Back to Journals » International Journal of Nanomedicine » Volume 13
PPARγ agonist-loaded PLGA-PEG nanocarriers as a potential treatment for Alzheimer’s disease: in vitro and in vivo studies
Authors Silva-Abreu M ![]() , Calpena AC
, Calpena AC ![]() , Andrés-Benito P, Aso E, Romero IA, Roig-Carles D, Gromnicova R
, Andrés-Benito P, Aso E, Romero IA, Roig-Carles D, Gromnicova R ![]() , Espina M
, Espina M ![]() , Ferrer I, García ML, Male D
, Ferrer I, García ML, Male D ![]()
Received 18 April 2018
Accepted for publication 30 May 2018
Published 20 September 2018 Volume 2018:13 Pages 5577—5590
DOI https://doi.org/10.2147/IJN.S171490
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Thomas Webster
Marcelle Silva-Abreu,1,2 Ana Cristina Calpena,1,2 Pol Andrés-Benito,3,4 Ester Aso,3,4 Ignacio A Romero,5 David Roig-Carles,5 Radka Gromnicova,5 Marta Espina,1,2 Isidre Ferrer,3,4 María Luisa García,1,2 David Male5
1Department of Pharmacy, Pharmaceutical Technology and Physical Chemistry, Faculty of Pharmacy and Food Sciences, University of Barcelona, Barcelona, Spain; 2Institute of Nanoscience and Nanotechnology (IN2UB), University of Barcelona, Barcelona, Spain; 3Servei d’Anatomia Patològica, Institut d’Investigació Biomèdica de Bellvitge-Hospital Universitari de Bellvitge, Universitat de Barcelona, L’Hospitalet de Llobregat, Spain; 4Centro de Investigación Biomédica en Red de Enfermedades Neurodegenerativas, Instituto de Salud Carlos III, Madrid, Spain; 5School of Life, Health and Chemical Sciences, Faculty of Science, The Open University, Walton Hall, Milton Keynes, UK
Objective: The first aim of this study was to develop a nanocarrier that could transport the peroxisome proliferator-activated receptor agonist, pioglitazone (PGZ) across brain endothelium and examine the mechanism of nanoparticle transcytosis. The second aim was to determine whether these nanocarriers could successfully treat a mouse model of Alzheimer’s disease (AD).
Methods: PGZ-loaded nanoparticles (PGZ-NPs) were synthesized by the solvent displacement technique, following a factorial design using poly (lactic-co-glycolic acid) polyethylene glycol (PLGA-PEG). The transport of the carriers was assessed in vitro, using a human brain endothelial cell line, cytotoxicity assays, fluorescence-tagged nanocarriers, fluorescence-activated cell sorting, confocal and transmission electron microscopy. The effectiveness of the treatment was assessed in APP/PS1 mice in a behavioral assay and by measuring the cortical deposition of β-amyloid.
Results: Incorporation of PGZ into the carriers promoted a 50x greater uptake into brain endothelium compared with the free drug and the carriers showed a delayed release profile of PGZ in vitro. In the doses used, the nanocarriers were not toxic for the endothelial cells, nor did they alter the permeability of the blood–brain barrier model. Electron microscopy indicated that the nanocarriers were transported from the apical to the basal surface of the endothelium by vesicular transcytosis. An efficacy test carried out in APP/PS1 transgenic mice showed a reduction of memory deficit in mice chronically treated with PGZ-NPs. Deposition of β-amyloid in the cerebral cortex, measured by immunohistochemistry and image analysis, was correspondingly reduced.
Conclusion: PLGA-PEG nanocarriers cross brain endothelium by transcytosis and can be loaded with a pharmaceutical agent to effectively treat a mouse model of AD.
Keywords: nanoparticle, Alzheimer’s disease, blood–brain barrier, brain endothelium, pioglitazone, APP/PS1 transgenic mouse
Corrigendum for this paper has been published.
Introduction
Alzheimer’s disease (AD) is a multifactorial brain disorder prevalent in elderly people.1,2 It is characterized by cognitive impairment, synaptic failure, aggregates of amyloid-beta (Aβ) and intraneuronal neurofibrillary tangles.3–5 However, AD is also referred to as a degenerative metabolic disease that is associated with physiological alterations such as hypercholesterolemia, metabolic syndrome, hypertension and diabetes type-2,6 a condition identified as a risk factor for the development of AD.
Pioglitazone (PGZ) (5-[[4-[2-(5-ethylpyridin-2-yl)ethoxy]phenyl]methyl]–1,3-thiazolidine-2,4-diona), an agonist of the peroxisome proliferator-activated receptor (PPARγ), is a thiazolidinedione used for the treatment of type-2 diabetes. The PPARγ receptors have various functions including anti-angiogenic, antifibrotic, anti-inflammatory and anti-tumor effects.7–10 PGZ is also neuroprotective in models of neurodegenerative disorders,10 highlighting PPARγ agonists as a promising treatment for AD.11 Thus, PGZ has been reported to reduce oxidative stress, normalize cerebral blood flow and glucose uptake, increase neuronal activity and exert positive effects on cerebrovascular functions in an animal model of AD.12 Furthermore, treatment with PGZ produced a significantly lower risk of dementia over the 5-year follow-up period in diabetic patients treated with PGZ compared with those who were not.13
Drug delivery to the brain is a challenge for the treatment of neurological disorders. The blood–brain barrier (BBB) is a physical interface between the central nervous system (CNS) and peripheral circulation that strictly controls which molecules can enter into the brain parenchyma.14 Only lipophilic molecules smaller than 400 Da can diffuse through the BBB; the passage of lipid insoluble or larger hydrophilic molecules is very limited.15,16 Different strategies to increase the transport of drugs through the BBB include liposomes, solid lipid nanoparticles (NPs), gold NPs and polymeric NPs.17–19 These drug-loaded polymeric NPs are a potential alternative to improve drug delivery to the brain.
The properties of polymeric NPs include drug encapsulation, stability, high loading capacity for many agents and controlled drug release kinetics. Moreover, they can be easily modified with a variety of surface-attached ligands.20,21 Different polymers which deliver a variety of molecules through the BBB have been studied.22–24 Poly (lactic-co-glycolic acid) (PLGA) systems have been the subject of great scientific interest due to their properties, specifically their biocompatibility and biodegradability. PLGA is an FDA-approved co-polymer used successfully for delivery to different tissues, including the brain.19,25–27
The attachment of specific ligands to the surface of NPs makes the delivery of drugs to CNS more targeted and may enhance the limited BBB penetration of therapeutic compounds. For instance, polyethylene glycol (PEG) can functionalize NPs to increase their plasma residence time, preventing their removal by mononuclear phagocytes. This allows the NPs to remain longer in circulation, thus increasing the probability of successful organ-targeted delivery and passage across the BBB.28–30
In this study, a formulation of PGZ-PLGA-PEG NPs was synthesized and their suitability for the treatment of AD was demonstrated. Physicochemical characterization and in vitro studies with a human brain microvascular endothelial cell line (hCMEC/D3) were carried out. An in vivo study for cognitive evaluation after treatment was performed in male APP/PS1 mice and wild-type-like (WT) littermates.
Materials and methods
Materials
PGZ was obtained from Capot Chemical (Hangzhou, People’s Republic of China) and Diblock copolymer PLGA-PEG (Evonik Ind., Resomer® Select 50:50 DLG mPEG 5,000–5 wt% PEG), was purchased from Evonik Corporation (Birmingham, AL, USA). Rhodamine 6G (Rhod), Tween® 80 (Tw 80), 70 kDA FITC-dextran and 24-well filter inserts (PET membrane, pore 1 μm) were acquired from Sigma-Aldrich (Gillingham, UK). The dialysis membrane MWCO 12,000–14,000 Da, was obtained from Medicell International Ltd. (London, UK). Trypsin-EDTA was purchased from Thermo Fisher (London, UK). Alamar Blue (AB) was obtained from Invitrogen Alfagene® (Carcavelos, Portugal).
Methods
Optimization and characterization of NPs
PGZ-NPs were obtained by the solvent displacement technique.31 The organic phase with PGZ (1 mg/mL) was solubilized in dimethyl sulfoxide (DMSO) 5%, to which a mixture of PLGA-PEG (9.5 mg/mL) and acetone (5 mL) was added. Once completely dissolved, the solution was added drop by drop with moderate stirring into 10 mL of an aqueous solution of 1.16% Tw 80. The pH was adjusted to 4.5 with HCl 0.1 M. Then, the solvents were evaporated and the NP dispersion was concentrated into a final volume of 10 mL under reduced pressure.
The conditions for NP production were optimized in a factorial design26 (Figure S1). Three independent variables (PGZ, PLGA-PEG and Tw 80 concentrations) and four dependent variables (average particle size [Zav], polydispersity index [PI], zeta potential [ZP] and entrapment efficiency [EE] were studied. A pH of 4.5 was kept constant for all the assays. A total of 16 experiments (eight factorial points, six axial points and two replicated center points) were required for the estimation of the pure error sum of squares using Statgraphics Plus 5.1 software (The Plains, VA, USA).
