Back to Journals » International Journal of Nanomedicine » Volume 16
Potentiality of Melittin-Loaded Niosomal Vesicles Against Vancomycin-Intermediate Staphylococcus aureus and Staphylococcal Skin Infection
Authors Sangboonruang S, Semakul N, Obeid MA, Ruano M ![]() , Kitidee K, Anukool U
, Kitidee K, Anukool U ![]() , Pringproa K, Chantawannakul P, Ferro VA
, Pringproa K, Chantawannakul P, Ferro VA ![]() , Tragoolpua Y
, Tragoolpua Y ![]() , Tragoolpua K
, Tragoolpua K ![]()
Received 25 June 2021
Accepted for publication 28 October 2021
Published 16 November 2021 Volume 2021:16 Pages 7639—7661
DOI https://doi.org/10.2147/IJN.S325901
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Ebrahim Mostafavi
Supplementary video of "Mel-loaded NISV against staphylococcal skin infection" [ID 325901].
Views: 136
Sirikwan Sangboonruang,1 Natthawat Semakul,2 Mohammad A Obeid,3 Marta Ruano,4 Kuntida Kitidee,5 Usanee Anukool,6,7 Kidsadagon Pringproa,8 Panuwan Chantawannakul,9 Valerie A Ferro,4 Yingmanee Tragoolpua,9 Khajornsak Tragoolpua6,7
1Biotechnology Section, Graduate School, Chiang Mai University, Chiang Mai, Thailand; 2Department of Chemistry, Faculty of Science, Chiang Mai University, Chiang Mai, Thailand; 3Department of Pharmaceutics and Pharmaceutical Technology, Faculty of Pharmacy, Yarmouk University, Irbid, Jordan; 4Strathclyde Institute of Pharmacy and Biomedical Sciences, University of Strathclyde, Glasgow, UK; 5Center for Research and Innovation, Faculty of Medical Technology, Mahidol University, Salaya, Nakhon Pathom, Thailand; 6Division of Clinical Microbiology, Department of Medical Technology, Faculty of Associated Medical Sciences, Chiang Mai University, Chiang Mai, Thailand; 7Infectious Diseases Research Unit (IDRU), Faculty of Associated Medical Sciences, Chiang Mai University, Chiang Mai, Thailand; 8Department of Veterinary Biosciences and Veterinary Public Health, Faculty of Veterinary Medicine, Chiang Mai University, Chiang Mai, Thailand; 9Division of Microbiology, Department of Biology, Faculty of Sciences, Chiang Mai University, Chiang Mai, Thailand
Correspondence: Khajornsak Tragoolpua
Division of Clinical Microbiology, Department of Medical Technology, Faculty of Associated Medical Sciences, Chiang Mai University, Chiang Mai, 50200, Thailand
Tel +66 5393 6039
Fax +66 5393 6042
Email [email protected]
Background: Staphylococcus aureus is an important human pathogen, especially causing skin and soft tissue infections (SSTIs). Over the decades, the infections caused by antibiotic-resistant strains have often become life-threatening. Consequently, exploration and development of competent approaches to combat these serious circumstances are urgently required.
Methods: The antibacterial activity of melittin (Mel) on S. aureus, methicillin-resistant S. aureus (MRSA) and clinical isolates of vancomycin-intermediate S. aureus (VISA) was investigated by minimum inhibitory concentration (MIC) and time-killing assays. The localization of Mel on the bacterial cell was visualized by confocal laser scanning microscopy and its effect on the membrane was indicated based on propidium iodide uptake. The non-ionic surfactant vesicle (NISV) or niosome nanocarrier was established for Mel loading (Mel-loaded NISV) by the thin-film hydration method. Physicochemical and in vitro biological properties of Mel-loaded NISVs were characterized. The cellular uptake of Mel-loaded NISVs was evaluated by holotomography analysis. In addition, an ex vivo study was conducted on a porcine ear skin model to assess the permeation ability of Mel-loaded NISVs and their potential to inhibit bacterial skin infection.
Results: The effective inhibitory activity of Mel on skin pathogens was demonstrated. Among the tested strains, VISA was most susceptible to Mel. Regarding to its function, Mel targeted the bacterial cell envelope and disrupted cell membrane integrity. Mel-loaded NISVs were successfully fabricated with a nano-size of 120– 200 nm and entrapment efficiency of greater than 90%. Moreover, Mel-loaded NISVs were taken up and accumulated in the intracellular space. Meanwhile, Mel was released and distributed throughout the cytosol and nucleus. Mel-loaded NISVs efficiently inhibited the growth of bacteria, particularly MRSA and VISA. Importantly, they not only penetrated epidermal and dermal skin layers, but also reduced the bacterial growth in infected skin.
Conclusion: Mel-loaded NISVs have a great potential to exhibit antibacterial activity. Therapeutic application of Mel-loaded NISVs could be further developed as an alternative platform for the treatment of skin infection via dermal and transdermal delivery.
Keywords: melittin, niosome, drug resistance, skin infection, Staphylococcus aureus, dermal and transdermal delivery
Graphical Abstract:

Introduction
Skin and soft tissue infections (SSTIs) are very common clinical manifestations that involve microbial invasion of the skin and underlying soft tissue. The symptoms of infections can range from superficial to severe. The infections can extend to subcutaneous tissue and require complex treatment.1–3 Staphylococcus aureus is one of main pathogens that is responsible for most cutaneous infections, and the treatment of staphylococcal infections has become more difficult owing to the widespread development of multidrug-resistant isolates. In particular, infections caused by methicillin-resistant Staphylococcus aureus (MRSA) have remained a serious public health concern.1,3–5 MRSA has become resistant to vancomycin, a mainstay of therapy for serious MRSA infections.6,7 The emergence of the more virulent strains with reduced clinical response to vancomycin has been documented and defined as vancomycin-intermediate or vancomycin-resistant Staphylococcus aureus (VISA or VRSA, respectively).6–9 Hence, additional choices for the treatment of multi-drug-resistant bacteria, including the application of alternative bioactive agents, are required.
Melittin (Mel), a principal component of honeybee (Apis mellifera) venom, is a cationic α-helical peptide consisting of 26 amino acids with amphipathic characteristics.10,11 Previously, Mel has been extensively investigated for its pharmaceutical and biological properties. A potent ability of Mel against various cancer cell types and broad-spectrum microorganisms has been successfully demonstrated in many studies.15–17 For its antimicrobial peptide (AMP) properties, Mel destroys the bacterial cell through the formation of ion channels or transmembrane pores.18 It penetrates the cell through the interaction with the negative charge of bacterial cell envelopes. The main targets of most AMPs are lipids in the bacterial cell wall. The peptides bind to anionic phospholipids and phosphate groups of teichoic and lipoteichoic acids composing the peptidoglycan layer of Gram-positive bacteria.19 The transmembrane pore produced by Mel can be explained with the toroidal model. Molecules of Mel perpendicularly insert into the lipid bilayer and cause the lipid monolayer to bend continuously from the top to the bottom; consequently, a toroidal shape is formed.18,20 The peptides anchor in the cytoplasmic membrane and alter the membrane structure of the microorganism, which facilitates the incorporation of peptides into the phospholipid dual layer of the cytoplasmic membrane.19,21 Apart from the membrane destruction, the peptides may exert their antibacterial activity by interactions with intracellular targets, which can be DNA, RNA and proteins. Thus, the intracellular processes are disrupted (Figure 1).18–20 An investigation into Mel’s activity on drug-resistant strains was conducted by Choi et al. The study demonstrated that Mel had an effective anti-MRSA activity in vitro and a protective effect in MRSA-infected mice in models of bacteremia and skin infection.17 Hitherto, the activity of Mel on VISA has not been reported. In this study, we aimed to examine the inhibitory activity of Mel toward VISA. In addition, even though the usefulness of Mel has been introduced, its therapeutic applications have been limited owing to its toxicity and hemolytic effects.22 To overcome these obstacles, encapsulation techniques and drug delivery systems are required.
