesis in the liver. Additionally, metformin yields improved hepatic insulin sensitivity and decreased fasting plasma insulin concentrations.10,42 Data also indicate that metformin has a beneficial effect on lipid metabolism secondary to improved insulin sensitivity and may help reverse hepatic steatosis,43,44 a condition closely associated with insulin resistance.42 Because 5-adenosine monophosphate-activated protein kinase actions also lead to reduced growth of human cancer cells, an antitumorigenic role has also been suggested for metformin,45 which is supported by a meta-analysis demonstrating a significant 50% reduction in hepatocellular carcinoma incidence in patients with diabetes receiving metformin versus not receiving metformin.46
Insulin sensitizers
Thiazolidinediones (TZDs), synthetic peroxisome proliferator-activated receptor-γ agonists,47 are potent insulin sensitizers in the liver, muscle, and adipose tissue48–50; evidence has shown TZDs to additionally alleviate lipotoxicity by transporting lipid deposits out of muscle, liver, and β-cells.10 Similar to that of metformin, the insulin-sensitizing effects of TZDs may counteract hepatic steatosis.44
Insulin secretagogues
Sulfonylureas (SUs) primarily increase insulin secretion by binding to the adenosine triphosphate-sensitive potassium (KATP) channel on pancreatic β-cell plasma membranes.51–53 Specifically, SUs bind to the SU receptor 1, a subunit on the KATP channel, and induce closure.53 As a result, concentrations of intracellular potassium rise, the β-cell membrane is depolarized, and insulin-containing secretory granules move to the cell surface for release into circulation.53,54
In contrast to the actions of SUs, the actions of incretin-based therapies are glucose-dependent.55 Incretin mimetics, or GLP-1 receptor agonists (GLP-1RAs), primarily increase insulin secretion and decrease glucagon secretion by activating the GLP-1 receptor.30,31 Delayed gastric emptying has also been associated with GLP-1RAs and shown to occur within minutes of administration.29 It is likely that this effect, as well as enhanced satiety and reduced food intake, involves interactions between the central and peripheral nervous systems.29
It is estimated that the half-life of biologically active GLP-1 in circulation is less than 2 minutes because of rapid degradation by DPP-4 activity.56,57 By inhibiting the DPP-4 enzyme and thereby increasing the half-life of endogenous incretin hormones in circulation, DPP-4 inhibitors indirectly increase stimulation of GLP-1 receptors.58 DPP-4 inhibitors are rapidly absorbed following oral administration and competitively and reversibly bind to the active site of DPP-4, reducing DPP-4 activity by 70% to 90% and, consequently, increasing GLP-1 concentrations approximately 1.5- to 4-fold at therapeutic doses.59 Preclinical and clinical evidence has demonstrated the benefits of DPP-4 pharmacologic inhibition, including increased concentrations of GIP and GLP-1, stimulation of insulin in circulation, improved glucose tolerance and insulin sensitivity, suppressed plasma glucagon, and prevention of weight gain.29 The clinical efficacy of DPP-4 inhibitors has been demonstrated in randomized controlled trials showing significant reductions from baseline in fasting plasma glucose, postprandial glucose, and A1C.59 DPP-4 inhibitors typically provide mean A1C reductions ranging from approximately 0.4% to 1.0% as monotherapy and from 0.7% to 1.2% in combination therapy regimens.59
Insulin independent agents
SGLT-2 inhibitors reduce elevated glucose concentrations through increased glucosuria by decreasing renal glucose reabsorption.34 The mechanism of action of SGLT-2 inhibitors is independent of insulin and may therefore complement a variety of other antidiabetes medications that are insulin dependent.34 It is also theorized that because SGLT-2 inhibitors target renal glucose handling, efficacy should not be compromised as further declines in β-cell function and increased insulin resistance occur.34
The α-glucosidase inhibitors (AGIs) primarily reduce postprandial plasma glucose concentrations through the competitive and reversible inhibition of α-glucosidase located in the brush border of the small intestine.60 Following nutrient ingestion, α-glucosidase hydrolyzes complex carbohydrates to monosaccharides for absorption. Thus, AGIs obstruct the degradation of starches and sucrose and delay carbohydrate reabsorption in a nonsystemic manner.60 However, a patient’s diet may alter efficacy. Studies indicate that AGIs may have a more potent effect in patients who consume a typical Eastern diet (whole grains, vegetables, fruits) compared with patients whose diet is characterized as Western (processed meat, high-fat dairy products, refined grains).60,61
Central nervous system
The development of therapies that target the central nervous system to reduce glucose concentrations is challenging.2 Bromocriptine, a quick-release sympatholytic D2-dopamine agonist, is currently the only approved antidiabetes medication that has a centrally mediated mechanism of action.2 Although the mechanism by which bromocriptine lowers plasma glucose concentrations is unknown, it is believed that early-morning treatment with bromocriptine resets circadian rhythms of hypothalamic dopamine and serotonin and thereby improves insulin sensitivity.62
Bile acid sequestrants
Bile acid sequestrants were initially developed to treat hypercholesterolemia; therefore, these agents have significant lipid-lowering properties.63 The mechanism by which colesevelam reduces plasma glucose concentrations is not fully understood, although study findings suggest that colesevelam reduces hyperglycemia by improving tissue glucose metabolism in both the fasting (increased plasma glucose clearance) and postprandial (increased glycolytic disposal of oral glucose) states.64 Colesevelam has also been associated with increased GLP-1 and GIP plasma concentrations, which likely contribute to observed improvements in β-cell function and increased glycolytic disposal.64
The American Diabetes Association currently does not favor the use of AGIs, colesevelam, or bromocriptine because of modest effects on glucose lowering, frequent dosing schedules, and occurrence of side effects.51 Therefore, discussion of the use of these agents in combination is limited in this review.