The experimental responses were the result of the individual influences and interactions of the three independent variables. In the surface of response, a low concentration of PGZ and PLGA-PEG produced smaller NPs and consequently greater homogeneity of the formulation. The formulation with the most appropriate physicochemical characteristics was selected for use in this study.
Transmission electron microscopy (TEM) was used to evaluate the morphology of PGZ-NPs. Before negative staining, the copper grids were activated with ultraviolet light and samples were positioned on the grid surface, diluted with water (1:3) and negative stained with a 2% (v/v) uranyl acetate solution. After being dried at room temperature, the samples were examined by TEM on a Jeol 1010 (Tec-nai Spirit TEM, FEI, Jeol, Welwyn Garden City, UK) at 80 kV.
The morphometric parameters (Zav and PI) of PGZ-NPs were determined by photon correlation spectroscopy (after 1:10 dilution with water) with a Zetasizer Nano ZS (Malvern Instruments, Malvern, UK) at 25°C. The surface charge or ZP was calculated from electrophoretic mobility. The values are the mean ± SD which are calculated for at least three different batches.32
The EE of PGZ-NPs was determined indirectly by measuring the concentration of the free drug in the dispersion medium. The non-entrapped drug was separated by a filtration/centrifugation technique (1:10 dilution) using Ultracell–100K (Amicon® Ultra; Millipore Corporation, Billerica, MA, USA) centrifugal filter devices at 12,000 rpm for 15 minutes. The EE was calculated using equation (1):
|
|
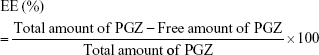
Samples were evaluated by HPLC using a validated analytical method.33 The mobile phase was: 0.1 M acetonitrile, ammonium acetate and glacial acetic acid (75:25:1 v/v), with a flow of 0.7 mL/min and a volume of injection of 10 μL. The reported values are the mean ± SD (n=6).
Release profile
In vitro PGZ release studies from NPs were performed in vertical Franz diffusion cells using membrane dialysis with MW 12–14 KDa cutoff under sink conditions27 at 37.0°C ± 0.5°C with moderate and continuous stirring. The PGZ-NPs and free drug at the same concentration (1 mg/mL) were dissolved in DMSO and PBS (60:40) and receptor solution (RS) at pH 7.4 for 23 hours. At specific time intervals, a volume of 0.2 mL of the formulations was placed in the donor compartment and the receptor compartment was filled with the same volume of RS. Amounts of PGZ released were measured by HPLC. Values are reported as the mean ± SD (n=3). PGZ released at each time point was evaluated and data were fitted into different kinetic models34 Akaike’s information criterion (AIC) and coefficient of determination (r2) were determined for each case as an indicator of the model’s suitability for each dataset.35
Syntheses of rhodamine NPs
To evaluate the cellular uptake of NPs by hCMEC/D3, Rhod was incorporated into the NPs. Rhod-NPs were synthesized by the same method as PGZ-NPs, but with addition of 100 μg/mL Rhod in 500 μL of methanol, mixed with 9.5 mg/mL PLGA-PEG in 5 mL of acetone (organic phase). After the preparation of Rhod-NPs, the EE was determined from the amount of the free-Rhod present in the aqueous phase of the formulations, obtained by filtration/centrifugation at 12,000 rpm for 15 minutes. The amount of Rhod was measured by fluorescence spectroscopy (λex 528 nm, λem 547 nm).
Cell culture
An immortalized (hCMEC/D3) line36 was cultured in endothelial basal medium-2, supplemented with 2.5% fetal bovine serum, hydrocortisone, vascular endothelial growth factor (VEGF), EGF, insulin-like growth factor I (IGF-I), human fibroblast growth factor, ascorbic acid and gentamicin sulphate according to the manufacturer’s formulation (Lonza, Basel, Switzerland). Cells were grown to confluence on tissue culture flasks or inserts coated with collagen Type one from calf skin (Sigma, Gillingham, UK) and incubated in 5% CO2 in air at 37°C.
Alamar blue (AB) was used to test for any cytotoxic effect of NPs on the hCMEC/D3 cell line. Cells were seeded in collagen-coated 96-well plates at 3×104 cells/well and were maintained for 24 hours. Then, cells were incubated with 100 μL/well of 0.5–10 μg/mL of either PGZ-NPs or Rhod-NPs diluted with endothelial culture medium for 24 hours. After the incubation, hCMEC/D3 cells were treated with 10% (v/v) of AB diluted in FBS-free endothelial culture medium. Absorbance was measured at 570 nm (reduced AB form) and 620 nm (oxidized AB form) between 4–5 hours after AB treatment using a FLUOstar OPTIMA microplate reader (BMG LABTECH, Ortenberg, Germany). The cell viability was calculated by the percentage of AB reduction, using the manufacturer’s protocol as described previously.37 All experiments were performed three times.
Cellular uptake of NPs
hCMEC/D3 cells were seeded in 12 well plates (120,000 cells/well) and grown for 2 days. At confluence, hCMEC/D3 cells were incubated with 1 μg/mL of Rhod-NPs for 15, 30 and 60 minutes, compared with untreated cells and analyzed by flow cytometry. The Free-Rhod and Free-Rhod-NPs (Rhod not encapsulated during NP production) were also studied to analyze whether the Rhod could internalize without NPs. Fluorescence-activated cell sorting analysis was performed by collecting cells with trypsin/EDTA and washing twice with Hanks balanced salt solution (HBSS) (Sigma Aldrich, UK). Washed cells were analyzed on a Becton-Dickinson (Reading, UK), FACS calibur (FL2 detector set at 410V). The experiments were done in triplicate and the values are reported as the mean ± SD (n=3) of the median fluorescence of 10,000 cells.
hCMEC/D3 monolayers were grown to confluence on fibronectin and collagen-coated 8-well Nunc La-Tek Chamber slides (Sigma Aldrich, UK). Confluent cells were treated with 1 μg/mL of Rhod-NPs for 3 hours at 37°C. Then, hCMEC/D3 cells were washed with HBSS and fixed in 4% formalin solution for 10 minutes at room temperature. The nuclei were counterstained with mounting medium DAPI (blue) (Vector Laboratories, Burlingame, CA, USA). Both nuclei staining and Rhod-NP-internalization were analyzed using confocal laser scanning microscopy (Leica TCS SP5, Leica Microsystems, Milton Keynes, UK).
Detection of internalization of NPs by TEM
The internalization of PGZ-NPs and Rhod-NPs in hCMEC/D3 cells was investigated by TEM. The cells were seeded at 120,000 cells per 1 cm2 on Transwell® inserts of polyester membrane (Costar, Corning, NY, USA). At confluence, the NPs (1 μg/mL) were applied to the apical side in endothelial culture medium for 6 hours. Then, both chambers were washed three times in HBSS and fixed in 2.5% glutaraldehyde for 1 hour at room temperature. The fixative was removed and chambers were washed three times with 0.1 M Sörenson’s phosphate (PB) and stored in this buffer at 4°C.
In order to process the samples for TEM, all incubations were applied to both sides of the chamber at room temperature. Firstly, the cells were post-fixed in 1% osmium tetroxide diluted with 0.1 M PB for 1 hour. Then, the insert was washed three times in 0.1 M PB and removed from the well. The membrane was cut out of the insert and gradually dehydrated in sequence with ethanol: 30% for 5 minutes, 50% for 5 minutes, 70% for 10 minutes, 100% for 10 minutes twice and 100% with a molecular sieve for 10 minutes. Then, the membranes were incubated in a mixture of 1:1 ratio of 100% ethanol and Epon resin overnight. They were embedded in Epon resin and the formed blocks were polymerized at 60°C for 48 hours. Resin blocks were micro sectioned at 80 nm thickness using a Diatome diamond knife. The sections were mounted onto pioloform-coated copper grids and counter stained in 3.5% uranyl acetate for 35 minutes followed by lead citrate for 10 minutes and finally washed three times before air-drying. The grids were imaged on TEM JEM 1010 (Jeol, Japan) at an acceleration voltage of 80 kV using a magnification of 25,000–40,000.
Transport and permeability assay
Transfer of NPs across brain endothelium was analyzed using permeable cell culture inserts and TEM. Briefly, 24-well cell-culture inserts (Millipore Millicell Hanging Cell Culture Insert, PET membrane; 1 μm) were coated with collagen and fibronectin and seeded with 8×104 cells per insert. At confluence, cells were washed with HBSS and cultured with endothelial culture medium but without the growth factors VEGF, IGF, EGF and maintained for 48 hours. Then, cells were treated with 1 μg/mL of PGZ-NPs or Rhod-NPs diluted in VEGF, IGF and EGF-free endothelial culture medium for 6 hours. The basolateral side-medium was collected and negative stained. NPs (as described above) were observed on TEM JEM-1400 operated at an accelerating voltage of 80 kV. The experiments were done in triplicate and the values are reported as the mean ± SD (n=3).