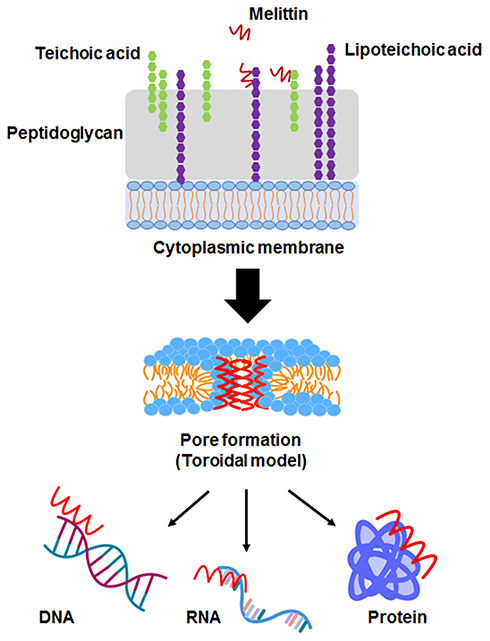 |
Figure 1 The proposed mechanism of Mel on bacterial cells. The possible mechanisms of most antimicrobial peptides (AMPs) target cell membranes through the negatively charged bacterial cell envelopes. Mel-induced pore formation is based on a toroidal model. Apart from the membrane destruction, the peptides may exert antibacterial activity by interactions with intracellular targets, which can be DNA, RNA and proteins, thus disrupting intracellular processes. |
Nanoparticle-based drug delivery systems can be used to encapsulate drugs or therapeutic substances and serve as a carrier that protects the drug or substance from interacting with unfavorable environments and reduces their toxicity.23–26 Previously, several delivery systems have been introduced to encapsulate Mel by different nanocarriers27,28 including lipid-based systems such as liposomes29–31 and niosomes.32 Because of the drawback of liposome-related unstable structures resulting in drug leakage,30,31 niosome-based drug delivery systems have become a promising strategy. Non-ionic surfactant vesicles (NISVs) or niosomes have similar functions and physicochemical properties to liposomes but differ in the composition of their structures.33,34 Niosomes are fabricated from non-ionic surfactants whereas liposomes are based on phospholipids, resulting in the improved properties and stability of niosomes. Moreover, niosomes also have various advantageous properties, including being biodegradable, biocompatible and non-immunogenic, and have a enhanced vesicular system for dermal and transdermal delivery.33,35,36 Several studies have successfully demonstrated the formulation of niosomes containing different proteins and peptides.35,37–39 Dabbagh Moghaddam et al fabricated a Mel-loaded niosome formulation for breast cancer treatment both in vitro and in vivo. Their results showed the superior inhibitory effect of Mel-loaded niosomes on breast cancer cells (4T1 and SKBR3 cells) compared to free Mel.32
In this study, we aimed to assess the antibacterial activity and cellular mechanisms of Mel against MRSA and VISA. Thus, niosomal vesicles loaded with Mel (Mel-loaded NISVs) were formulated for the purpose of dermal and transdermal delivery. The physicochemical characteristics and antibacterial activity of Mel-loaded NISVs were evaluated. Finally, investigations into the properties of Mel-loaded NISVs, in terms of permeation ability and abatement of bacterial skin infection, were conducted in an ex vivo porcine skin model.
Materials and Methods
Ethics
This study received ethical approval from the Institutional Ethics Committee, Chiang Mai University (approval no.: AMSEC 62EM-008).
Materials
Synthetic high-purity Mel (GIGAVLKVLTTGLPALISWIKRKRQQG) from A. mellifera venom was synthesized by Synpeptide (Shanghai, China) and fluorescein isothiocyanate (FITC)-labeled Mel was manufactured by GenScript (Piscataway, NJ, USA). All peptides were prepared at 20 mg/mL stock concentration and stored at −20°C until used. Sorbitan monostearate (Span 60), polysorbate (Tween 80) and cholesterol were purchased from Sigma-Aldrich (St Louis, MO, USA). All other chemicals used were of analytical grade.
Cell Culture
Normal embryonic mouse fibroblasts (NIH 3T3) (kindly obtained from Asst Prof. Dr Fahsai Kantawong, Department of Medical Technology, Faculty of Associated Medical Sciences, Chiang Mai University, Chiang Mai, Thailand) and the human malignant melanoma (A375) cell line (ATCC, MD, USA) were cultured in Dulbecco’s modified Eagle’s medium (DMEM) (Gibco, New York, USA) supplemented with 10% (v/v) fetal bovine serum (FBS), penicillin (100 units/mL) and streptomycin (100 µg/mL), and maintained in a humidified atmosphere of 5% CO2 at 37°C.
Bacterial Strains
Bacterial strains used in this study were S. aureus ATCC 25923, MRSA ATCC 43300 and 155 clinical isolates of MRSA (obtained from the Clinical Microbiology Laboratory, Maharaj Nakorn Chiang Mai Hospital), which were identified by standard laboratory methods. The obtained MRSA clinical isolates were further confirmed for methicillin resistance using oxacillin discs (30 μg). In brief, the broth culture of bacterial strains with turbidity was adjusted to the 0.5 McFarland standard and lawn culture was performed on Mueller–Hinton agar (MHA) plates. Then, an oxacillin disk was placed on the lawn culture and incubated at 37°C in ambient air for 18 hours. The strains showing a zone of inhibition diameter of ≤22 mm were considered MRSA.
For screening of VRSA or VISA, 155 clinical isolates of MRSA were tested for vancomycin susceptibility using a vancomycin agar screen method according to Clinical Laboratory Standards Institute (CLSI) guidelines (M07-A9 and M100S-S22). In brief, a direct colony was resuspended in brain–heart infusion (BHI) broth equivalent to the 0.5 McFarland standard, and a 10 μL of suspension was dropped onto the BHI agar containing 6 μg/mL of vancomycin and incubated at 37°C in ambient air for 24 hours. After the incubation time, the plates were carefully examined for evidence of colonies. The resistant isolates were further subjected to a vancomycin minimum inhibitory concentration (MIC) test. The interpretive criteria for vancomycin and S. aureus were ≤2 μg/mL for susceptible, 4–8 μg/mL for intermediate and ≥16 μg/mL for resistant.