Basal insulin
Many patients will eventually require insulin to maintain glycemic control.51 The primary mechanism of action of insulin and its analogs is the activation of insulin receptors.51 Thus, insulin analogs lower plasma glucose concentrations by stimulating peripheral glucose uptake and inhibiting HGP.65,66 Although various formulations of insulin are available, guidance from the AACE recommends initial insulin therapy with long-acting basal insulin analogs because basal formulations are associated with less glycemic variability and a lower risk for hypoglycemia compared with neutral protamine Hagedorn, an intermediate-acting insulin.3 Several types of long-acting basal insulin analogs are available in the US, including insulin glargine (Lantus® [Sanofi-Aventis, Bridgewater, NJ, USA] and Toujeo® [Sanofi-Aventis]), insulin detemir (Levemir® [Novo Nordisk, Bagsværd, Denmark]), and insulin degludec (Tresiba® [Novo Nordisk]).51,66,67 Although both formulations of insulin glargine provide prolonged activity and thus allow once-daily dosing, Toujeo is a concentrated glargine, and its duration of action exceeds that of Lantus (approximately 36 vs 24 hours).68 In addition, Toujeo has been shown to significantly reduce nighttime low blood sugar events compared with Lantus (P=0.0002).69 Insulin detemir or Levemir is also associated with a constant duration of action, but similar to that of Lantus (approximately 24 hours), and may be administered once or twice daily in divided doses.66 Of the currently available long-acting basal insulin analogs, Tresiba provides the longest duration of effect, lasting at least 42 hours, primarily because of delayed absorption of insulin degludec from the subcutaneous tissue into systemic circulation.67
Potential combinations to improve pathophysiologic dysfunction
Enhancing endogenous insulin action
There are many combination regimens that have the potential to address multiple sites of pathophysiologic dysfunction of T2D, and those that may exert complementary effects are outlined in Table 2. The combination of metformin and a TZD has been suggested10 because metformin reduces HGP by inhibiting gluconeogenesis in the liver but is only a weak sensitizer in the muscle,42,70,71 and TZDs are potent insulin sensitizers in liver, muscle, and adipose tissue.48 Metformin and TZDs individually have a robust effect on A1C, and each exerts its actions through different pathways, providing an additive effect on glucose lowering.10 Metformin and TZDs are also associated with a low risk for hypoglycemia,3 making these agents appealing for use in combination. However, because metformin primarily relies on renal function for elimination,72 it is contraindicated in patients with renal disease or dysfunction.73 Metformin is also associated with gastrointestinal adverse effects that may lead to discontinuation for some patients but may dissipate with continued use or with dosing adjustments.74 TZDs have been shown to have beneficial effects on cognitive disorders related to insulin resistance in the central nervous system in studies in mice75 and to exert favorable effects on cardiovascular (CV) risk markers in patients with T2D.76 However, TZDs are associated with an increased risk for weight gain and fluid retention, which may worsen or lead to congestive heart failure (HF).77,78
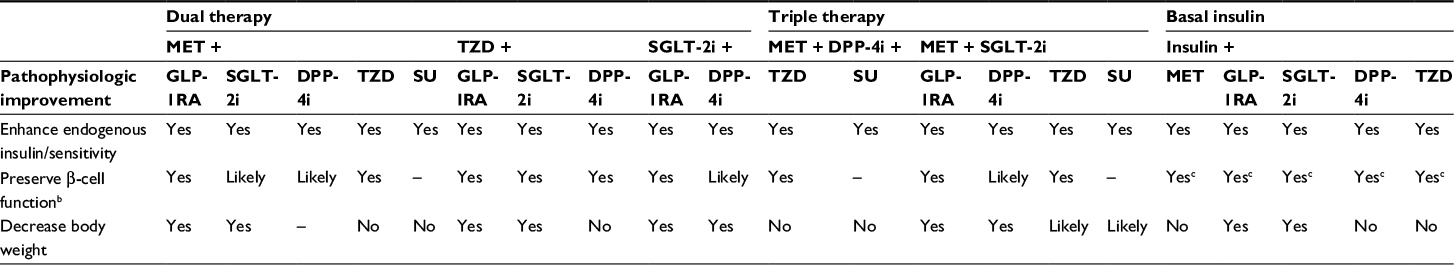 | Table 2 Potential regimens for targeted combination therapy to address major pathophysiologic defectsa Notes: aPotential improvements based on individual therapeutic profiles. Not inclusive of all potential combination regimens (– indicates improvement not likely). bAvailable data indicate beneficial effects on β-cell function with DPP-4 inhibitors and SGLT-2 inhibitors, although effects are not as well established as those associated with GLP-1RAs or TZDs. cEarly insulin therapy may exert protective effects on the β-cells. Abbreviations: DPP-4i, dipeptidyl peptidase-4 inhibitor; GLP-1RA, glucagon-like peptide-1 receptor agonist; MET, metformin; SGLT-2i, sodium-glucose cotransporter-2 inhibitor; SU, sulfonylurea; TZD, thiazolidinedione. |
Insulin secretagogues, such as SUs or incretin-based therapies, may also act in a complementary manner to metformin by facilitating insulin secretion. SUs are associated with moderate glycemic efficacy, although these agents are recommended for patients with a disease duration of less than 5 years55 because the mechanism of action is dependent on functioning β-cells.53 In addition, because SUs act independently of plasma glucose concentrations, the main risk is hypoglycemia, which may be exacerbated by coadministration with other drugs, especially those that inhibit SU metabolism via the cytochrome P450 2C9 isozyme.79 To avoid hypoglycemia, the addition of a DPP-4 inhibitor or a GLP-1RA to metformin may be considered instead. Although GLP-1RAs are associated with a more robust effect on A1C lowering compared with DPP-4 inhibitors,80 each has been shown to improve glycemic control without increasing the risk for hypoglycemia.51 GLP-1RAs are also associated with gastrointestinal side effects, particularly transient nausea, which may be attributed to delayed gastric emptying.80 Preclinical and clinical studies also demonstrate additional complementary actions between metformin and a DPP-4 inhibitor or a GLP-1RA. In vivo, metformin increased circulating concentrations of active GLP-1 but did not affect the activity of the DPP-4 enzyme.81 Thus, as demonstrated in healthy subjects, metformin and a DPP-4 inhibitor increased GLP-1 concentrations through two separate pathways, providing an additive increase in GLP-1 concentrations.82 Similar benefits were observed with metformin in combination with exogenous GLP-1 administration in patients with T2D, although findings in this study suggested that metformin enhanced injected active GLP-1 concentrations by inhibiting DPP-4 activity.83
Preserving β-cell function
Maintaining β-cell function is a key aspect in delaying disease progression,84 and as noted above, the efficacy of drugs that depend on β-cell function for their actions, such as the SUs, declines as β-cell function declines.10 Furthermore, results of in vitro studies have suggested that prolonged exposure to SUs may in fact cause disturbances in islet cell function85 and may induce β-cell apoptosis as a secondary effect to the closure of the KATP channel.86
In contrast, substantial evidence has demonstrated that TZDs positively affect β-cells by improving insulin secretory capacity and function, preserving structure and mass, and exerting protective effects from oxidative stress.87 Preclinical studies have also shown that GLP-1RAs and DPP-4 inhibitors improve or preserve β-cell mass by promoting β-cell proliferation and inhibiting apoptosis.30,88 Clinical trials in patients with T2D further demonstrate that GLP-1RAs preserve β-cell function and sensitivity to glucose with a durable effect,89,90 and DPP-4 inhibitors sustain β-cell function during 2 years of treatment.91 More recently, SGLT-2 inhibitors have been shown to improve β-cell function in patients with T2D, despite having no direct effect on pancreatic β-cells, through the reversal of glucotoxicity and improved insulin sensitivity.92 Therefore, the combination of a TZD with a DPP-4 inhibitor, GLP-1RA, or SGLT-2 inhibitor may provide both insulin-sensitizing and insulin secretory effects, in addition to helping preserve β-cell function.
Promoting weight loss
Treatment-related effects on weight are also important to consider because weight gain can worsen insulin resistance and thereby diminish efficacy.89 TZDs are associated with weight gain, in part due to fluid retention and fat cell proliferation,93 as are SUs, as a secondary effect on adipocyte KATP channels.94 Metformin is associated with weight loss or is weight neutral and has been shown to maintain efficacy across all body weight categories, including patients who are overweight or obese,55 providing a rationale for use in combination. Because AGIs slow intestinal carbohydrate digestion and absorption,51 weight gain is not expected, and treatment has been shown to minimize weight gain when used in combination with SUs.95 However, because AGIs have limited potency in lowering A1C and are also commonly associated with gastrointestinal side effects, their use in combination regimens may not always be suitable.3