The effect of the transport of NPs on the hCMEC/D3 cells was measured by a paracellular permeability assay as described previously by Tai et al.38 hCMEC/D3 treated with 10 ng/mL of TNFα and IFNγ for 24 hours was used as a positive control.39 At the end of these experiments, the apical side-culture medium was removed and 400 μL of assay buffer (0.1% BSA in DMEM without phenol red) containing 2 mg/mL 70 kDa FITC-dextran was added. The fluorescence that crossed to the basolateral side was measured every 10 minutes for 1 hour using a BMG plate reader, and the permeability coefficient Pe derived.38
In vivo assay with APP/PS1 mice
The experiments were carried out in 7 month male APP/PS1 mice and WT littermates on a C57/Bl6J genetic background. The generation of mice, expressing the human mutated APPswe and PS1dE9, has been described elsewhere.40 Animals were maintained under standard animal housing conditions in a 12-hour dark-light cycle with free access to food and water. Genotypes were confirmed from 1 cm tail clips by PCR using conditions recommended by Jackson Laboratory. Mice were randomly assigned to treatment groups and the experiments were analyzed blind. The study was carried out following the guidelines of the European Communities Council Directive 2010/63/EU and with the approval of the local ethical committee of the University of Barcelona.
Free-PGZ and PGZ-NPs (10 mg/kg) were dissolved in 5% DMSO and administered orally in a volume of 10 mL/kg body weight. Animals were treated once a day, 5 days per week, for 4 weeks with the compounds or with the vehicle alone. The number of animals included in each group was 3–4. After a 3-day wash-out, animals were subjected to behavioral evaluation.
Memory performance was evaluated with the two-object recognition test. On day 1, mice were placed for 9 minutes in a Y-maze, in which two identical objects were situated at the ends of the arms; the time that the mice spent exploring each object was recorded. Then, 24 hours after the training session, animals were placed again for 9 minutes in the V-maze, with one of the two familiar objects replaced by a novel object. The time that the animals spent exploring the two objects was recorded and an object recognition index (RI) was calculated, as the difference between the time spent exploring the novel object (TN) and the familiar object (TF) divided by the total time spent exploring the two objects [RI=(TN−TF)/(TN+TF)]. At the end of the behavioral testing, the animals were killed and their brains rapidly removed from the skull, fixed in 4% paraformaldehyde and processed for immunohistochemistry.
Aβ immunohistochemistry
Fixed tissue samples were embedded in paraffin. Consecutive de-waxed 4 μ coronal sections were incubated with 98% formic acid (20 M, 3 minutes) and treated with 10 mM sodium citrate buffer, pH 6.0, for 20 minutes to enhance antigenicity. Endogenous peroxidases were blocked with 10% methanol−1% H2O2 for 15 minutes and then blocked with 3% normal horse serum in PBS. They were incubated (4°C overnight) with primary antibody against Aβ (clone 6F/3D 1:50, Dako, Denmark) and a peroxidase-conjugated secondary antibody, visualized with 7 mM diaminobenzidine in PBS (5 minutes). Sections were lightly counterstained with hematoxylin. The cortical total Aβ burden was calculated as the percentage of the area of amyloid deposition in plaques with respect to the total cortical area (0.6 mm2) in nine pictures taken from three different sections (−0.1, −1.5 and −2.0 mm from bregma) of each animal brain (three pictures per section corresponding to cingulate/retrosplenial and motor cortex, somatosensory cortex and piriform/entorhinal cortex). The selected areas were the main regions of the cerebral cortex in which Aβ is deposited in APP/PS1 mice. A researcher who did not know the treatments performed the quantifications. Aβ quantification was calculated using the Analysis tool of Adobe® Photoshop® CS4 (Maidenhead, UK).
Statistical analysis
The sample size for experimentation was computed using the Power and Precision software (Biostat, Englewood, NJ, USA), assuming a power of 95% and no missing data. Data were analyzed using GraphPad Prism version 6.0 software. Statgraphics Plus 5.1 software was used to analyze the surface of response. Student’s t-test or one way ANOVA, followed by Tukey’s multiple comparison or Dunnett’s multiple comparison test were used to analyze the in vitro assays. Statistical analysis for the in vivo assay was performed with the SPSS® Statistics v21.0 software (IBM, New York, NY, USA). Memory data were analyzed with two-way ANOVA (genotype and treatment as between factors), followed by Tukey’s post hoc. Aβ area was analyzed with one-way ANOVA (treatment as between factors).
Results
PGZ nanocarrier characterization and release profile
After a detailed analysis of the parameters affecting NP synthesis (Figure S1), a single formulation of NPs was chosen for the biological studies (Figure S2). These PGZ-NPs had a mean size of 155.0 ± 1.8 nm, a polydispersity index distribution of 0.1, negative ZP of −13.0 ± 0.5 mV and an EE of 92.5%. Smaller size facilitates passage through the BBB,16 and may improve the delivery of drugs. Moreover, low PGZ concentrations produced NPs with greater negative ZP that could help prevent particle aggregation, thereby increasing the stability of the dispersion.41 Importantly, the lower concentration of Tw also decreased the toxicity for hCMEC/D3 cells, besides facilitating interaction with endothelial surface molecules and hence, transport across the BBB.42
Different kinetic models of drug release were tested with both formulations to select the best fit. The formulations showed dissimilar profiles and the best fits were “One phase exponential association” for Free-NPs and “hyperbola” for PGZ-NPs, with respect to the AIC and coefficient of determination (r2) values obtained (Figure 1). After 10 hours, the free drug achieved 76.2% transfer whereas NPs released 57.1% of the initially bound drug. In addition, Free-PGZ showed a faster release than PGZ entrapped in the particles (comparing Ymax to Bmax). Free-PGZ had a constant of dissolution (K) of 0.46 hour−1 whereas NP-entrapped PGZ had a constant of 1.72 hours. This result indicates that NPs are still achieving a sustained release of the drug.
 | Figure 1 Release profile of PGZ from PGZ-NPs and Free-PGZ. |
Cytotoxicity assay
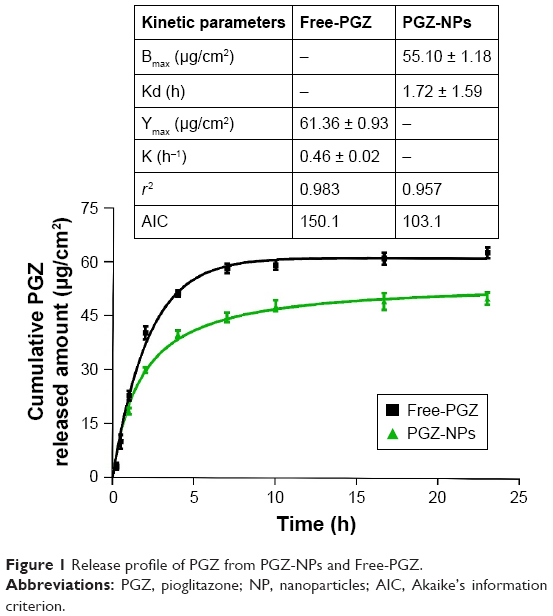
The in vitro cytotoxicity of PGZ-NPs and Rhod-NPs for hCMEC/D3 cells was assessed by determining the cell viability using the AB assay (Figure 2). Cells exposed to increasing concentrations of PGZ-NPs for 24 hours showed decreased cell viability from 5.1 nM (=2 μg/mL) (76.56% ± 2.72%). Treatment with Rhod-NPs also produced significant toxicity at 5.1 nM and Rhod-NPs were more toxic than PGZ-NPs at concentrations from 5.1 nM (P<0.05). On the basis of the cytotoxicity assays, the dose of 1 μg/mL was selected to investigate the characteristics of both types of NPs in vitro.
 | Figure 2 hCMEC/D3 cell viability measured by AB assay when exposed to 1.2–25 nM (0.5–10 μg/mL) PGZ-NPs and Rhod-NPs for 24 hours. |
Transport of NPs by brain endothelium
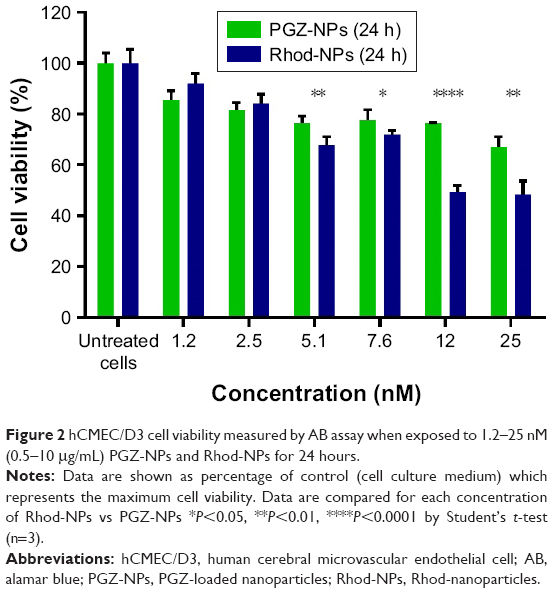
Rhod was used to track the cellular uptake of NPs by hCMEC/D3 cells. NPs prepared using Rhod showed high EE (around 99%) and remained associated with the cells for extended periods of time. The cellular uptake of Rhod-NPs into hCMEC/D3 was measured at 15, 30 and 60 minutes by FACS. The values of fluorescence of Rhod-NPs increased in a dose and time-dependent manner (P<0.05) when compared to untreated cells (Figure 3). The uptake was time-dependent with significant uptake of NPs within the first 15 minutes of exposure. The fluorescence in Rhod-NPs treated hCMEC/D3 cells was >50x higher than cells exposed to Free-Rhod (Figure 3C).