Determination of Minimum Inhibitory Concentration (MIC) and Minimum Bactericidal Concentration (MBC) of Mel
The inhibitory effect of Mel on bacterial growth was determined by the broth microdilution method. Mel was dissolved in BHI broth at various concentrations and added to each well of a microplate (100 μL/well). Then, the bacterial suspension, previously adjusted to the 0.5 McFarland standard, containing approximately 1–2×108 colony-forming units (CFU)/mL, was appropriately diluted to obtain a final concentration of 105 CFU/mL. The microtiter plate containing Mel was loaded with 100 μL of bacterial suspension to yield the final test concentration of bacteria, approximately 104 CFU/well, and incubated at 37°C for 24 hours. After incubation, the MIC was observed and defined as the lowest concentration that could inhibit bacterial growth.
For MBC analysis, viable cells from all wells with no visible bacterial growth were determined on the BHI agar plates. After 24 hours’ incubation, the MBC endpoint was defined as the lowest concentration at which the bacterial population was killed by 99.9%.
Time-Kill Assay
To assess the rate of bacterial killing by Mel, time-kill assays were performed. The bacterial suspension was adjusted to the 0.5 McFarland standard and diluted to obtain a final concentration of 105 CFU/mL (104 CFU/well), and treated with Mel at concentrations corresponding to 0.5× MIC and 1× MIC. Bacterial growth was quantified after 0, 1, 2, 4 and 6 hours’ incubation by determination of viable cells on an agar plate.
Studies on the Cellular Mechanism of Mel on Bacterial Cells
Localization of Mel
The localization of Mel was investigated using a confocal laser scanning microscope (CLSM). Bacteria were grown to mid-logarithmic phase and collected by centrifugation at 12,000×g for 5 minutes. Approximately 107 CFU were resuspended in FITC-labeled Mel at a concentration corresponding to 1× MIC. Following incubation for 30 minutes at 37°C, bacterial cells were washed with PBS. The nuclei were then counterstained with 4ʹ,6-diamidino-2-phenylindole (DAPI) (Invitrogen, Thermo Fisher Scientific, MA, USA) and mounted in ProLong Gold antifade reagent (Invitrogen, Thermo Fisher Scientific, MA USA). Images were acquired using a CLSM (LSM 800 with Airyscan; Zeiss, Oberkochen, Germany).
Study of Membrane Integrity
The effects of Mel on the membrane integrity of bacterial cells were investigated by propidium iodide (PI) staining and the fluorescence intensity was monitored.40 Approximately 107 CFU were treated with Mel at concentrations corresponding to 0.5× MIC and 1× MIC. The bacterial cells were treated with 2.5% (v/v) Triton X-100 (TX-100) as a positive control. After 30 minutes’ incubation time, the bacterial cell suspension was washed with PBS and stained with PI, followed by visualization under a fluorescence microscope (Axioscope 5; Zeiss, Oberkochen, Germany). Then, 200 μL of stained bacterial suspension was added to a flat-bottomed, 96-well, black microtiter plate and the fluorescence intensity was measured at 617 nm using a fluorescence microplate reader (SynergyTM H4 hybrid microplate reader; BioTek Instruments, Winooski, VT, USA).
Formulation of Mel-Loaded NISVs by Thin-Film Hydration Method
NISV formulations were prepared according to the thin-film hydration (TFH) method41,42 using different ratios of non-ionic surfactants (Span 60 and Tween 80) and cholesterol, as indicated in Table 1. In brief, the surfactants and cholesterol were dissolved in 10 mL chloroform in a round-bottomed flask. The organic solvent was evaporated using a rotary evaporator under vacuum at 60°C and 100 rpm to produce a thin lipid film. The thin film was then hydrated with 10 mL PBS (pH 7.4) for empty NISV or PBS containing Mel for Mel-loaded NISV at 50°C under mechanical stirring. The resulting NISV dispersion was sonicated for size reduction using an ultrasonic probe homogenizer (Hielscher UP50H; Hielscher, NJ, USA) at 80% amplitude for 30 minutes in an ice bath. A schematic representation of a Mel-loaded NISV is shown in Figure 2.
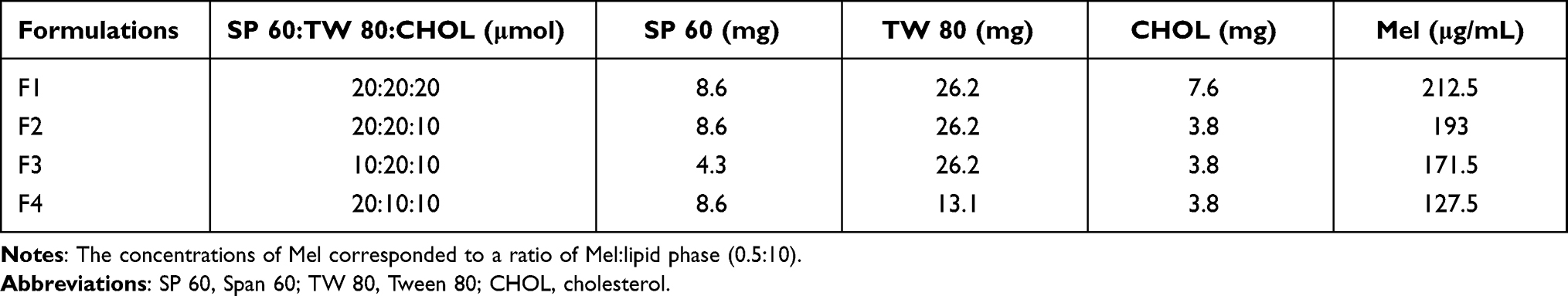 |
Table 1 Compositions of Different NISV Formulations |
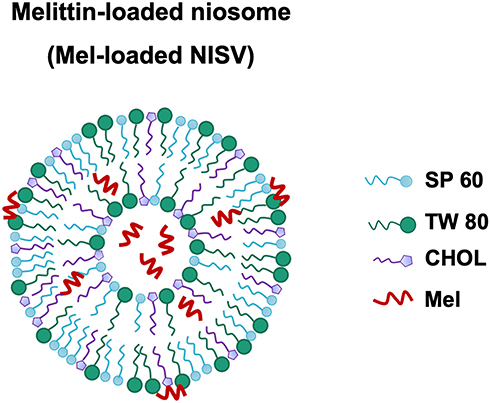 |
Figure 2 Schematic representation of a Mel-loaded NISV. |
Physiochemical Characterization of Mel-Loaded NISVs
Particle Size, Polydispersity Index (PDI) and Surface Charge Analysis
The average particle size, PDI and zeta (ζ) potential (ZP) of the surface charge of NISV formulations were determined using a Malvern Zetasizer Nano ZSP system (Malvern Instruments, Worcestershire, UK). The measurements were carried out for empty NISV and Mel-loaded NISV formulations in aqueous medium at a 1/10 dilution. Each sample was measured based on at least three measurements in three individual runs.