For patients in whom weight loss is difficult, the use of a GLP-1RA with an SGLT-2 inhibitor may be particularly beneficial because GLP-1RAs slow gastric emptying, increase satiety, and reduce food intake,29 and SGLT-2 inhibitors decrease weight through increased caloric loss via glucosuria.3 Additionally, GLP-1RA s and SGLT-2 inhibitors reduce blood pressure in patients with T2D, possibly as a secondary effect of weight loss and other factors, such as the mild osmotic diuretic effect of the SGLT-2 inhibitors.34,96 In a recent retrospective analysis, patients had a two-fold greater weight loss with GLP-1RA and SGLT-2 inhibitor combination therapy (−7.2 kg) compared with patients taking an SGLT-2 inhibitor alone (−3.0 kg).97 Because the actions of GLP-1RAs are distinct from those of SGLT-2 inhibitors, the reduction in weight is likely additive when used in combination. DPP-4 inhibitors may also complement SGLT-2 inhibitors in this regard. Inhibition of the DPP-4 enzyme is associated with prevention of weight gain,29 and clinical study findings have shown an initial combination therapy with a DPP-4 inhibitor and an SGLT-2 inhibitor in treatment-naïve patients to significantly reduce weight compared with a DPP-4 inhibitor administered alone.98
Triple oral combination therapy
Much of the previous discussion has focused on potential dual therapy combinations; however, some patients with high A1C may require initial triple oral pharmacotherapy or intensified treatment with a third agent if dual therapy regimens do not provide adequate glycemic control. In patients with more recent disease, triple oral therapy with metformin, an SU, and a DPP-4 inhibitor may help address key disease aspects, including decreased insulin secretion and increased HGP. SUs in this population may also provide greater efficacy because β-cell function may still be intact. In patients with inadequate glycemic control despite metformin and SU dual therapy, the addition of a DPP-4 inhibitor, an SGLT-2 inhibitor, or a TZD has been shown to improve glycemic control.99,100 However, as previously mentioned, SUs increase the risk for hypoglycemia,3 making a TZD in combination with metformin and a DPP-4 inhibitor or an SGLT-2 inhibitor an alternative choice if avoiding hypoglycemia is a primary treatment goal.101,102 TZDs in these combinations should also provide additive benefits on β-cell function and mass, although adverse effects on body weight may limit use. Triple therapy with metformin, a DPP-4 inhibitor, and an SGLT-2 inhibitor has also been shown to be efficacious in reducing A1C without increasing the risk for hypoglycemia or weight gain.103–107 Based on the respective mechanisms of action, this particular combination allows improved insulin sensitivity and enhanced insulin production, with additional clinical benefits on weight and blood pressure. In addition, SGLT-2 inhibitors are associated with a rise in fasting plasma glucagon concentrations and endogenous glucose production, which could limit efficacy.108,109 However, preclinical studies have shown the suppressive effects of metformin on HGP to counteract the rise in endogenous glucose production and thereby enhance the glucose-lowering properties of SGLT-2 inhibitors.110 DPP-4 inhibitors, which suppress glucagon secretion and therefore inhibit endogenous glucose production,111 have been shown to reduce SGLT-2 inhibitor-induced increases in glucagon when used concomitantly.112
Combination with basal insulin
In general, the initiation of insulin should be considered for patients with initial A1C >9% and symptomatic hyperglycemia or for patients with A1C >8.0% despite dual therapy and/or long-standing T2D, because adding a third antidiabetes drug in this scenario will be less likely to achieve glycemic targets (Figure 1).3 Although previous guidance has suggested delaying the initiation of insulin therapy until after several rounds of treatment failure, more recent recommendations suggest initiating insulin earlier, often in combination with DPP-4 inhibitors, SGLT-2 inhibitors, or GLP-1RAs.3 Research also supports the early use of insulin to protect β-cell function through the rapid reversal of glucolipotoxicity.113
Weight gain is common with insulin therapy and has been attributed to the correction of severe hyperglycemia leading to rebuilding of muscle and fat mass, as well as increased caloric retention resulting from decreased glucosuria.93 In addition, patients experiencing hypoglycemia may increase their food intake.93 For patients requiring increasing insulin doses, TZDs have been suggested as adjunct therapy to help reduce insulin daily doses and establish glycemic control.51 Although TZDs may additionally help to preserve β-cell function,87 weight gain and fluid retention associated with TZD therapy may be exacerbated with concomitant insulin use,114 limiting use in some patients. In patients taking SU therapy, it is generally recommended that the dose be reduced or treatment stopped after starting basal insulin.3
In patients with T2D receiving basal insulin, controlled clinical trials support the use of incretin-based therapies or SGLT-2 inhibitors. For example, in patients on background insulin glargine with or without OADs, add-on exenatide for 30 weeks was associated with significantly greater reductions from baseline in A1C (−1.74% vs −1.04%, P<0.001) and weight (−1.8 vs +1.0 kg) versus add-on placebo, with no increased risk for hypoglycemia.115 Additional clinical studies of other GLP-1RAs added to existing basal insulin regimens have demonstrated similar results.116,117 Clinical trials of add-on DPP-4 inhibitor therapy to basal insulin (or other insulin formulations) have also demonstrated significant improvements in A1C without an increased risk for weight gain or hypoglycemia versus controls.118–121 Importantly, findings observed from clinical trials of basal insulin in combination with incretin-based therapies are consistent with those from real-world observational studies.122,123 Available clinical trial data have also shown that the addition of an SGLT-2 inhibitor to basal insulin or other formulations (with or without other OADs) effectively reduces A1C and weight and maintains a low risk for hypoglycemia.124–126 Of interest, insulin detemir may have a more favorable effect on weight compared with other basal insulin formulations, and patients treated with detemir plus other OADs have been shown to consistently experience weight loss.127,128
Based on clinical trial findings, the AACE suggests a DPP-4 inhibitor, GLP-1RA, or SGLT-2 inhibitor in combination with basal insulin to decrease basal and postprandial glucose and weight gain, without increasing the risk for hypoglycemia and weight gain.3 With more complex insulin regimens, SGLT-2 inhibitors are also suggested to reduce the amount of insulin needed.51 Consistently, clinical trials have shown that patients treated with GLP-1RAs, DPP-4 inhibitors, and SGLT-2 inhibitors in combination with insulin (basal and other formulations) require fewer increases in daily insulin doses compared with controls.115,116,118,119,124–126 DPP-4 inhibitors and SGLT-2 inhibitors have the advantage of oral administration; however, because patients are already receiving insulin injections, the route of administration may not be a primary factor in the treatment selection.129 In addition, injection pens with single administration of combination basal insulin and GLP-1RA (insulin degludec and liraglutide and insulin glargine and lixisenatide fixed ratio) are currently in development.130,131