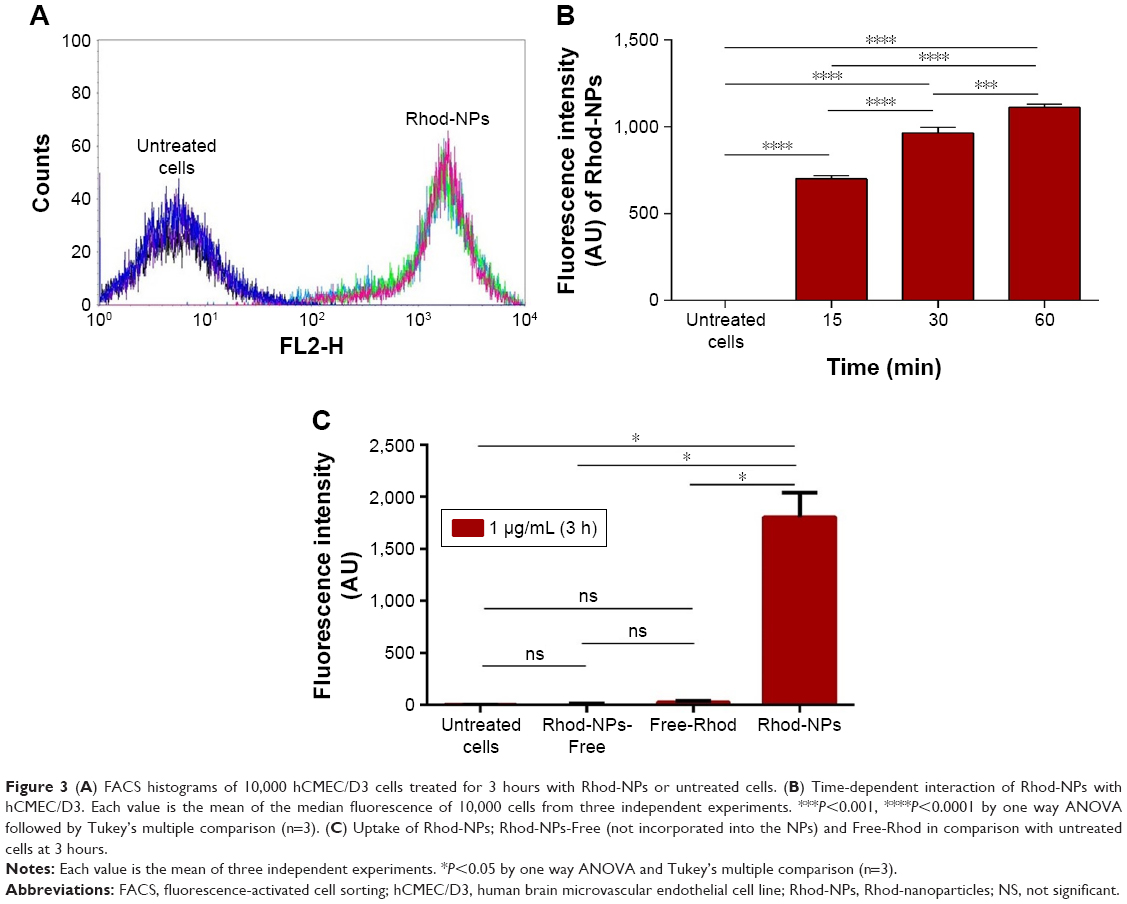 | Figure 3 (A) FACS histograms of 10,000 hCMEC/D3 cells treated for 3 hours with Rhod-NPs or untreated cells. (B) Time-dependent interaction of Rhod-NPs with hCMEC/D3. Each value is the mean of the median fluorescence of 10,000 cells from three independent experiments. ***P<0.001, ****P<0.0001 by one way ANOVA followed by Tukey’s multiple comparison (n=3). (C) Uptake of Rhod-NPs; Rhod-NPs-Free (not incorporated into the NPs) and Free-Rhod in comparison with untreated cells at 3 hours. |
Routes of transcytosis
To determine the subcellular localization of the NPs, hCMEC/D3 cells were treated with Rhod-NPs and examined by confocal microscopy (Figure 4). Red punctate fluorescence was observed in all cells; the size corresponded to individual NPs localized primarily in the cell body, close to the nucleus. These observations confirm the results obtained by FACS which demonstrated that all cells take up the Rhod-NPs (Figure 3A) and that the fluorescence signal was much higher when using Rhod-NPs in comparison to Free-Rhod. It has been proposed that internalization of NPs of this size occurs predominantly by adsorption to the cell surface followed by vesicular endocytosis. The results are consistent with NP-uptake into either caveolae or clathrin-coated vesicles (CCVs).
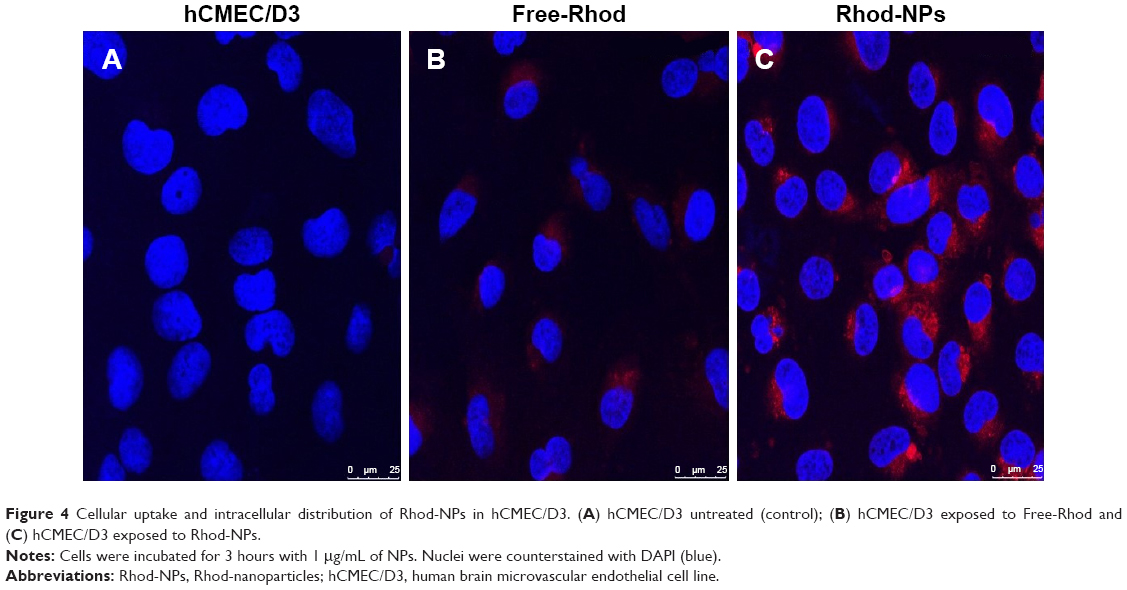 | Figure 4 Cellular uptake and intracellular distribution of Rhod-NPs in hCMEC/D3. (A) hCMEC/D3 untreated (control); (B) hCMEC/D3 exposed to Free-Rhod and (C) hCMEC/D3 exposed to Rhod-NPs. |
Confocal microscopy has insufficient resolution to determine exactly where the NPs are localized within the cells. To obtain a better understanding of the mechanism of NPs uptake, the cells were examined by TEM. The images show individual PGZ-NPs and Rhod-NP (100–150 nm) in the cytoplasm (Figure 5); the size corresponds to their initial physical characterization (Figure S2). No NPs were seen in intercellular junctions. This implies that the NPs have been taken up individually by endothelial cells and have not aggregated inside the cells. It is not possible to see a distinct vesicle membrane or electron-dense coat proteins around the NPs. However, it should be noted that both caveolae and CCVs are normally smaller (50–100 nm) than these NPs; hence a caveolus or CCV would contain just one NP and the closely apposed vesicular membrane would be difficult to visualize by TEM. Notably, NPs were not seen in larger pinocytotic vesicles or intercellular junctions. Nor was there any evidence of disturbance of the plasma membrane. These observations exclude paracellular movement as the route of movement and direct trans-membrane transfer to the cytosol is also unlikely. Hence, the most likely route of uptake across the endothelium is via endocytosis from the apical surface into a single CCV or caveolus. This is in accordance with the observed clustering of the NPs in the perinuclear region seen by confocal fluorescence microscopy, which is a characteristic of caveolae.