Entrapment Efficiency (EE)
Free Mel was separated from Mel-loaded NISVs by ultracentrifugation with a 10 kDa molecular weight cut-off at 8000×g and 4°C for 6 hours. The niosomal pellets were resuspended in 2.5 mL of PBS. The filtrates were collected for determination of the free Mel using a UV-Vis spectrophotometer at 210 nm. The amount of loaded Mel was obtained by subtracting the amount of free drug from the total drug incorporated in 10 mL niosomal dispersion. The following equation was used to determine the EE of Mel in NISVs.
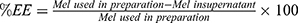
Scanning Electron Microscopy (SEM)
The morphology of empty and loaded NISV formulations was examined using a field emission scanning electron microscope (JEOL JSM-6335F; JEOL, Tokyo, Japan). A drop of the formulations was air dried on copper tape and coated with 30 nm of gold for 30 seconds under argon at a pressure of 0.2 atm. After preparation, the shape and size of particles were studied using SEM at an accelerating voltage of 15 kV.
Fourier Transform Infrared (FT-IR) Spectrometry
To ensure that Mel loaded in the NISV had been successfully prepared, the functional groups of chemical substances in NISV formulations were studied by infrared spectroscopy. Mel, NISV and Mel-loaded NISV samples were lyophilized prior to the analysis. Potassium bromide (KBr) was dried at 110°C for 24 hours and kept in a desiccator before use. Each sample was mixed with dried KBr to construct pellets at a pressure of 2 kgf cm−2. FT-IR spectra were recorded in the range of 4000–400 cm−1 at a resolution of 2 cm−1 using the transmittance mode of the FT-IR spectrometer (Bruker Tensor 27 FTIR; Bruker, Germany).
In Vitro Drug Release
A drug release assay was performed according to Sadeghi et al,38 with some modifications, to assess the amount of Mel released from NISVs in PBS adjusted to pH 5.5 and 7.4 using a dialysis membrane with a molecular weight cut-off of 3.5 kDa (D-TubeTM Dialyzer Midi; Merck, Darmstadt, Germany). A sample of Mel-loaded NISVs (800 μL) was dialyzed against 50 mL of PBS at 37°C. At specific time intervals (0, 1, 2, 4, 6, 12, 18, 24, 36 and 48 hours), 3 mL of PBS was collected and replaced with the same volume of fresh buffer. The buffer samples were analyzed using a UV-Vis spectrophotometer at 210 nm to determine the concentration of released Mel. Each experiment was repeated in triplicate in three individual experiments.
Cellular Uptake
The uptake of FITC-labeled Mel-loaded NISVs was visualized by 3D holotomographic microscopy according to Jantakee et al,43 with some modifications. A375 cells (5×104 cells) were seeded in a 50 mm Tomo dish and incubated at 37°C in a humidified atmosphere of 5% CO2 until they reached 70% confluence. For the purpose of visualizing of NISVs, a 500 μL NISV sample was stained with 40 μL of lipophilic fluorescent marker Nile Red (NR) solution (0.25 mg/mL) in PBS for 20 minutes in the dark, at room temperature. Following washing with PBS, the pellet was resuspended and the NISV suspension was subjected to the cells. Then, the monolayer of cells was treated with NR-labeled NISVs loaded with FITC-labeled Mel for 4 hours and live cell imaging was observed using 3D holotomographic microscopy (Tomocube HT-1H; Tomocube, Daejeon, South Korea).
In Vitro Studies
Cytotoxicity Assay
To examine the toxicity of NISVs, cytotoxicity analysis was conducted on NIH-3T3 and A375 cells by the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay. In brief, the cells (3×104 per well) were seeded in a 96-well tissue culture plate, cultured for 24 hours, and treated with various dilutions of empty NISVs. After 24 hours’ incubation time, 20 μL of MTT solution (5 mg/mL) was added to the treated cells and incubated at 37°C for 4 hours. The supernatant was removed and 200 μL of dimethyl sulfoxide (DMSO) was added to dissolve the dark-blue crystals of MTT–formazan produced by living cells. The absorbance was then quantified with a microplate reader using a test wavelength of 540 nm and a reference wavelength of 630 nm, and cell viability was examined. All experiments were performed in triplicate and repeated in three independent experiments.
Antibacterial Activity
Antibacterial activity of Mel-loaded NISVs was determined against S. aureus ATCC 25923, MRSA ATCC 43300 and VISA No. 4, 36 and 87 for 24 hours using CFU determination. The stock solution of either empty NISVs or Mel-loaded NISVs was diluted to the proper concentrations. One-hundred microliter NISV samples were loaded into a 96-well microtiter plate, and then 100 μL of bacterial suspension (105 CFU/mL) was added to each well and incubated at 37°C for 24 hours. For CFU counts, the treated bacteria were further diluted and plated on MHA plates, which were incubated for 24 hours at 37°C.
Ex Vivo Studies
Preparation and Differentiation of Porcine Ear Skin Model
We received permission for the use of porcine ear skin samples, with an animal carcass consent form from the authorized slaughterhouse, Chiang Mai-Lamphun Co-operative Slaughterhouse Ltd (License number: P3-4(1)-15/48LP and P 05 46010/2549), to collect and use the pig head carcass sample for research purposes. The animal carcass sample was collected by the authorized veterinarian, Ms Ratchadaporn Boripun (Veterinary License Number 01-11675/2559), according to the Cruelty Prevention and Welfare of Animal Act, B.E. 2557 (2014), and the regulations and procedures of the slaughterhouse.
Porcine ear skin samples were used immediately or frozen at −20°C until use within 2 weeks. Skin sections free of injuries or redness were prepared by rinsing with sterile deionized water. The surface was disinfected with a solution of 70% (v/v) ethanol and allowed to dry in a laminar flow cabinet. The skin was sectioned into 1 cm2 segments before its use in further experiments.
To differentiate and evaluate the thickness of the epidermis and dermis layers of the porcine ear skin, periodic acid–Schiff (PAS) staining was performed. In brief, the skin samples were fixed with 10% (v/v) formalin for 24 hours at room temperature, followed by a paraffin-embedding process. The paraffin-embedded tissue blocks were sectioned at 10 μm on a microtome. Afterwards, the paraffin-embedded tissue sections were deparaffinized in two changes of xylene (10 minutes each), rehydrated in two changes of 100%, 95% and 70% (v/v) ethanol (5 minutes each), and washed briefly in water. Then, the sample was incubated with Schiff reagent for 15 minutes, followed by washing in running tap water and counterstaining with Mayer’s hematoxylin for 2 minutes. After washing in running tap water, the sample was dehydrated through two changes of 95% and 100% (v/v) ethanol (5 minutes each), and a coverslip was placed with a mounting medium.