More recently, there has been concern about whether SGLT-2 inhibitors are associated with an increased risk of diabetic ketoacidosis (DKA), particularly in patients with a recent reduction in concomitant insulin dose.132 Based on postmarketing reports of 73 cases in patients treated with SGLT-2 inhibitors (March 2013–June 2015), the US Food and Drug Administration required labeling of SGLT-2 inhibitors to include warnings of an increased risk of DKA.133 More expansive reviews of data have shown that most patients had only slightly elevated plasma glucose concentrations (euglycemic DKA), and some cases occurred in patients with type 1 diabetes.132 In addition, data from the clinical development programs for the three US-approved SGLT-2 inhibitors have shown no clear association between DKA and SGLT-2 inhibitor treatment.132 For example, in the EMPA-REG OUTCOME trial in 7,020 patients, the incidence of DKA was ≤0.1% and did not differ between empagliflozin and placebo groups.134 A similar incidence rate of <0.1% for DKA was also reported from the clinical development programs for canagliflozin (N=17,596) and dapagliflozin (N>18,000).132,135 The AACE has also undertaken a rigorous examination of this potential risk and concluded that the occurrence of DKA in treated patients is infrequent, and the risk–benefit ratio remains in favor of continued SGLT-2 inhibitor use, with no changes in current recommendations required.136 Regulatory agencies and pharmaceutical companies will continue to monitor this potential risk, and attention to risk factors for euglycemic DKA, such as reductions in concomitant insulin doses, has been advised when treating patients with SGLT-2 inhibitors.132
CV safety
The effect of antidiabetes therapies on CV outcomes remains a major concern. Research has reported mixed findings as to whether SUs or insulin are associated with an increased risk for CV events.137–140 Prescribing information for glipizide, glyburide, and glimepiride includes a warning of an increased risk of CV mortality with the first-generation SU, tolbutamide, versus insulin as add-on to diet or diet alone.141–143 Although the use of SUs in patients with CV risk appears to be declining in clinical practice,144 recent findings of an increased risk for mortality and CV events with treatment warrant continued caution.145,146 For example, initial SU monotherapy in a German cohort of patients with T2D significantly increased the risk of major CV events, overall mortality, and T2D-related hospitalization compared with initial metformin monotherapy (all P<0.001).146 Findings from a systematic review additionally found an increased risk of all-cause mortality and CV-related mortality with SUs compared with other antidiabetes drugs and a significantly higher risk of myocardial infarction (vs DPP-4 inhibitors) in randomized clinical trials, which was also confirmed in observational studies.145 Further, the risk of stroke was significantly higher in patients treated with SUs in clinical trials versus those treated with DPP-4 inhibitors, GLP-1RAs, TZDs, or insulin.145
TZDs have been associated with HF, attributed to treatment-induced fluid retention.3,51 These agents are therefore contraindicated in patients with preexisting HF, and the prescribing information for pioglitazone and rosiglitazone contains warnings for congestive HF.77,78 Although there is not yet conclusive evidence to establish CV benefit with this treatment,147 fewer patients with T2D had a CV disease event with bromocriptine quick-release as add-on to standard of care compared with placebo in a 1-year trial.148
CV outcome trials
Findings have been published from three CV outcome trials for DPP-4 inhibitors showing no increased risk for major CV events with alogliptin, saxagliptin, or sitagliptin, respectively, versus placebo as add-on to standard of care.149–151 However, a potential for an increased risk of hospitalization for HF versus placebo was observed in patients treated with saxagliptin149 and in a subgroup of patients with no history of HF at baseline treated with alogliptin.152 A recent observational cohort study using data from a US insurance claims database evaluated the risk of hospitalization for HF between patients with T2D treated with DPP-4 inhibitors or an SU and between patients treated with saxagliptin or sitagliptin.153 In patients with no baseline CV disease, DPP-4 inhibitors were associated with a significantly lower risk of hospitalization for HF compared with those treated with an SU (P=0.013). The risk of hospitalization for HF between saxagliptin and sitagliptin in patients with or without baseline CV disease and between DPP-4 inhibitors and SUs in patients with baseline CV disease was not significantly different.153 Additional CV outcome studies for linagliptin are ongoing.154,155 Until additional findings are known, it is recommended that DPP-4 inhibitors be used with caution, or not at all, in patients with preexisting HF.4
In the EMPA-REG OUTCOME CV outcomes trial in patients with T2D and established CV disease, empagliflozin as add-on to standard of care was associated with significantly lower rates of major CV events (ie, death from CV causes, nonfatal myocardial infarction, nonfatal stroke), primarily driven by a significant reduction in death from CV causes, as well as death from any cause and hospitalization for HF compared with add-on placebo.134 CV outcome trials for dapagliflozin and canagliflozin are underway and are expected to be completed in 2017 (canagliflozin) and 2019 (dapagliflozin). Each trial includes patients with T2D at high risk for CV events.156,157 Most of the GLP-1RA CV trials are ongoing, although one study for lixisenatide recently reported preliminary findings.158 In this study, no increased risk for major CV events was observed in patients with a recent acute coronary syndrome event treated with lixisenatide versus placebo.158
Conclusion
The multitude of therapeutic options for T2D adds to the complexity of managing patients with this disease but also allows clinicians to readjust and rebalance treatment over time as physiologic changes occur throughout the course of the disease. As understanding of the pathophysiology of T2D grows, it is becoming apparent that early use of targeted combination therapy has the potential to slow disease progression, potentially through the correction of known pathologies and the preservation of β-cell function. Obesity plays a central role in the development and worsening of T2D, and thus it can be argued that treatments that facilitate weight loss may be most beneficial. Whereas the underlying pathophysiology of T2D should be a key factor in choosing which medications to combine, the treatment choice must also be individualized to patients’ needs and tolerability. Based on individual therapeutic profiles, the newer incretin-based therapies and SGLT-2 inhibitors are appropriate for use in combination with one another or in combination with more traditional agents (eg, metformin, TZDs, insulin) to reduce A1C without increasing the risk of hypoglycemia. In addition, because these newer agents act on different pathways, the beneficial actions on glycemia and weight are likely additive when used in combination. Although clinical inertia remains an issue, emerging research helps to support and advocate for the use of early combination therapy as a practical approach to preempt the development of long-term complications and disease progression.
Acknowledgments
Medical writing support for the preparation of this manuscript was provided by Meg Shurak, MS, and Janet E Matsuura, PhD, from Complete Healthcare Communications, LLC (Chadds Ford, PA, USA), with funding from AstraZeneca.
Disclosure