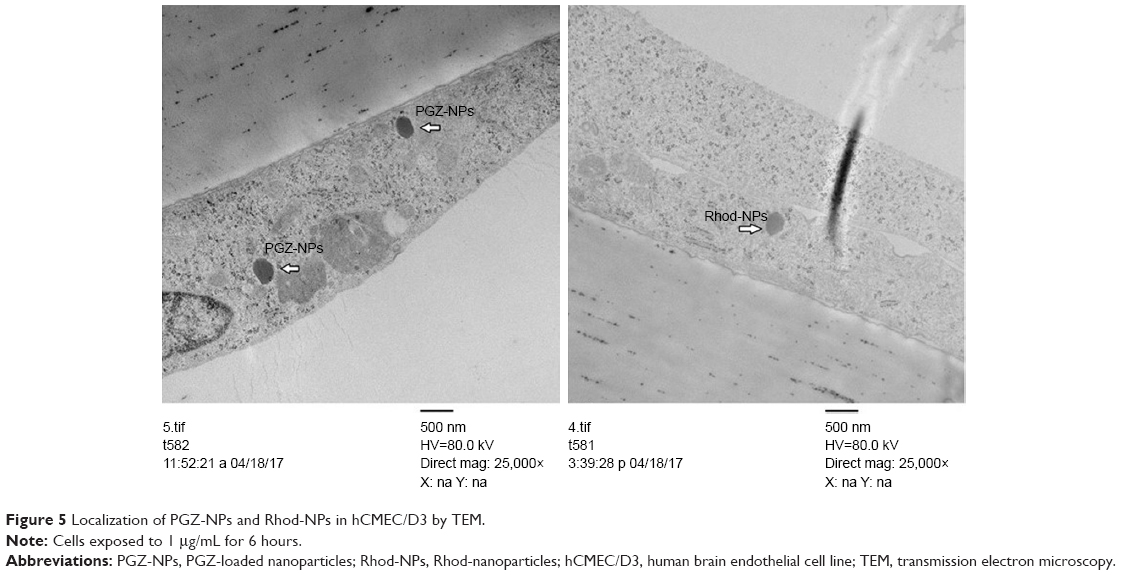 | Figure 5 Localization of PGZ-NPs and Rhod-NPs in hCMEC/D3 by TEM. |
Transport and permeability assays
To determine whether NPs are released from the basolateral side of the endothelium, hCMEC/D3 cells were grown in confluent monolayers on filters in tissue-culture inserts. PGZ-NPs and Rhod-NPs were applied to the apical surface and the medium harvested from the basolateral side. After 6 hours, NPs had crossed the endothelial monolayer (Figures 6A and S3). The NPs were similar in size (100–200 nm) to those initially applied, suggesting they had crossed the endothelium intact.
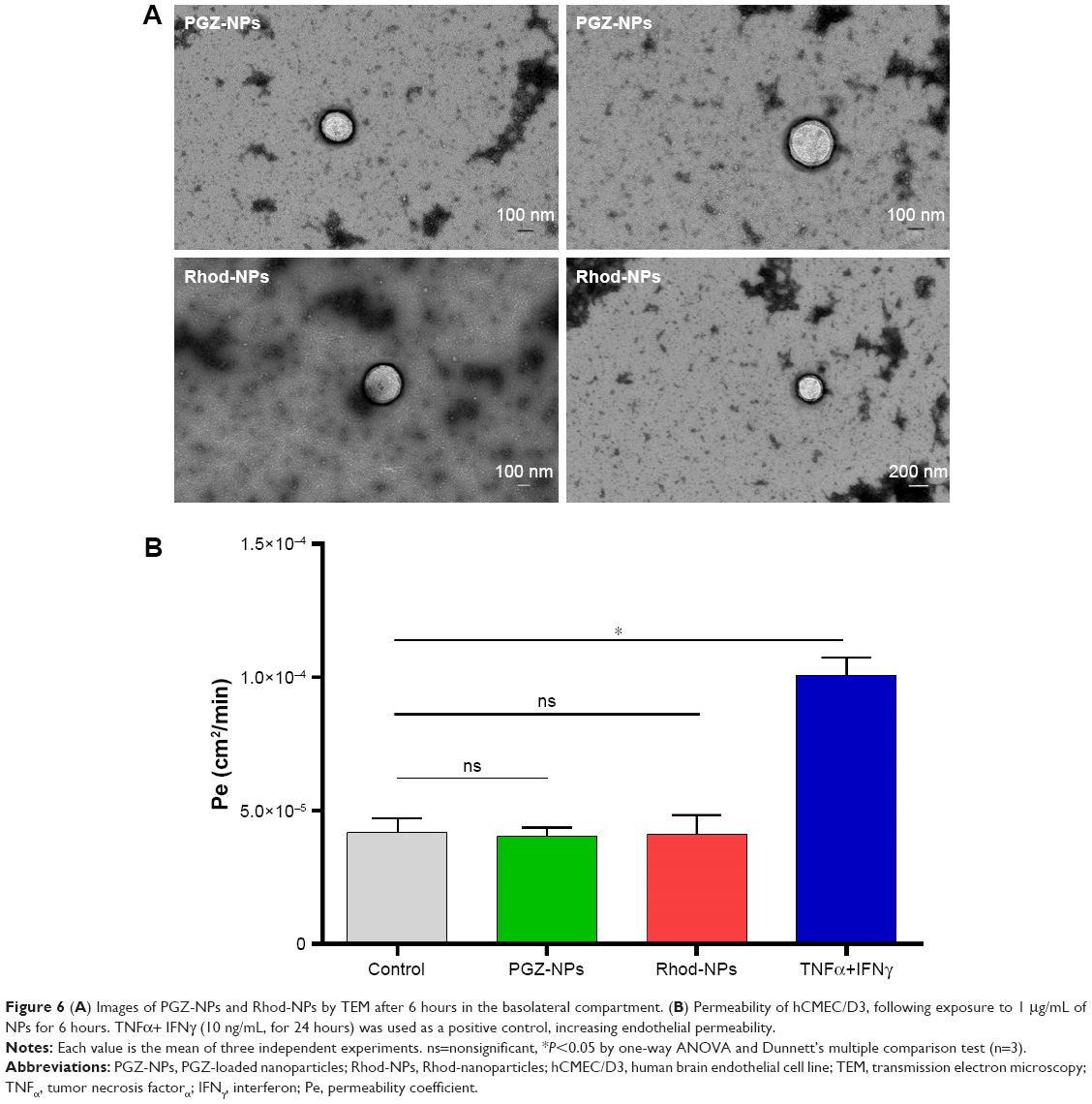 | Figure 6 (A) Images of PGZ-NPs and Rhod-NPs by TEM after 6 hours in the basolateral compartment. (B) Permeability of hCMEC/D3, following exposure to 1 μg/mL of NPs for 6 hours. TNFα+ IFNγ (10 ng/mL, for 24 hours) was used as a positive control, increasing endothelial permeability. |
The integrity of the cell monolayer after exposure to both types of NPs was assessed using a paracellular tracer, FITC-dextran (MW 70 kDa). Changes in the permeability coefficient were calculated and the data show that treatment of hCMEC/D3 cells with PGZ-NPs and Rhod-NPs for 6 hours does not increase the paracellular permeability of the endothelial monolayer (Figure 6B). This demonstrates that the NPs had not altered paracellular permeability of the endothelial monolayer, and further confirms that the NPs seen in Figure 6A had crossed the endothelium by transcytosis.
Effect of treatment on memory and neuropathology in APP/PS1 mice
In order to test the in vivo effect of the PGZ-NPs, a comparison was made with Free-PGZ in APP/PS1 mice. Oral administration was chosen as this reflects the route of PGZ administration in humans. Previous work has shown that exposure of these NPs to acid (0.1 M HCl) for 15 minutes (reflecting gastric conditions) followed by neutralization does not affect their size, ZP, PI or EE. Daily administration of Free-PGZ and PGZ-NPs (10 mg/kg) for 4 weeks reduced the memory impairment observed in vehicle-treated APP/PS1 mice, as revealed by the two-object recognition test (Figure 7A). Two-way ANOVA revealed a significant genotype [F(1, 16)=5.413, P<0.05] and treatment effect [F(2, 16)=12.717, P<0.001] and interaction between the two factors [F(2, 16)=3.551, P=0.053]. Subsequent Tukey’s post hoc tests revealed that APP/PS1 mice treated with vehicle exhibited a memory impairment when compared to corresponding WT littermates (P<0.01) and that Free-PGZ (P<0.05) and PGZ-NPs (P<0.01) increased the recognition index in APP/PS1 mice when compared to vehicle. No significant difference in the total exploration time during the memory acquisition session or the memory test was observed between groups, discounting any possible impact of the treatments on the anxiety levels or the activity of the mice.