Permeation Study
For the purpose of visualizing the skin-penetration behavior of Mel-loaded NISVs, the NISVs loaded with FITC-labeled Mel were prestained with NR, as described in the subsection ”Cellular Uptake”, above. The NR-labeled NISVs loaded with FITC-labeled Mel were then applied on the porcine skin for 2 and 4 hours at 37°C. Afterwards, the tissue sample was embedded in optimal cutting temperature (OCT) compound prior to frozen sectioning on a microtome–cryostat. The sample was orthogonally sectioned (in the z-axis) to the surface (10 μm thickness) and mounted with ProLong Gold antifade reagent containing DAPI (1.5 μg/mL). The skin sections were observed by CLSM (FluoView FV1000; Olympus Optical, Tokyo, Japan) and the penetration of NISVs into the porcine skin was traced and visualized by the presence of FITC and NR.
Preparation of Infected Skin Models
In this study, the skin infection model was divided into two groups: undamaged and burn wound skin models. For the burn wound model, the wound was created and modified according to Alves et al.44 In brief, the angular tip of a teasing needle, with 5 mm length and 1 mm diameter, was flamed for 30 seconds and placed in contact with the skin for 60 seconds to create a grid of burn wounds with width × length × depth dimensions of 1 mm × 1 × mm × 5 mm.
Prior to inoculation of bacteria on skin sections, FITC-labeled S. aureus was prepared as previously described,45 with some modifications. In brief, a bacterial suspension was previously adjusted to 1×1010 CFU/mL and washed with PBS. The pellet was resuspended in 0.1 M carbonate buffer containing 0.1 mg/mL FITC isomer (Sigma-Aldrich, MO, USA) and incubated for 1 hour in the dark at room temperature, followed by washing with PBS. Then, the pellet was resuspended in PBS for further experiments.
Treatment of Bacterial Skin Infection
The undamaged or burn wound skin samples were inoculated with 10 μL of FITC-labeled S. aureus (108 CFU) and incubated at 37°C in a humidified atmosphere of 5% CO2 for 20 hours. Then, NR-labeled Mel-loaded NISVs (50 μL of stock solution) were introduced to the infected skin samples for 4 hours, while the control samples were treated with PBS only. After the incubation time, the skin samples were immediately frozen and embedded in OCT compound prior to frozen sectioning on a microtome–cryostat. The sample was orthogonally sectioned to the surface (10 μm thickness) and mounted with ProLong Gold antifade reagent containing DAPI (1.5 μg/mL). The skin sections were observed under a CLSM (FluoView FV1000; Olympus Optical, Tokyo, Japan).
For the quantitative analysis, the infected skin samples were treated with Mel-loaded NISVs. After the incubation period, the skin samples were weighed and immersed in broth medium at a ratio of 1:10 (w/v). Subsequently, the samples were mixed by a Stomacher automatic homogenizer (AES-Chemunex, Cranbury, NJ, USA) for 20 minutes. The resulting supernatant was subjected to serial dilution and plated on agar plates for CFU counting.
Statistical Analysis
All results are represented as mean ± standard error of the mean (SEM). Comparisons between groups were made using one-way analysis of variance. Notably, p<0.05 was considered statistically significant. All calculations were performed using SPSS Statistics 23.0 software (IBM Corp., Armonk, NY, USA).
Results
Antibiotic Susceptibility Testing
All 155 clinical isolates were verified for methicillin resistance and further examined for vancomycin susceptibility. Among 155 MRSA isolates, 16 isolates were positive for vancomycin screening and subjected to vancomycin MIC determination. The MICs of 10 isolates after treatment with vancomycin were <4 μg/mL, indicating that they were vancomycin susceptible, while six isolates (3.87%) were identified as VISA with MICs of 4–8 μg/mL, according to recent CLSI breakpoints for vancomycin. None of the isolates was found to be VRSA by MIC determination.
Determination of MIC and MBC of Mel on Skin Pathogenic Bacteria
The antimicrobial effects of Mel on skin pathogenic bacteria, including clinical isolates of antibiotic-resistant bacteria, were investigated and described as MIC and MBC. MIC indicated the inhibitory potential of Mel, while MBC showed the killing effect on tested microorganisms. The bacterial strains, including S. aureus ATCC 25923, MRSA ATCC 43300 and VISA clinical isolates, were treated with Mel at various concentrations. The MIC and MBC values of Mel against the tested pathogenic bacteria are shown in Table 2.
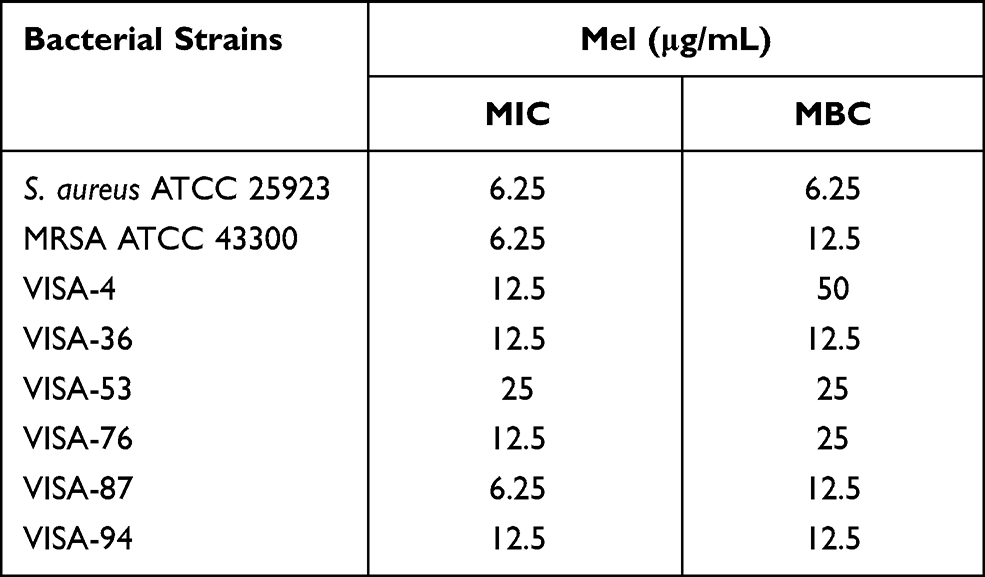 |
Table 2 Minimum Inhibitory Concentration (MIC) and Minimum Bactericidal Concentration (MBC) of Mel Against Skin Pathogenic Bacteria |
All tested microorganisms were susceptible to Mel, with MIC and MBC in the range of 6.25–25 μg/mL and 6.25–50 μg/mL, respectively. This result indicated that Mel exhibited a bactericidal effect against these pathogenic bacteria.
Time-Kill Kinetics of Mel
The time course of bacterial viability was determined after the tested bacteria were treated with Mel at the concentrations corresponding to 0.5× and 1× MIC. The time-kill kinetics profile of Mel against S. aureus ATCC 25923, MRSA ATCC 43300 and VISA clinical isolates (VISA-4, -36 and -87) showed a reduction in the number of viable cells at the concentration of 1× MIC by 2 log10 or 99% reduction within 1 hour, and complete killing was observed after 6 hours. In the treatment with the concentration corresponding to 0.5× MIC, the bacterial growth rate slightly increased in S. aureus and MRSA, whereas the number of VISA strains remained constant during the treatment period (Figure 3). This indicated a greater susceptibility of VISA to Mel.