PL has received a research grant and is a speaker and consultant for AstraZeneca, Sanofi-Aventis, LLC, and Novo Nordisk, Inc. He has also received a research grant and is a speaker for Eli Lilly and Company and Boehringer Ingelheim, GmbH; is a speaker and consultant for Janssen Pharmaceuticals, Inc.; and is a speaker for GlaxoSmithKline, plc. The author reports no other conflicts of interest in this work.
References
International Diabetes Federation, Global burden. Sixth edition; 2014. Available from: http://www.idf.org/diabetesatlas/5e/the-global-burden. Accessed February 9, 2015. | ||
Kahn SE, Cooper ME, Del Prato S. Pathophysiology and treatment of type 2 diabetes: perspectives on the past, present, and future. Lancet. 2014;383(9922):1068–1083. | ||
Garber AJ, Abrahamson MJ, Barzilay JI, et al. Consensus statement by the American Association of Clinical Endocrinologists and American College of Endocrinology on the comprehensive type 2 diabetes management algorithm - 2016 executive summary. Endocr Pract. 2016;22(1):84–113. | ||
American Diabetes Association. 7. Approaches to glycemic treatment. Diabetes Care. 2016;39(Suppl 1):S52–S59. | ||
Berkowitz SA, Krumme AA, Avorn J, et al. Initial choice of oral glucose-lowering medication for diabetes mellitus: a patient-centered comparative effectiveness study. JAMA Intern Med. 2014;174(12):1955–1962. | ||
Khunti K, Wolden ML, Thorsted BL, Andersen M, Davies MJ. Clinical inertia in people with type 2 diabetes: a retrospective cohort study of more than 80,000 people. Diabetes Care. 2013;36(11):3411–3417. | ||
Levin PA, Wei W, Zhou S, Xie L, Baser O. Outcomes and treatment patterns of adding a third agent to 2 OADs in patients with type 2 diabetes. J Manag Care Spec Pharm. 2014;20(5):501–512. | ||
Brown JB, Nichols GA, Perry A. The burden of treatment failure in type 2 diabetes. Diabetes Care. 2004;27(7):1535–1540. | ||
Garber AJ, Abrahamson MJ, Barzilay JI, et al. American Association of Clinical Endocrinologists’ comprehensive diabetes management algorithm 2013 consensus statement--executive summary. Endocr Pract. 2013;19(3):536–557. | ||
DeFronzo RA. Banting Lecture. From the triumvirate to the ominous octet: a new paradigm for the treatment of type 2 diabetes mellitus. Diabetes. 2009;58(4):773–795. | ||
Triplitt CL. Examining the mechanisms of glucose regulation. Am J Manag Care. 2012;18(1 suppl):S4–S10. | ||
Ravnskjaer K, Hogan MF, Lackey D, et al. Glucagon regulates gluconeogenesis through KAT2B- and WDR5-mediated epigenetic effects. J Clin Invest. 2013;123(10):4318–4328. | ||
Triplitt C. Understanding the kidney’s role in blood glucose regulation. Am J Manag Care. 2012;18:S11–S16. | ||
Gerich JE, Meyer C, Woerle HJ, Stumvoll M. Renal gluconeogenesis: its importance in human glucose homeostasis. Diabetes Care. 2001;24(2):382–391. | ||
Basu R, Chandramouli V, Dicke B, Landau B, Rizza R. Obesity and type 2 diabetes impair insulin-induced suppression of glycogenolysis as well as gluconeogenesis. Diabetes. 2005;54(7):1942–1948. | ||
Barthel A, Schmoll D. Novel concepts in insulin regulation of hepatic gluconeogenesis. Am J Physiol Endocrinol Metab. 2003;285(4):E685–E692. | ||
Guilherme A, Virbasius JV, Puri V, Czech MP. Adipocyte dysfunctions linking obesity to insulin resistance and type 2 diabetes. Nat Rev Mol Cell Biol. 2008;9(5):367–377. | ||
Wellen KE, Hotamisligil GS. Inflammation, stress, and diabetes. J Clin Invest. 2005;115(5):1111–1119. | ||
Wensveen FM, Valentic S, Sestan M, Turk Wensveen T, Polic B. The “Big Bang” in obese fat: Events initiating obesity-induced adipose tissue inflammation. Eur J Immunol. 2015;45(9):2446–2456. | ||
Xu H, Barnes GT, Yang Q, et al. Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. J Clin Invest. 2003;112(12):1821–1830. | ||
Weisberg SP, McCann D, Desai M, Rosenbaum M, Leibel RL, Ferrante AW, Jr. Obesity is associated with macrophage accumulation in adipose tissue. J Clin Invest. 2003;112(12):1796–1808. | ||
Poitout V, Robertson RP. Glucolipotoxicity: fuel excess and beta-cell dysfunction. Endocr Rev. 2008;29(3):351–366. | ||
Russo GT, Giorda CB, Cercone S, Nicolucci A, Cucinotta D; BetaDecline Study Group. Factors associated with beta-cell dysfunction in type 2 diabetes: the BETADECLINE study. PLoS One. 2014;9(10):e109702. | ||
DeFronzo RA, Tripathy D. Skeletal muscle insulin resistance is the primary defect in type 2 diabetes. Diabetes Care. 2009;32(suppl 2):S157–S163. | ||
Cnop M, Vidal J, Hull RL, et al. Progressive loss of beta-cell function leads to worsening glucose tolerance in first-degree relatives of subjects with type 2 diabetes. Diabetes Care. 2007;30(3):677–682. | ||
Hu FB, van Dam RM, Liu S. Diet and risk of Type II diabetes: the role of types of fat and carbohydrate. Diabetologia. 2001;44(7):805–817. | ||
Guenard F, Deshaies Y, Cianflone K, Kral JG, Marceau P, Vohl MC. Differential methylation in glucoregulatory genes of offspring born before vs. after maternal gastrointestinal bypass surgery. Proc Natl Acad Sci U S A. 2013;110(28):11439–11444. | ||
Nauck MA, Baller B, Meier JJ. Gastric inhibitory polypeptide and glucagon-like peptide-1 in the pathogenesis of type 2 diabetes. Diabetes. 2004;53(suppl 3):S190–S196. | ||
Drucker DJ. The biology of incretin hormones. Cell Metab. 2006;3(3):153–165. | ||
Drucker DJ, Nauck MA. The incretin system: glucagon-like peptide-1 receptor agonists and dipeptidyl peptidase-4 inhibitors in type 2 diabetes. Lancet. 2006;368(9548):1696–1705. | ||
Drucker DJ, Sherman SI, Gorelick FS, Bergenstal RM, Sherwin RS, Buse JB. Incretin-based therapies for the treatment of type 2 diabetes: evaluation of the risks and benefits. Diabetes Care. 2010;33(2):428–433. | ||
Nauck MA, Heimesaat MM, Orskov C, Holst JJ, Ebert R, Creutzfeldt W. Preserved incretin activity of glucagon-like peptide 1 [7-36 amide] but not of synthetic human gastric inhibitory polypeptide in patients with type-2 diabetes mellitus. J Clin Invest.1993;91(1):301–307. | ||
Nauck MA, Vardarli I, Deacon CF, Holst JJ, Meier JJ. Secretion of glucagon-like peptide-1 (GLP-1) in type 2 diabetes: what is up, what is down? Diabetologia. 2011;54(1):10–18. | ||
Nauck MA. Update on developments with SGLT2 inhibitors in the management of type 2 diabetes. Drug Des Devel Ther. 2014;8:1335–1380. | ||
Meyer C, Woerle HJ, Dostou JM, Welle SL, Gerich JE. Abnormal renal, hepatic, and muscle glucose metabolism following glucose ingestion in type 2 diabetes. Am J Physiol Endocrinol Metab. 2004;287(6):E1049–E1056. | ||
Abdul-Ghani MA, Norton L, Defronzo RA. Role of sodium-glucose cotransporter 2 (SGLT 2) inhibitors in the treatment of type 2 diabetes. Endocr Rev. 2011;32(4):515–531. | ||
Murphy KG, Bloom SR. Gut hormones in the control of appetite. Exp Physiol. 2004;89(5):507–516. | ||
Thaler JP, Schwartz MW. Minireview: Inflammation and obesity pathogenesis: the hypothalamus heats up. Endocrinology. 2010;151(9):4109–4115. | ||
Thaler JP, Yi CX, Schur EA, et al. Obesity is associated with hypothalamic injury in rodents and humans. J Clin Invest. 2012;122(1):153–162. | ||
Shi SQ, Ansari TS, McGuinness OP, Wasserman DH, Johnson CH. Circadian disruption leads to insulin resistance and obesity. Curr Biol. 2013;23(5):372–381. | ||
Gale JE, Cox HI, Qian J, Block GD, Colwell CS, Matveyenko AV. Disruption of circadian rhythms accelerates development of diabetes through pancreatic beta-cell loss and dysfunction. J Biol Rhythms. 2011;26(5):423–433. | ||
Viollet B, Guigas B, Sanz Garcia N, Leclerc J, Foretz M, Andreelli F. Cellular and molecular mechanisms of metformin: an overview. Clin Sci (Lond). 2012;122(6):253–270. | ||
Wang CC, Lin SK, Tseng YF, et al. Elevation of serum aminotransferase activity increases risk of carotid atherosclerosis in patients with non-alcoholic fatty liver disease. J Gastroenterol Hepatol. 2009;24(8):1411–1416. | ||
Idilman R, Mizrak D, Corapcioglu D, et al. Clinical trial: insulin-sensitizing agents may reduce consequences of insulin resistance in individuals with non-alcoholic steatohepatitis. Aliment Pharmacol Ther. 2008;28(2):200–208. | ||