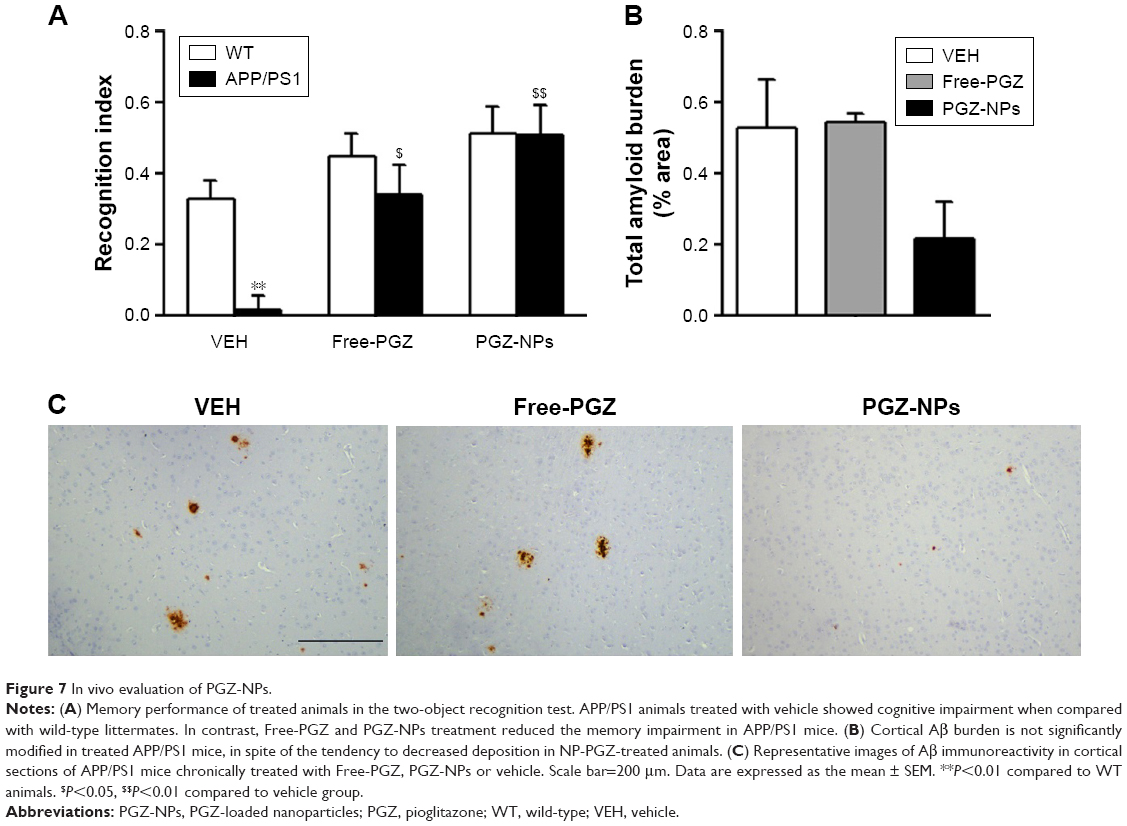 | Figure 7 In vivo evaluation of PGZ-NPs. |
Chronic treatment with Free-PGZ or PGZ-NPs did not significantly modify [F(2, 8)=2.993, P=0.04] the total Aβ burden in the cortex with respect to vehicle-treated mice (Figure 7B and C). The vehicle employed in this study did not induce any modification in Aβ burden when compared to untreated mice (data not shown). Notably, the level of Aβ deposition in mice treated with PGZ-NPs was half of that in mice treated with Free-PGZ (Figure 7); however there was considerable variation between individual animals. These results demonstrate that a PPARγ agonist improves memory in APP/PS1 mice and suggest that encapsulation of PGZ in NPs can reduce the Aβ burden.
Discussion
In this study, we have demonstrated that PGZ-NPs developed by a displacement technique are appropriate for the treatment of a model of AD. The release profile from NPs was slower than Free-PGZ. Moreover, the NPs were not cytotoxic for human brain endothelium (hCMEC/D3) at the doses used and produced no alteration in permeability of the endothelial monolayer. These results are in accordance with a previous study using mouse Bend-3 cells, which indicated that PLGA-PEG NPs did not damage endothelial cells.19
Theoretically, endocytosis of NPs can occur by passive transfer across the cell membrane into the cytoplasm or by active endocytosis into vesicles, including CCVs and caveolae. NP size affects the principal entry route; the smallest NPs (<10 nm) can directly cross the plasma membrane whereas endocytosis is the principal entry route for larger NPs.43
The route of endocytosis depends greatly on how the NPs interact with different domains of the plasma membrane, which is affected by size, coating, surfactant and surface charge as well as any specific targeting molecule on the NPs. These factors may also affect the ability of the NPs to cross the BBB in vivo.44 The subcellular localization of NPs in this study, determined by confocal and electron microscopy, indicated that they cross the endothelium intact by vesicular transcytosis.
Most endothelial cells have large numbers of caveolae which mediate transport of nutrients to the tissue.45 However, brain endothelium in vivo has relatively few caveolae compared with endothelium in other tissues and consequently there is less internalization by this route. However, PEG on the surface of NPs can improve their internalization by brain endothelium46 and it has been suggested that PEGylated PLGA NPs could enter these cells in CCVs47 as an alternative to caveolar transcytosis. CCVs have previously been shown to play a key role in the transportation of PEGylated NPs in which energy-dependent endocytosis is involved.48 Moreover, particles up to 200 nm in diameter can be efficiently taken up into CCVs.49 The 50-fold higher rate of uptake of Rhod-NPs compared with free rhodamine also implies that the NPs are taken up by a different mechanism than the free tracer.
These observations indicate that the PGZ-NPs enter and can potentially cross brain endothelium directly. Transcytosis of NPs within vesicles shields any cargo molecule such as PGZ or rhodamine from ABC-transporters (eg, pgp/ABCB1), located in the apical plasma membrane, which act on substrates in the cytoplasm and/or the membrane. Even if the NPs enter the cytoplasm, the delayed release of cargo means that less drug can be removed by multi-drug transporters.
The experiments in vivo demonstrated the potential for PGZ, and particularly PGZ-NPs, to reduce memory deficit and neuropathology in APP/PS1 mice. These results are in accordance with Searcy et al50 who demonstrated that Free-PGZ improved reversal learning in a triple transgenic mouse model of AD mice. Moreover, other studies showed that APP/PS1 mice treated for nine days with PGZ reversed non-cognitive behavioral deficits and restored distance and speed traveled in an open field51 and, improved partially, the cognitive impairments in the Morris water maze test.52 A number of other studies have reported neuroprotective properties of PGZ.9,10,50,52,53 Moreover, the Phase III clinical trial also demonstrated a role for PGZ in slowing cognitive decline in people with mild cognitive impairment due to AD.54
In this study, the overall reduction in Aβ burden in PGZ-NPs treated mice was striking although individual results were variable. Previous work has also produced divergent results. In one other study, the amyloidogenic APP processing and Aβ production were not affected by treatment with pioglitazone.51 In contrast, an acute 2-week treatment with combined leptin and PGZ resulted in a reduction of spatial memory deficits (Y maze) and brain β-amyloid levels (soluble β-amyloid and amyloid plaque burden) relative to vehicle-treated animals.55
Conclusion
The overall conclusion from our study is that PGZ-NPs reduce memory impairment and neuropathology in APP/PS1 mice. However, it is not certain whether the release of PGZ from NPs in vivo occurs inside the CNS or outside the CNS. The results indicate that any difference between the effect of Free-PGZ and PGZ-NPs is most likely due to either the slower release profile from PGZ-NPs (Figure 1) or the improved rate of transcytosis across brain endothelium (Figures 3–6) with the potential for evading the action of multi-drug transporters at the blood–brain barrier.
The data all confirm that NPs cross endothelial cells in vitro without affecting cellular integrity. Moreover, PGZ encapsulated into polymeric NPs (PLGA-PEG) improved the cognitive deficit in APP/PS1 male mice in a similar way to Free-PGZ, and showed a clear tendency to reduce beta amyloid deposition in the cerebral cortex, suggesting that PGZ-NPs are a new alternative to treat AD, both improving drug delivery into the brain and providing for a more sustained drug release.
Acknowledgments
This work was supported by the Coordination for the Improvement of Higher Education Personnel (CAPES) – Brazil; the Spanish Ministry of Science and Innovation (MAT2014-59134R). Studies at the Open University were supported by the Santander Research Foundation and the BBSRC (BB/K009184/1). The authors would like to thank Ms Shereen Nizari for her help with confocal microscopy.
Author contributions
MS-A carried out the experiments, analyzed the data/results and wrote the paper. ACC helped analyze the results of the release profile and statistics. PA-B and EA carried out in vivo experiments and analyzed in vivo results. IAR designed the in vitro permeability assay. DR-C helped with uptake assays and analysis of the permeability assay. RG carried out localization of NPs by transmission electronic microscopy. ME analyzed the design factorial. IF provided the APP-PS1 mice and facilities to carry out the behavioral experiments. MLG helped with physicochemical characterization of NPs, DM conceived and designed all the in vitro experiments with hCMEC/D3 cells and edited the paper.