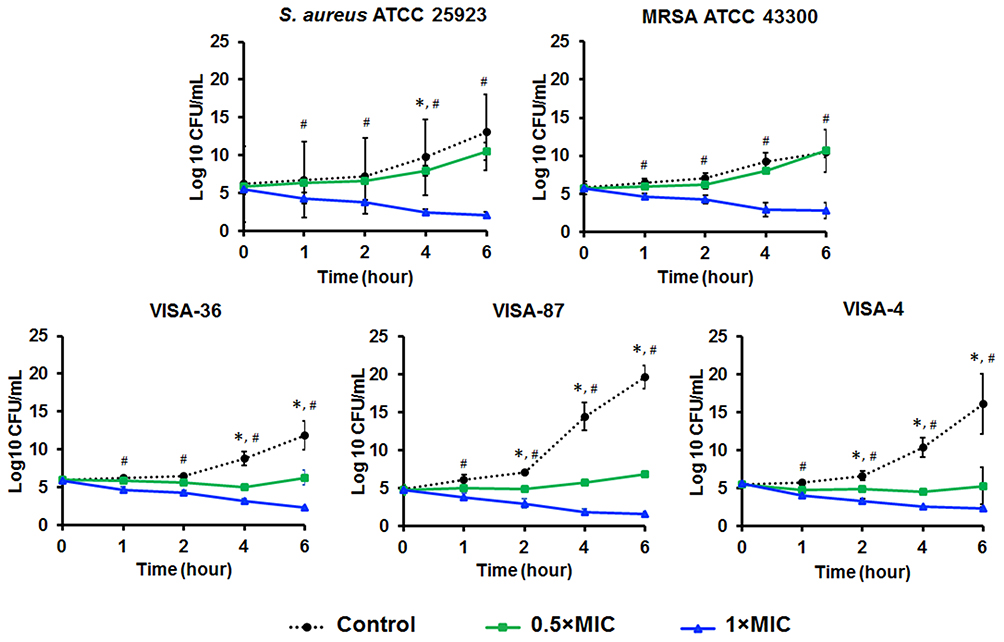 |
Figure 3 Time-kill kinetics of Mel against S. aureus ATCC 25923, MRSA ATCC 43300, and VISA-4, -36 and -87. The microorganisms were treated with Mel at 0.5× or 1× MIC. The viability of bacteria was measured at the indicated time points. Data are represented as the mean ± SEM of three independent experiments performed in triplicate. *p<0.05, significant between 0.5× MIC and control, #p<0.05, significant between 1× MIC and control. |
Target and Biological Properties of Mel
The study of the localization of Mel in the bacterial cells was performed on VISA-87, and Mel was traced through the fluorescence of FITC. VISA-87 was treated with FITC-labeled Mel (green) corresponding to 0.5× MIC, and the localization of Mel was visualized using CLSM. The results showed that, upon Mel treatment, VISA-87 appeared as a hollow round shape with fluorescence clearly defining the bacterial surface, and the nucleus was stained with DAPI (blue), suggesting that Mel was accumulated on the membrane (Figure 4). To further understand the biological activity of Mel on the membrane integrity, a PI uptake assay was performed (Figure 5). The results showed that the relative fluorescence intensity (RFI) was increased in the treated microorganisms compared to untreated strains. The RFI of membrane integrity was induced on tested microorganisms upon the addition of Mel in a dose-dependent manner and significantly (p<0.05) increased on VISA-87. Noticeably, VISA-87 was more susceptible to Mel than S. aureus ATCC 25923, MRSA 43300. This result indicated that Mel disrupted bacterial membrane integrity, leading to bacterial cell death, and exhibited a specific activity against VISA.
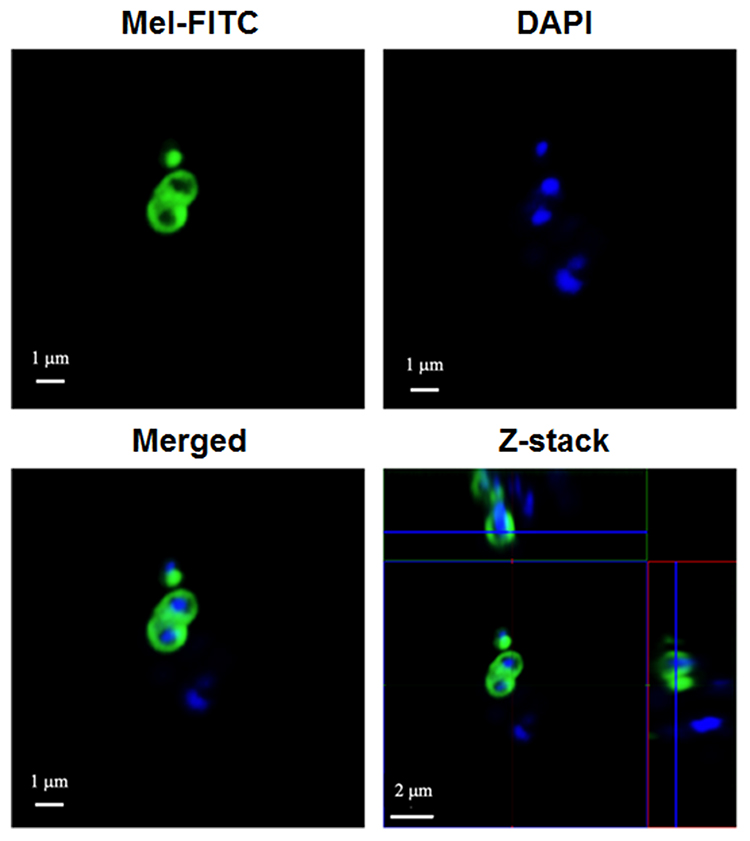 |
Figure 4 Localization of FITC-labeled Mel on VISA-87. Approximately 107 CFU of VISA-87 were incubated with FITC-labeled Mel at 1× MIC (green) for 30 minutes. The bacteria were washed and stained with DAPI (blue). Images were taken using a CLSM. |
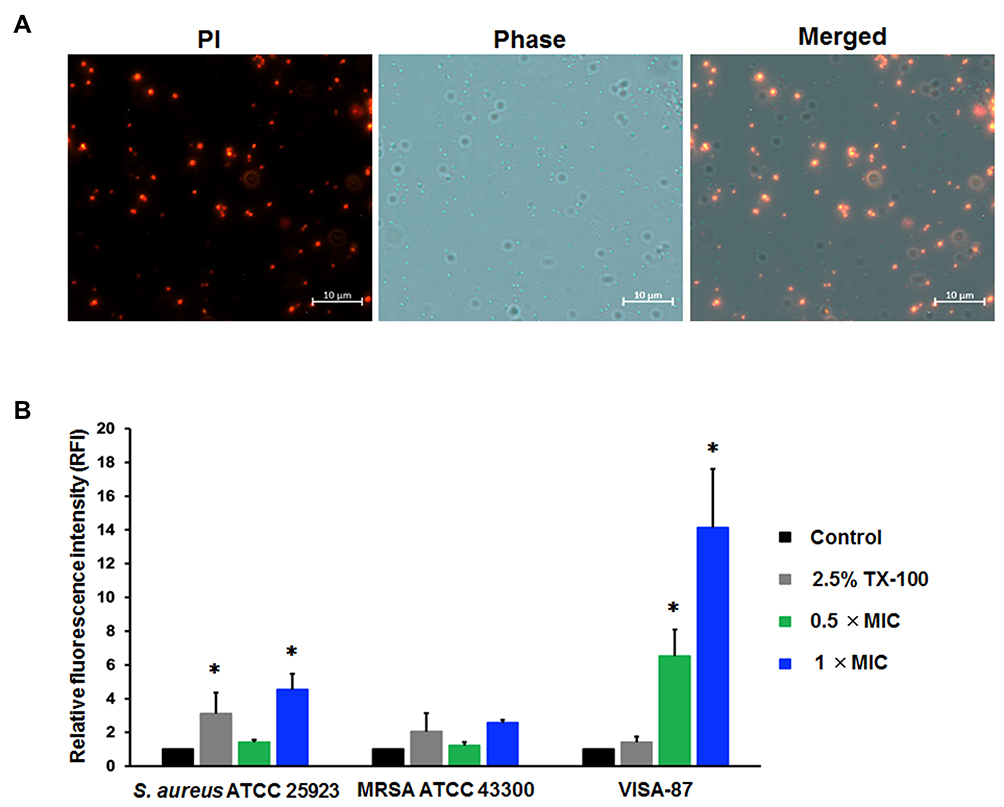 |