Facciorusso A. The influence of diabetes in the pathogenesis and the clinical course of hepatocellular carcinoma: recent findings and new perspectives. Curr Diabetes Rev. 2013;9(5):382–386. | ||
Singh S, Singh PP, Singh AG, Murad MH, Sanchez W. Anti-diabetic medications and the risk of hepatocellular cancer: a systematic review and meta-analysis. Am. J. Gastroenterol. 2013;108(6):881–891; quiz 892. | ||
Zhao D, Shi Z, Warriner AH, et al. Molecular mechanism of thiazolidinedione-mediated inhibitory effects on osteoclastogenesis. PLoS One. 2014;9(7):e102706. | ||
Miyazaki Y, Mahankali A, Matsuda M, et al. Improved glycemic control and enhanced insulin sensitivity in type 2 diabetic subjects treated with pioglitazone. Diabetes Care. 2001;24(4):710–719. | ||
Miyazaki Y, Matsuda M, DeFronzo RA. Dose-response effect of pioglitazone on insulin sensitivity and insulin secretion in type 2 diabetes. Diabetes Care. 2002;25(3):517–523. | ||
Miyazaki Y, Glass L, Triplitt C, et al. Effect of rosiglitazone on glucose and non-esterified fatty acid metabolism in Type II diabetic patients. Diabetologia. 2001;44(12):2210–2219. | ||
American Diabetes Association. Standards of Medical Care in Diabetes-2016. Diabetes Care. 2016;39 Suppl 1:S1–S119. | ||
Proks P, Reimann F, Green N, Gribble F, Ashcroft F. Sulfonylurea stimulation of insulin secretion. Diabetes. 2002;51(suppl 3):S368–S376. | ||
Aquilante CL. Sulfonylurea pharmacogenomics in type 2 diabetes: the influence of drug target and diabetes risk polymorphisms. Expert Rev Cardiovasc Ther. 2010;8(3):359–372. | ||
Reis AF, Velho G. Sulfonylurea receptor -1 (SUR1): genetic and metabolic evidences for a role in the susceptibility to type 2 diabetes mellitus. Diabetes Metab. 2002;28(1):14–19. | ||
Handelsman Y, Bloomgarden ZT, Grunberger G, et al. American Association of Clinical Endocrinologists and American College of Endocrinology clinical practice guidelines for developing a diabetes mellitus comprehensive care plan-2015. Endocr Pract. 2015;21(suppl 1):1–87. | ||
Baggio L, Drucker DJ. Biology of incretins: GLP-1 and GIP. Gastroenterology. 2007;132:2131–2157. | ||
Deacon CF, Johnsen AH, Holst JJ. Degradation of glucagon-like peptide-1 by human plasma in vitro yields an N-terminally truncated peptide that is a major endogenous metabolite in vivo. J Clin Endocrinol Metab. 1995;80(3):952–957. | ||
Nauck MA, El-Ouaghlidi A. The therapeutic actions of DPP-IV inhibition are not mediated by glucagon-like peptide-1. Diabetologia. 2005;48(4):608–611. | ||
Baetta R, Corsini A. Pharmacology of dipeptidyl peptidase-4 inhibitors: similarities and differences. Drugs. 2011;71(11):1441–1467. | ||
He K, Shi JC, Mao XM. Safety and efficacy of acarbose in the treatment of diabetes in Chinese patients. Ther Clin Risk Manag. 2014;10:505–511. | ||
Zhu Q, Tong Y, Wu T, Li J, Tong N. Comparison of the hypoglycemic effect of acarbose monotherapy in patients with type 2 diabetes mellitus consuming an Eastern or Western diet: a systematic meta-analysis. Clin Ther. 2013;35(6):880–899. | ||
Holt RI, Barnett AH, Bailey CJ. Bromocriptine: old drug, new formulation and new indication. Diabetes Obes Metab. 2010;12(12):1048–1057. | ||
Handelsman Y. Role of bile acid sequestrants in the treatment of type 2 diabetes. Diabetes Care. 2011;34(suppl 2):S244–S250. | ||
Beysen C, Murphy EJ, Deines K, et al. Effect of bile acid sequestrants on glucose metabolism, hepatic de novo lipogenesis, and cholesterol and bile acid kinetics in type 2 diabetes: a randomised controlled study. Diabetologia. 2012;55(2):432–442. | ||
Lantus® (insulin glargine [rDNA origin] injection). [prescribing information]. Bridgewater, NJ: sanofi-aventis; 2009. | ||
Levemir® (insulin detemir) [prescribing information]. Bagsvaerd, Denmark: Novo Nordisk A/S; 2015. | ||
Tresiba® (insulin degludec injection) [prescribing information]. Bagsvaerd, Denmark: Novo Nordisk A/S; 2015. | ||
Toujeo® (insulin glargine) [prescribing Information], Summary of Product Characteristics. Frankfurt, Germany: Sanofi-Aventis Deutschland, GmbH; 2015. | ||
Sanofi. Press Release: Sanofi reports positive phase 3 results for Toujeo® (insulin glargine [rDNA origin] injection, 300 U/mL); 2015. Available from: http://www.news.sanofi.us/2014-06-14-Sanofi-Reports-Positive-Phase-3-Results-for-Toujeo-insulin-glargine-rDNA-origin-injection-300-U-mL. Accessed December 7, 2015. | ||
Hallsten K, Virtanen KA, Lonnqvist F, et al. Rosiglitazone but not metformin enhances insulin- and exercise-stimulated skeletal muscle glucose uptake in patients with newly diagnosed type 2 diabetes. Diabetes. 2002;51(12):3479–3485. | ||
Malin SK, Gerber R, Chipkin SR, Braun B. Independent and combined effects of exercise training and metformin on insulin sensitivity in individuals with prediabetes. Diabetes Care. 2012;35(1):131–136. | ||
Graham GG, Punt J, Arora M, et al. Clinical pharmacokinetics of metformin. Clin. Pharmacokinet. 2011;50(2):81–98. | ||
Glucophage® (metformin hydrochloride) and Glucophage XR® (metformin hydrochloride extended-release) [prescribing information]. Princeton, NJ: Bristol-Myers Squibb Company; 2009. | ||
Kim CH, Han KA, Oh HJ, et al. Safety, tolerability, and efficacy of metformin extended-release oral antidiabetic therapy in patients with type 2 diabetes: an observational trial in Asia. J Diabetes.2012;4(4):395–406. | ||
Yu Y, Li X, Blanchard J, et al. Insulin sensitizers improve learning and attenuate tau hyperphosphorylation and neuroinflammation in 3xTg-AD mice. J Neural Transm (Vienna). 2015;122(4):593–606. | ||
Genovese S, De Berardis G, Nicolucci A, et al. Effect of pioglitazone versus metformin on cardiovascular risk markers in type 2 diabetes. Adv Ther. 2013;30(2):190–202. | ||
Actos® (pioglitazone hydrochloride) [prescribing information]. Deerfield, IL: Takeda Pharmaceuticals America, Inc.; 2012. | ||
Avandia® (rosiglitazone maleate) [prescribing information]. Research Triangle Park, NC: GlaxoSmithKline; 2011. | ||
Amin M, Suksomboon N. Pharmacotherapy of type 2 diabetes mellitus: an update on drug-drug interactions. Drug Saf. 2014;37(11):903–919. | ||
Reid T. Choosing GLP-1 receptor agonists or DPP-4 inhibitors: weighing the clinical trial evidence. Clin Diabetes. 2012;30(1):3–12. | ||
Mulherin AJ, Oh AH, Kim H, Grieco A, Lauffer LM, Brubaker PL. Mechanisms underlying metformin-induced secretion of glucagon-like peptide-1 from the intestinal L cell. Endocrinology. 2011;152(12):4610–4619. | ||
Migoya EM, Bergeron R, Miller JL, et al. Dipeptidyl peptidase-4 inhibitors administered in combination with metformin result in an additive increase in the plasma concentration of active GLP-1. Clin Pharmacol Ther. 2010;88(6):801–808. | ||
Cuthbertson J, Patterson S, O’Harte FP, Bell PM. Addition of metformin to exogenous glucagon-like peptide-1 results in increased serum glucagon-like peptide-1 concentrations and greater glucose lowering in type 2 diabetes mellitus. Metabolism. 2011;60(1):52–56. | ||
Kahn SE, Lachin JM, Zinman B, et al. Effects of rosiglitazone, glyburide, and metformin on beta-cell function and insulin sensitivity in ADOPT. Diabetes. 2011;60(5):1552–1560. | ||
Del Guerra S, Marselli L, Lupi R, et al. Effects of prolonged in vitro exposure to sulphonylureas on the function and survival of human islets. J Diabetes Complications. 2005;19(1):60–64. | ||
Maedler K, Carr RD, Bosco D, Zuellig RA, Berney T, Donath MY. Sulfonylurea induced beta-cell apoptosis in cultured human islets. J Clin Endocrinol Metab. 2005;90(1):501–506. | ||
Campbell IW, Mariz S. Beta-cell preservation with thiazolidinediones. Diabetes Res Clin. Pract. 2007;76(2):163–176. | ||
Vilsboll T. The effects of glucagon-like peptide-1 on the beta cell. Diabetes Obes Metab. 2009;11(suppl 3):11–18. | ||
Garber AJ. Incretin effects on beta-cell function, replication, and mass: the human perspective. Diabetes Care. 2011;34 Suppl 2:S258–S263. | ||
Garber A, Henry R, Ratner R, et al. Liraglutide versus glimepiride monotherapy for type 2 diabetes (LEAD-3 Mono): a randomised, 52-week, phase III, double-blind, parallel-treatment trial. Lancet. 2009;373(9662):473–481. | ||