Disclosure
The authors report no conflicts of interest in this work.
References
Anand R, Gill KD, Mahdi AA. Therapeutics of Alzheimer’s disease: Past, present and future. Neuropharmacology. 2014;76 Pt A:27–50. | ||
Toba J, Nikkuni M, Ishizeki M, et al. PPARγ agonist pioglitazone improves cerebellar dysfunction at pre-Aβ deposition stage in APPswe/PS1dE9 Alzheimer’s disease model mice. Biochem Biophys Res Commun. 2016;473(4):1039–1044. | ||
Md S, Bhattmisra SK, Zeeshan F, et al. Nano-carrier enabled drug delivery systems for nose to brain targeting for the treatment of neurodegenerative disorders. J Drug Deliv Sci Technol. 2018;43:295–310. | ||
Anand A, Patience AA, Sharma N, Khurana N. The present and future of pharmacotherapy of Alzheimer’s disease: A comprehensive review. Eur J Pharmacol. 2017;815:364–375. | ||
Sun D, Li N, Zhang W, et al. Design of PLGA-functionalized quercetin nanoparticles for potential use in Alzheimer’s disease. Colloids Surf B Biointerfaces. 2016;148:116–129. | ||
Calvo-Ochoa E, Arias C. Cellular and metabolic alterations in the hippocampus caused by insulin signalling dysfunction and its association with cognitive impairment during aging and Alzheimer’s disease: studies in animal models. Diabetes Metab Res Rev. 2015;31(1):1–13. | ||
Pan H, Chen J, Xu J, Chen M, Ma R. Antifibrotic effect by activation of peroxisome proliferator-activated receptor-gamma in corneal fibroblasts. Mol Vis. 2009;15:2279–2286. | ||
Yamamoto A, Kakuta H, Miyachi H, Sugimoto Y. Involvement of the Retinoid X Receptor Ligand in the Anti-Inflammatory Effect Induced by Peroxisome Proliferator-Activated Receptor γ Agonist In Vivo. PPAR Res. 2011;2011:1–8. | ||
Chen J, Li S, Sun W, Li J. Anti-diabetes drug pioglitazone ameliorates synaptic defects in AD transgenic mice by inhibiting cyclin-dependent kinase5 activity. PLoS One. 2015;10(4):e0123864–12. | ||
El-Sahar AE, Safar MM, Zaki HF, Attia AS, Ain-Shoka AA. Neuroprotective effects of pioglitazone against transient cerebral ischemic reperfusion injury in diabetic rats: Modulation of antioxidant, anti-inflammatory, and anti-apoptotic biomarkers. Pharmacol Rep. 2015;67(5):901–906. | ||
Zolezzi JM, Inestrosa NC. Peroxisome proliferator-activated receptors and Alzheimer’s disease: hitting the blood-brain barrier. Mol Neurobiol. 2013;48(3):438–451. | ||
Nicolakakis N, Aboulkassim T, Ongali B, et al. Complete rescue of cerebrovascular function in aged Alzheimer’s disease transgenic mice by antioxidants and pioglitazone, a peroxisome proliferator-activated receptor gamma agonist. J Neurosci. 2008;28(37):9287–9296. | ||
Chou PS, Ho BL, Yang YH. Effects of pioglitazone on the incidence of dementia in patients with diabetes. J Diabetes Complications. 2017;31(6):1053–1057. | ||
Liu D, Lin B, Shao W, Zhu Z, Ji T, Yang C. In Vitro and in Vivo Studies on the Transport of PEGylated Silica Nanoparticles across the Blood–Brain Barrier. ACS Appl Mater Interfaces. 2014;6(3):2131–2136. | ||
Masserini M. Nanoparticles for Brain Drug Delivery. ISRN Biochem. 2013;2013(6):1–18. | ||
Mikitsh JL, Chacko AM. Pathways for small molecule delivery to the central nervous system across the blood-brain barrier. Perspect Medicin Chem. 2014;6:PMC.S13384. | ||
Al Asmari AK, Ullah Z, Tariq M, Fatani A. Preparation, characterization, and in vivo evaluation of intranasally administered liposomal formulation of donepezil. Drug Des Devel Ther. 2016;10:205–215. | ||
Gromnicova R, Yilmaz CU, Orhan N, et al. Localization and mobility of glucose-coated gold nanoparticles within the brain. Nanomedicine. 2016;11(6):617–625. | ||
Sánchez-López E, Ettcheto M, Egea MA, et al. New potential strategies for Alzheimer’s disease prevention: pegylated biodegradable dexibuprofen nanospheres administration to APPswe/PS1dE9. Nanomedicine Nanotechnology. Biol Med. 2017;13(3):1171–1182. | ||
Patel T, Zhou J, Piepmeier JM, Saltzman WM. Polymeric nanoparticles for drug delivery to the central nervous system. Adv Drug Deliv Rev. 2012;64(7):701–705. | ||
Tosi G, Bortot B, Ruozi B, et al. Potential use of polymeric nanoparticles for drug delivery across the blood-brain barrier. Curr Med Chem. 2013;20(17):2212–2225. | ||
Salvalaio M, Rigon L, Belletti D, et al. Targeted Polymeric Nanoparticles for Brain Delivery of High Molecular Weight Molecules in Lysosomal Storage Disorders. PLoS One. 2016;11(5):e0156452. | ||
Guccione C, Oufir M, Piazzini V, et al. Andrographolide-loaded nanoparticles for brain delivery: Formulation, characterisation and in vitro permeability using hCMEC/D3 cell line. Eur J Pharm Biopharm. 2017;119:253–263. | ||
Shadap MD, Ali A, Bhatnagar A, et al. Design, Development, Optimization and Characterization of Donepezil Loaded Chitosan Nanoparticles for Brain Targeting to Treat Alzheimer’s Disease. Sci Adv Mater. 2014;6(4):720–735. | ||
Araújo J, Vega E, Lopes C, Egea MA, Garcia ML, Souto EB. Effect of polymer viscosity on physicochemical properties and ocular tolerance of FB-loaded PLGA nanospheres. Colloids Surf B Biointerfaces. 2009;72(1):48–56. | ||
Silva-Abreu M, Calpena AC, Espina M, et al. Optimization, Biopharmaceutical Profile and Therapeutic Efficacy of Pioglitazone-loaded PLGA-PEG Nanospheres as a Novel Strategy for Ocular Inflammatory Disorders. Pharm Res. 2018;35(1):11. | ||
Abrego G, Alvarado H, Souto EB, et al. Biopharmaceutical profile of pranoprofen-loaded PLGA nanoparticles containing hydrogels for ocular administration. Eur J Pharm Biopharm. 2015;95(Pt B):261–270. | ||
Xu Y, Du Y. Effect of molecular structure of chitosan on protein delivery properties of chitosan nanoparticles. Int J Pharm. 2003;250(1):215–226. | ||
Resende AP, Silva B, Braz BS, Nunes T, Gonçalves L, Delgado E. Ex vivo permeation of erythropoietin through porcine conjunctiva, cornea, and sclera. Drug Deliv Transl Res. 2017;7(5):625–631. | ||