Figure 5 Membrane-targeting property of Mel. The effect of Mel on bacterial membrane integrity was investigated by PI staining. The tested bacteria (S. aureus ATCC 25923, MRSA 43300 and VISA-87) were treated with Mel corresponding to 0.5× MIC or 1× MIC for 30 minutes. (A) Bacterial cells were then stained with PI and (B) the fluorescence intensity was measured at 617 nm. Data are represented as the mean ± SEM of three independent experiments performed in triplicate. *p<0.05 as determined by one-way analysis of variance. Scale bar represents 10 μm. |
Physicochemical Characterization of Mel-Loaded NISVs
Particle Size, Polydispersity Index (PDI), Zeta Potential (ZP), Entrapment Efficiency (EE) and Morphology
Empty NISVs and Mel-loaded NISVs were prepared by the TFH method with differing ratios of non-ionic surfactants and cholesterol. Dynamic light scattering (DLS) revealed that all formulations showed a monomodal distribution. The mean particle size of all empty NISV formulations was approximately 100–180 nm, while, loading with Mel led to an increase in size ranging from 120 to 200 nm for formulations 1–3 (F1–F3) and from 200 to 450 nm for formulation 4 (F4), with a PDI of 0.31–0.49 (Supplementary data 1; Figure S1), and the ZP of surface charge was negative (−7 to −11 mV). All formulations showed a great ability to incorporate high amounts of Mel, with drug EE reaching more than 90% (Table 3).
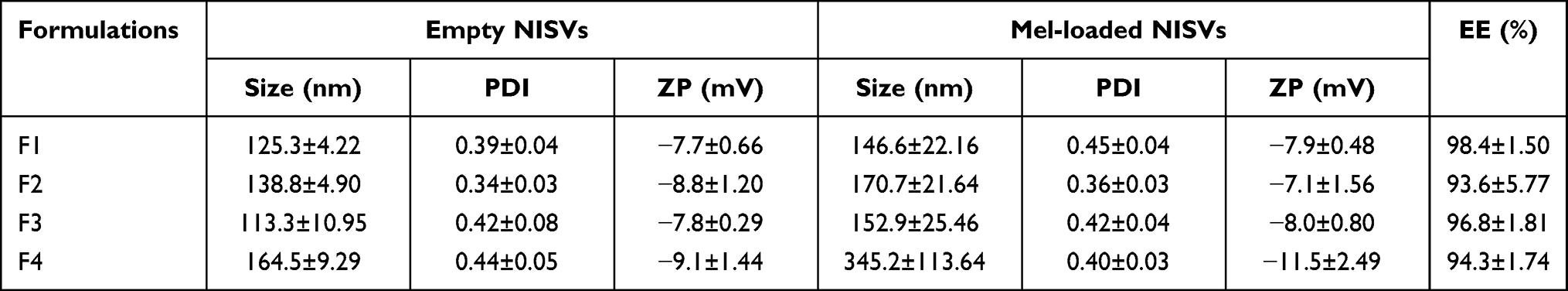 |
Table 3 Particle Size, Polydispersity Index (PDI), ζ Potential (ZP) and Entrapment Efficiency (EE) of NISV Formulations |
Based on the physicochemical properties of Mel-loaded NISV, F1 was shown to achieve the highest EE and PDI values. F3 had the next highest values of EE and PDI, while F4 showed the largest particle size and ZP and a high value of PDI, leading to easy aggregation. Formulation F2 had a particle size of less than 200 nm, with the lowest PDI and a relatively high EE. Thus, this formulation, F2, was suitable for further investigation into its biological properties.
Moreover, SEM images of empty NISVs and Mel-loaded NISVs showed a relatively uniform nanoparticle size with smooth and spherical structures (Figure 6A). This result is concordant with DLS analysis, which demonstrated slightly larger Mel-loaded NISVs and homogeneously sized vesicle populations.