Leibowitz G, Cahn A, Bhatt DL, et al. Impact of treatment with saxagliptin on glycaemic stability and beta-cell function in the SAVOR-TIMI 53 study. Diabetes Obes Metab. 2015;17(5):487–494. | ||
Merovci A, Mari A, Solis C, et al. Dapagliflozin lowers plasma glucose concentration and improves beta cell function. J Clin Endocrinol Metab. 2015:100(5):1927–1932. | ||
Fonseca V. Effect of thiazolidinediones on body weight in patients with diabetes mellitus. Am J Med. 2003;115(suppl 8A):42S–48S. | ||
Shi H, Moustaid-Moussa N, Wilkison WO, Zemel MB. Role of the sulfonylurea receptor in regulating human adipocyte metabolism. FASEB J. 1999;13(13):1833–1838. | ||
Acarbose [prescribing information]. Morgantown, WV: Mylan Pharmaceuticals, Inc.; 2015. | ||
Wang B, Zhong J, Lin H, et al. Blood pressure-lowering effects of GLP-1 receptor agonists exenatide and liraglutide: a meta-analysis of clinical trials. Diabetes Obes Metab. 2013;15(8):737–749. | ||
McGovern A, Dutta N, Watters K, Munro N, Feher M. Additive weight loss effect with a combination of an oral sodium-glucose cotransporter 2 (SGLT2) inhibitor and a glucagon-like peptide 1 (GLP-1) agonist in type 2 diabetes. A6 (P143). Paper presented at: Diabetes UK Professional Conference; March 11–13, 2015; London, UK. | ||
Lewin A, DeFronzo R, Patel S, et al. Initial combination of empagliflozin and linagliptin in subjects with type 2 diabetes. Diabetes Care. 2015;38(3):394–402. | ||
Matthaei S, Bowering K, Rohwedder K, Grohl A, Parikh S; Study 05 Group. Dapagliflozin improves glycemic control and reduces body weight as add-on therapy to metformin plus sulfonylurea: a 24-week randomized, double-blind clinical trial. Diabetes Care. 2015;38(3):365–372. | ||
Liu SC, Chien KL, Wang CH, Chen WC, Leung CH. Efficacy and safety of adding pioglitazone or sitagliptin to patients with type 2 diabetes insufficiently controlled with metformin and a sulfonylurea. Endocr Pract. 2013:1–30. | ||
DeFronzo RA, Burant CF, Fleck P, Wilson C, Mekki Q, Pratley RE. Efficacy and tolerability of the DPP-4 inhibitor alogliptin combined with pioglitazone, in metformin-treated patients with type 2 diabetes. J Clin Endocrinol Metab. 2012;97(5):1615–1622. | ||
Kovacs CS, Seshiah V, Swallow R, et al. Empagliflozin improves glycaemic and weight control as add-on therapy to pioglitazone or pioglitazone plus metformin in patients with type 2 diabetes: a 24-week, randomized, placebo-controlled trial. Diabetes Obes Metab. 2014;16(2):147–158. | ||
Rosenstock J, Hansen L, Zee P, et al. Dual add-on therapy in type 2 diabetes poorly controlled with metformin monotherapy: a randomized double-blind trial of saxagliptin plus dapagliflozin addition versus single addition of saxagliptin or dapagliflozin to metformin. Diabetes Care. 2014;38(3):376–383. | ||
DeFronzo RA, Lewin A, Patel S, et al. Combination of empagliflozin and linagliptin as second-line therapy in subjects with type 2 diabetes inadequately controlled on metformin. Diabetes Care. 2015;38(3):384–393. | ||
Jabbour SA, Hardy E, Sugg J, Parikh S; Study 10 Group. Dapagliflozin is effective as add-on therapy to sitagliptin with or without metformin: a 24-week, multicenter, randomized, double-blind, placebo-controlled study. Diabetes Care. 2014;37(3):740–750. | ||
Matthaei S, Catrinoiu D, Celinski A, et al. Randomized, double-blind trial of triple therapy with saxagliptin add-on to dapagliflozin plus metformin in patients with type 2 diabetes. Diabetes Care. 2015;38(11):2018–2024. | ||
Mathieu C, Ranetti AE, Li D, et al. Randomized, double-blind, phase 3 trial of triple therapy with dapagliflozin add-on to saxagliptin plus metformin in type 2 diabetes. Diabetes Care. 2015;38(11):2009–2017. | ||
Merovci A, Solis-Herrera C, Daniele G, et al. Dapagliflozin improves muscle insulin sensitivity but enhances endogenous glucose production. J Clin Invest. 2014;124(2):509–514. | ||
Ferrannini E, Muscelli E, Frascerra S, et al. Metabolic response to sodium-glucose cotransporter 2 inhibition in type 2 diabetic patients. J Clin Invest. 2014;124(2):499–508. | ||
Neschen S, Scheerer M, Seelig A, et al. Metformin supports the antidiabetic effect of a sodium glucose cotransporter 2 inhibitor by suppressing endogenous glucose production in diabetic mice. Diabetes. 2015;64(1):284–290. | ||
Balas B, Baig MR, Watson C, et al. The dipeptidyl peptidase IV inhibitor vildagliptin suppresses endogenous glucose production and enhances islet function after single-dose administration in type 2 diabetic patients. J Clin Endocrinol Metab. 2007;92(4):1249–1255. | ||
Hansen L, Iqbal N, Ekholm E, Cook W, Hirshberg B. Postprandial dynamics of plasma glucose, insulin, and glucagon in patients with type 2 diabetes treated with saxagliptin plus dapagliflozin add-on to metformin therapy. Endocr Pract. 2014;20(11):1187–1197. | ||
Raz I, Mosenzon O. Early insulinization to prevent diabetes progression. Diabetes Care. 2013;36(suppl 2):S190–S197. | ||
Scheen AJ. Combined thiazolidinedione-insulin therapy: should we be concerned about safety? Drug Saf. 2004;27(12):841–856. | ||
Buse JB, Bergenstal RM, Glass LC, et al. Use of twice-daily exenatide in basal insulin-treated patients with type 2 diabetes: a randomized, controlled trial. Ann Intern Med. 2011;154(2):103–112. | ||
Ahmann A, Rodbard HW, Rosenstock J, et al. Efficacy and safety of liraglutide versus placebo added to basal insulin analogues (with or without metformin) in patients with type 2 diabetes: a randomized, placebo-controlled trial. Diabetes Obes Metab. 2015;17(11):1056–1064. | ||
Rosenstock J, Fonseca VA, Gross JL, et al. Advancing basal insulin replacement in type 2 diabetes inadequately controlled with insulin glargine plus oral agents: A comparison of adding albiglutide, a weekly GLP-1 receptor agonist, versus thrice-daily prandial insulin lispro. Diabetes Care. 2014;37(8):2317–2325. | ||
Barnett AH, Charbonnel B, Donovan M, Fleming D, Chen R. Effect of saxagliptin as add-on therapy in patients with poorly controlled type 2 diabetes on insulin alone or insulin combined with metformin. Curr Med Res Opin. 2012;28(4):513–523. | ||
Yki-Jarvinen H, Rosenstock J, Duran-Garcia S, et al. Effects of adding linagliptin to basal insulin regimen for inadequately controlled type 2 diabetes: a ≥52-week randomized, double-blind study. Diabetes Care. 2013;36(12):3875–3881. | ||
Rosenstock J, Rendell MS, Gross JL, Fleck PR, Wilson CA, Mekki Q. Alogliptin added to insulin therapy in patients with type 2 diabetes reduces HbA(1C) without causing weight gain or increased hypoglycaemia. Diabetes Obes Metab. 2009;11(12):1145–1152. | ||
Vilsboll T, Rosenstock J, Yki-Jarvinen H, et al. Efficacy and safety of sitagliptin when added to insulin therapy in patients with type 2 diabetes. Diabetes Obes Metab. 2010;12(2):167–177. | ||
Yoon NM, Cavaghan MK, Brunelle RL, Roach P. Exenatide added to insulin therapy: a retrospective review of clinical practice over two years in an academic endocrinology outpatient setting. Clin Ther. 2009;31(7):1511–1523. | ||
Seufert J, Pegelow K, Bramlage P. Efficacy and safety of insulin glargine added to a fixed-dose combination of metformin and a dipeptidyl peptidase-4 inhibitor: results of the GOLD observational study. Vasc Health Risk Manag. 2013;9:711–717. | ||
Rosenstock J, Jelaska A, Zeller C, Kim G, Broedl UC, Woerle HJ. Impact of empagliflozin added on to basal insulin in type 2 diabetes inadequately controlled on basal insulin: a 78-week randomized, double-blind, placebo-controlled trial. Diabetes Obes Metab. 2015;17(10):936–948. | ||
Wilding JP, Woo V, Soler NG, et al. Long-term efficacy of dapagliflozin in patients with type 2 diabetes mellitus receiving high doses of insulin: a randomized trial. Ann Intern Med. 2012;156(6):405–415. | ||
Neal B, Perkovic V, de Zeeuw D, et al. Efficacy and safety of canagliflozin, an inhibitor of sodium-glucose cotransporter 2, when used in conjunction with insulin therapy in patients with type 2 diabetes. Diabetes Care. 2015;38(3):403–411. | ||