Grabrucker AM, Ruozi B, Belletti D, et al. Nanoparticle transport across the blood brain barrier. Tissue Barriers. 2016;4(1):e1153568. | ||
Fessi H, Puisieux F, Devissaguet JP, Ammoury N, Benita S. Nanocapsule formation by interfacial polymer deposition following solvent displacement. Int J Pharm. 1989;55(1):R1–R4. | ||
Vega E, Gamisans F, García ML, Chauvet A, Lacoulonche F, Egea MA. PLGA nanospheres for the ocular delivery of flurbiprofen: drug release and interactions. J Pharm Sci. 2008;97(12):5306–5317. | ||
Silva-Abreu M, Espinoza LC, Rodríguez-Lagunas MJ, et al. Human Skin Permeation Studies with PPARγ Agonist to Improve Its Permeability and Efficacy in Inflammatory Processes. Int J Mol Sci. 2017;18(12):2548. | ||
Mallandrich M, Fernández-Campos F, Clares B, et al. Developing Transdermal Applications of Ketorolac Tromethamine Entrapped in Stimuli Sensitive Block Copolymer Hydrogels. Pharm Res. 2017;34(8):1728–1740. | ||
Yamaoka K, Nakagawa T, Uno T. Application of Akaike’s information criterion (AIC) in the evaluation of linear pharmacokinetic equations. J Pharmacokinet Biopharm. 1978;6(2):165–175. | ||
Weksler BB, Subileau EA, Perrière N, et al. Blood-brain barrier-specific properties of a human adult brain endothelial cell line. Faseb J. 2005;19(13):1872–1874. | ||
Andreani T, Kiill CP, de Souza AL, et al. Surface engineering of silica nanoparticles for oral insulin delivery: characterization and cell toxicity studies. Colloids Surf B Biointerfaces. 2014;123:916–923. | ||
Tai LM, Holloway KA, Male DK, Loughlin AJ, Romero IA. Amyloid-beta-induced occludin down-regulation and increased permeability in human brain endothelial cells is mediated by MAPK activation. J Cell Mol Med. 2010;14(5):1101–1112. | ||
Lopez-Ramirez MA, Wu D, Pryce G, et al. MicroRNA-155 negatively affects blood-brain barrier function during neuroinflammation. Faseb J. 2014;28(6):2551–2565. | ||
Borchelt DR, Ratovitski T, van Lare J, et al. Accelerated amyloid deposition in the brains of transgenic mice coexpressing mutant presenilin 1 and amyloid precursor proteins. Neuron. 1997;19(4):939–945. | ||
dal Magro R, Ornaghi F, Cambianica I, et al. ApoE-modified solid lipid nanoparticles: A feasible strategy to cross the blood-brain barrier. J Control Release. 2017(249):103–110. | ||
Kreuter J, Ramge P, Petrov V, et al. Direct evidence that polysorbate-80-coated poly(butylcyanoacrylate) nanoparticles deliver drugs to the CNS via specific mechanisms requiring prior binding of drug to the nanoparticles. Pharm Res. 2003;20(3):409–416. | ||
Gromnicova R, Kaya M, Romero IA, et al. Transport of Gold Nanoparticles by Vascular Endothelium from Different Human Tissues. PLoS One. 2016;11(8):e0161610. | ||
Shilo M, Sharon A, Baranes K, Motiei M, Lellouche JP, Popovtzer R. The effect of nanoparticle size on the probability to cross the blood-brain barrier: an in-vitro endothelial cell model. J Nanobiotechnology. 2015;13(1):19. | ||
Fröhlich E. The role of surface charge in cellular uptake and cytotoxicity of medical nanoparticles. Int J Nanomedicine. 2012;7:5577–5591. | ||
Neves AR, Queiroz JF, Weksler B, Romero IA, Couraud PO, Reis S. Solid lipid nanoparticles as a vehicle for brain-targeted drug delivery: two new strategies of functionalization with apolipoprotein E. Nanotechnology. 2015;26(49):495103. | ||
Wohlfart S, Gelperina S, Kreuter J. Transport of drugs across the blood-brain barrier by nanoparticles. J Control Release. 2012;161(2):264–273. | ||
Smith MW, Gumbleton M. Endocytosis at the blood-brain barrier: from basic understanding to drug delivery strategies. J Drug Target. 2006;14(4):191–214. | ||
Rejman J, Oberle V, Zuhorn IS, Hoekstra D. Size-dependent internalization of particles via the pathways of clathrin- and caveolae-mediated endocytosis. Biochem J. 2004;377(Pt 1):159–169. | ||
Searcy JL, Phelps JT, Pancani T, et al. Long-Term Pioglitazone Treatment Improves Learning and Attenuates Pathological Markers in a Mouse Model of Alzheimer’s Disease. Journal of Alzheimer’s Disease. 2012;30(4):943–961. | ||
Mandrekar-Colucci S, Karlo JC, Landreth GE. Mechanisms underlying the rapid peroxisome proliferator-activated receptor-γ-mediated amyloid clearance and reversal of cognitive deficits in a murine model of Alzheimer’s disease. J Neurosci. 2012;32(30):10117–10128. | ||
Yin QQ, Pei JJ, Xu S, et al. Pioglitazone improves cognitive function via increasing insulin sensitivity and strengthening antioxidant defense system in fructose-drinking insulin resistance rats. PLoS One. 2013;8(3):e59313. | ||
Papadopoulos P, Rosa-Neto P, Rochford J, Hamel E. Pioglitazone improves reversal learning and exerts mixed cerebrovascular effects in a mouse model of Alzheimer’s disease with combined amyloid-β and cerebrovascular pathology. PLoS One. 2013;8(7):e68612. | ||
Takeda. Extension study of the safety and efficacy of pioglitazone to slow cognitive decline in participants with mild cognitive impairment due to Alzheimer disease. Available from: https://clinicaltrials.gov/ct2/show/NCT02284906?term=pioglitazone&cond=Alzheimer+Disease&rank=3. NLM identifier: NCT02284906. Accessed February 15, 2018. | ||
Fernandez-Martos CM, Atkinson RAK, Chuah MI, King AE, Vickers JC. Combination treatment with leptin and pioglitazone in a mouse model of Alzheimer’s disease. Alzheimers Dement. 2017;3(1):92–106. |
Supplementary materials
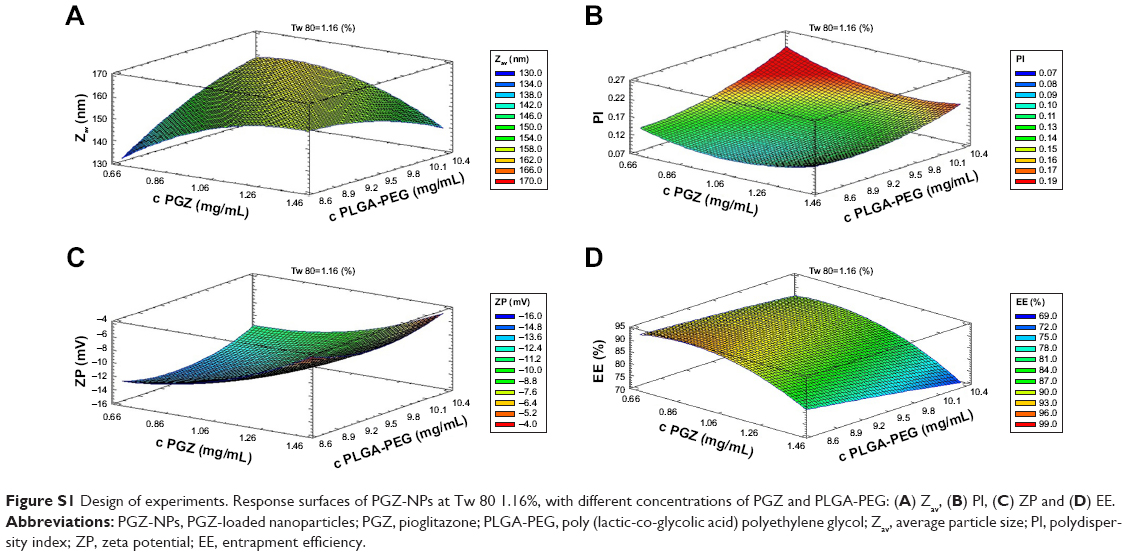 | Figure S1 Design of experiments. Response surfaces of PGZ-NPs at Tw 80 1.16%, with different concentrations of PGZ and PLGA-PEG: (A) Zav, (B) PI, (C) ZP and (D) EE. |
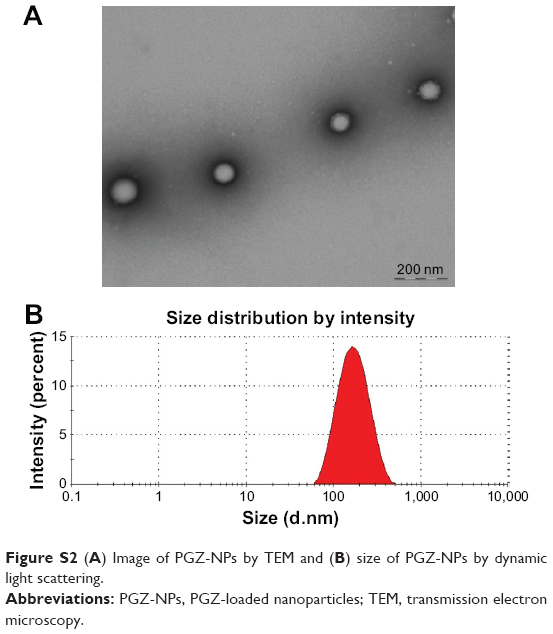 | Figure S2 (A) Image of PGZ-NPs by TEM and (B) size of PGZ-NPs by dynamic light scattering. |
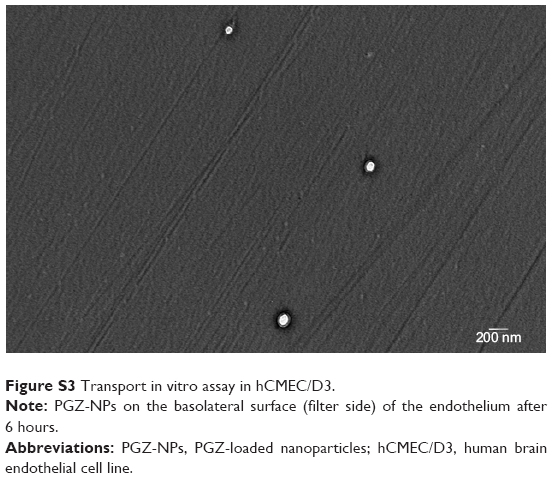 | Figure S3 Transport in vitro assay in hCMEC/D3. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.