Damci T, Emral R, Svendsen AL, Balkir T, Vora J. Lower risk of hypoglycaemia and greater odds for weight loss with initiation of insulin detemir compared with insulin glargine in Turkish patients with type 2 diabetes mellitus: local results of a multinational observational study. BMC Endocr Disord. 2014;14:61. | ||
Yale JF, Damci T, Kaiser M, et al. Initiation of once daily insulin detemir is not associated with weight gain in patients with type 2 diabetes mellitus: results from an observational study. Diabetol Metab Syndr. 2013;5(1):56. | ||
Vora J. Combining incretin-based therapies with insulin: realizing the potential in type 2 diabetes. Diabetes Care. 2013;36(suppl 2):S226–S232. | ||
American Diabetes Association. Fast Facts Data and Statistics about Diabetes; 2015. Available from: http://professional.diabetes.org/ResourcesForProfessionals.aspx?cid=91777&loc=dorg-statistics. Accessed March 23, 2015. | ||
Gough SC, Bode B, Woo V, et al. Efficacy and safety of a fixed-ratio combination of insulin degludec and liraglutide (IDegLira) compared with its components given alone: results of a phase 3, open-label, randomised, 26-week, treat-to-target trial in insulin-naive patients with type 2 diabetes. Lancet Diabetes Endocrinol. 2014;2(11):885–893. | ||
Rosenstock J, Ferrannini E. Euglycemic diabetic ketoacidosis: a predictable, detectable, and preventable safety concern with SGLT2 inhibitors. Diabetes Care. 2015;38(9):1638–1642. | ||
FDA Drug Safety Communication: FDA revises labels of SGLT2 inhibitors for diabetes to include warnings about too much acid in the blood and serious urinary tract infections; 2015. Available from: http://www.fda.gov/Drugs/DrugSafety/ucm475463.htm. Accessed January 29, 2016. | ||
Zinman B, Wanner C, Lachin JM, et al. Empagliflozin, cardiovascular outcomes, and mortality in type 2 diabetes. N Engl J Med. 2015;373(22):2117–2128. | ||
Erondu N, Desai M, Ways K, Meininger G. Diabetic ketoacidosis and related events in the canagliflozin type 2 diabetes clinical program. Diabetes Care. 2015;38(9):1680–1686. | ||
AACE/ACE scientific and clinical review: association of SGLT2 inhibitors and DKA; 2015. Available from: http://resources.aace.com/in-the-news/aaceace-scientific-and-clinical-review-association-sglt2-inhibitors-and-dka. Accessed January 29, 2016. | ||
Hsu PF, Sung SH, Cheng HM, et al. Association of clinical symptomatic hypoglycemia with cardiovascular events and total mortality in type 2 diabetes: a nationwide population-based study. Diabetes Care. 2013;36(4):894–900. | ||
ORIGIN Trial Investigators, Gerstein HC, Bosch J, et al. Basal insulin and cardiovascular and other outcomes in dysglycemia. N Engl J Med. 2012;367(4):319–328. | ||
UK Prospective Diabetes Study (UKPDS) Group. Intensive blood-glucose control with sulphonylureas or insulin compared with conventional treatment and risk of complications in patients with type 2 diabetes. Lancet. 1998;352(9131):837–853. | ||
Roumie CL, Hung AM, Greevy RA, et al. Comparative effectiveness of sulfonylurea and metformin monotherapy on cardiovascular events in type 2 diabetes mellitus: a cohort study. Ann Intern Med. 2012;157(9):601–610. | ||
Glucotrol® (glipizide tablets) [prescribing information]. New York, NY: Pfizer Inc; 2010. | ||
Micronase® (glyburide tablets) [full prescribing information]. New York, NY: Pfizer Inc. 2010. | ||
Amaryl® (glimepiride) [prescribing information]. Bridgewater, NJ: sanofi-aventis; 2013. | ||
Kachroo S, Kawabata H, Colilla S, et al. Use of sulfonylureas in patients with multiple risk factors for CV disease or those with existing CV disease - CREST Study: are we following current ADA/EASD guidelines? [A1112]. Paper presented at: 51st Annual Meeting, European Association for the Study of Diabetes; September 14–18,2015; Stockholm, Sweden. | ||
Baxter C, Das R, Langerman H, et al. Increased risk of cardiovascular-related events associated with sulfonylureas compared to other antihyperglycaemic drugs: a Bayesian meta-analysis of survival data. [A128]. Paper presented at: 51st Annual Meeting, European Association for the Study of Diabetes; September 14–18,2015; Stockholm, Sweden. | ||
Berg B, Wilke T, Groth A, et al. Treatment characteristics and outcomes associated with sulphonylurea versus metformin therapy in incident type 2 diabetes mellitus patients: results of the German CREST study. [A130]. Paper presented at: 51st Annual Meeting, European Association for the Study of Diabetes; September 14–18,2015; Stockholm, Sweden. | ||
Cycloset® (bromocriptine mesylate) [prescribing information]. Tiverton, RI: VeroScience, LLC; 2010. | ||
Gaziano JM, Cincotta AH, O’Connor CM, et al. Randomized clinical trial of quick-release bromocriptine among patients with type 2 diabetes on overall safety and cardiovascular outcomes. Diabetes Care. 2010;33(7):1503–1508. | ||
Scirica BM, Bhatt DL, Braunwald E, et al. Saxagliptin and cardiovascular outcomes in patients with type 2 diabetes mellitus. N Engl J Med. 2013;369(14):1317–1326. | ||
White WB, Cannon CP, Heller SR, et al. Alogliptin after acute coronary syndrome in patients with type 2 diabetes. N Engl J Med. 2013;369(14):1327–1335. | ||
Green JB, Bethel MA, Armstrong PW, et al. Effect of sitagliptin on cardiovascular outcomes in type 2 diabetes. N Engl J Med. 2015;373(3):232–242. | ||
Zannad F, Cannon C, Cushman WC, et al. Heart failure and mortality outcomes in patients with type 2 diabetes taking alogliptin versus placebo in EXAMINE: a multicentre, randomised, double-blind trial. Lancet. 2015;385(9982):2067–2076. | ||
Fu AZ, Johnston SS, Ghannam A, Tsai K, Cappell K, Fowler R, Riehle E, Cole AL, Kalsekar I, Sheehan J. Association between hospitalization for heart failure and dipeptidyl peptidase 4 inhibitors in patients with type 2 diabetes: an observational study. Diabetes Care. 2016;39(5):726–734. | ||
Rosenstock J, Marx N, Kahn SE, et al. Cardiovascular outcome trials in type 2 diabetes and the sulphonylurea controversy: rationale for the active-comparator CAROLINA trial. Diab Vasc Dis Res. 2013;10(4):289–301. | ||
Cardiovascular and renal microvascular outcome study with linagliptin in patients with type 2 diabetes mellitus at high vascular risk (CARMELINA); 2015. Available from: http://clinicaltrials.gov/ct2/show/NCT01897532?term=NCT01897532&rank=1. Accessed March 16, 2015. | ||
Multicenter trial to evaluate the effect of dapagliflozin on the incidence of cardiovascular events (DECLARE-TIMI58); 2015. Available from: http://clinicaltrials.gov/ct2/show/NCT01730534?term=declare&rank=2. Accessed September 29, 2015. | ||
Neal B, Perkovic V, de Zeeuw D, et al. Rationale, design, and baseline characteristics of the Canagliflozin Cardiovascular Assessment Study (CANVAS)--a randomized placebo-controlled trial. Am. Heart J. 2013;166(2):217–223.e11. | ||
American Diabetes Association. First CVD outcome trial of a GLP-1 agonist finds no cardiac risk or benefit; 2015. Available from: http://www.diabetes.org/newsroom/press-releases/2015/elixa.html?referrer=http://google.diabetes.org/search?site=Diabetes&client=diabetes&entqr=3&oe=ISO-8859-1&ie=ISO-8859-1&ud=1&proxystylesheet=diabetes&output=xml_no_dtd&proxyreload=1&q=ELIXA. Accessed June 18, 2015. | ||
Jiang Y, Huang W, Wang J, et al. Metformin plays a dual role in MIN6 pancreatic beta cell function through AMPK-dependent autophagy. Int J Biol Sci. 2014;10(3):268–277. | ||
Chao EC. SGLT-2 inhibitors: a new mechanism for glycemic control. Clin Diabetes. 2014;32(1):4–11. | ||
Barnett AH. Redefining the role of thiazolidinediones in the management of type 2 diabetes. Vasc Health Risk Manag. 2009;5(1):141–151. | ||
Green JB, Feinglos MN. Are sulfonylureas passe? Curr Diab Rep. 2006;6(5):373–377. | ||
Harrison LB, Adams-Huet B, Raskin P, Lingvay I. Beta-cell function preservation after 3.5 years of intensive diabetes therapy. Diabetes Care. 2012;35(7):1406–1412. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.