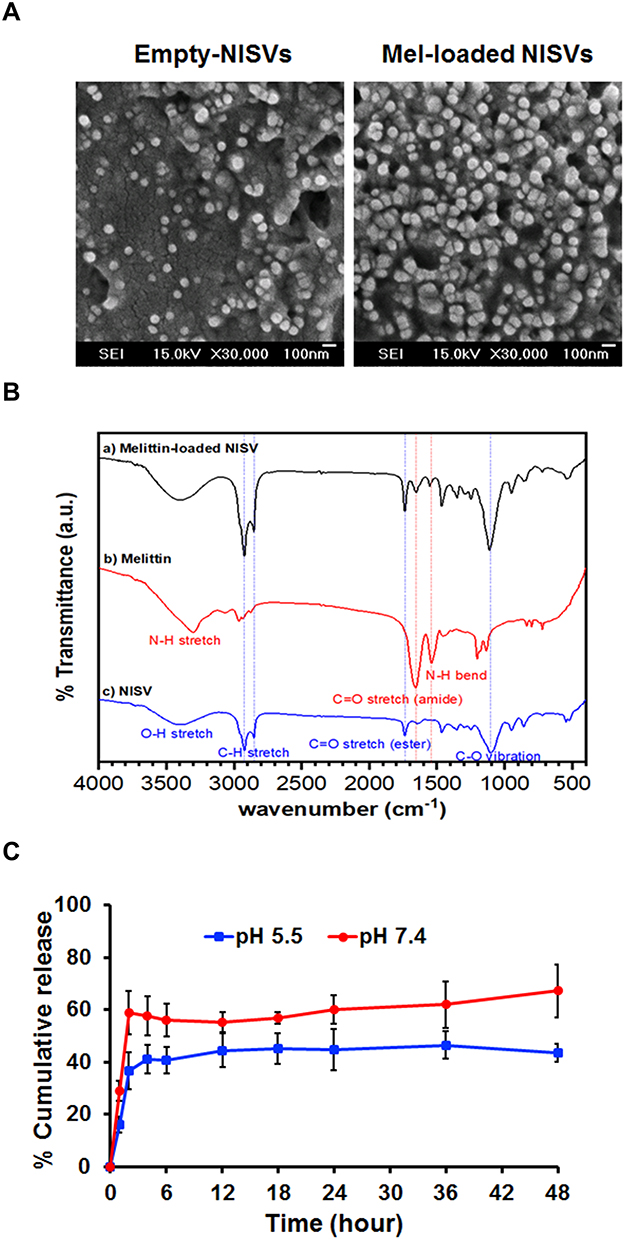 |
Figure 6 Characteristics of Mel-loaded NISVs from F2. (A) Morphology of empty NISV and Mel-loaded NISV images based on field emission SEM. The scale bar represents 100 nm. (B) FT-IR spectra of Mel, empty NISVs and Mel-loaded NISVs. (C) In vitro release profile of Mel from NISVs at 37°C, at pH 5.5 and pH 7.4 for 48 hours. Data are represented as the mean ± SEM of three independent trials. |
Fourier Transform Infrared (FT-IR) Spectroscopy
To ensure that Mel was successfully loaded in the NISV, the key functional groups of target compounds were investigated by infrared spectroscopy. The FT-IR spectra of Mel, empty NISVs and Mel-loaded NISVs are shown in Figure 6B. Mel showed characteristic absorption peaks at 1657 and 1539 cm−1, corresponding to the stretching of the C=O and N–H bonds of the amide group, respectively.46 Mel-loaded NISVs showed absorption signals at wavelengths of 1651 and 1555 cm−1, which were slightly shifted from the characteristic signals of Mel. This presumably was due to the interaction between the amide groups of Mel and the NISVs.
Furthermore, Mel-loaded NISVs also showed major peaks at 1738 and 1113 cm−1 within the C=O stretching and C–O vibration ester functional group,47 which were slightly shifted from the characteristic peaks of empty NISVs at 1724 and 1107 cm−1. Mel-loaded NISVs also showed stretching bands at 2922 and 2853 cm−1 within the C–H stretching region.47 From this, it can be inferred that Mel was successfully encapsulated by the niosomal vesicle system.
In Vitro Release of Mel from NISVs
The in vitro release profile of Mel from NISVs was determined during a 48-hour experiment in two different buffer solutions, at pH 5.5 and pH 7.4 (Figure 6C). The results showed a maximum release of Mel from the NISVs after 2 and 4 hours at pH 7.4 and pH 5.5, respectively, and a Mel-sustained release from NISVs occurred up to 48 hours. This result suggested the capability of the niosomal carrier for encapsulation of Mel, with a release of the Mel from NISVs in a sustained manner.
Cellular Uptake of Mel-Loaded NISVs
To visualize the cellular uptake of Mel-loaded NISVs, FITC-labeled Mel-loaded NISVs were formulated, followed by labeling with NR. After incubation with A375 cells for 4 hours, the cellular uptake and live cell imaging were analyzed by 3D holotomography (Figure 7 and Supplementary video 1). Cellular uptake was observed in A375 cells and accumulated within the cytoplasm, while tracking of Mel by FITC (green) showed the distribution throughout the cell. This suggested that Mel-loaded NISVs were taken up into the cytoplasm and release of Mel was dispersed within the cell, not only in the cytoplasm but also penetrating the nucleus (blue).
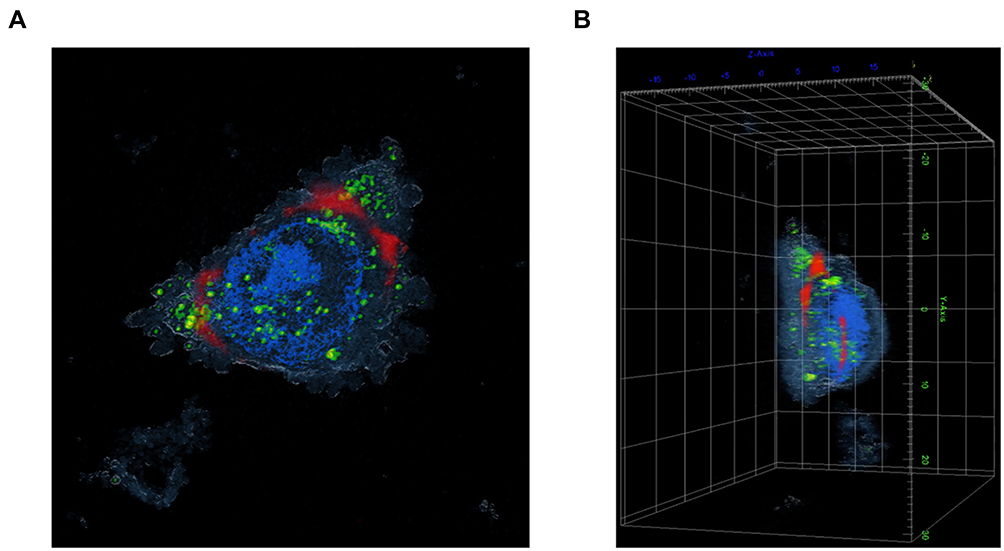 |
Figure 7 Live cell imaging using holotomography of A375 cells treated with Mel-loaded NISVs from F2. (A) XY image and (B) 3D rendered holotomographs of FITC-labeled Mel (green) loaded into niosomes. The formulation was labeled with lipophilic NR (red) for 4 hours. The cellular nuclei were counterstained by DAPI (blue). |
Cytotoxicity of NISVs
Since this niosomal formulation was intended to be applied onto the skin as a topical dosage form, a cytotoxicity assay was carried out on cell lines related to skin tissue. In this study, NIH-3T3 and A375 cell lines were used to determine the toxicity of the NISV formulation by the MTT assay. Cytotoxicity of the NISVs was not observed in a dilution range from 100× to 10×. However, at a high concentration (2× dilution), viability was significantly (p<0.05) reduced. This result suggested that empty NISVs had no cytotoxicity on NIH-3T3 and A375 cells in the dilution range from 100× to 10× (Figure 8A).
 |
Figure 8 In vitro cytotoxicity study and antibacterial activity of Mel-loaded NISVs. (A) Investigation of cytotoxicity of empty NISVs on NIH-3T3 and A375 cells. The cells were treated with various dilutions of empty NISVs and cell viability was evaluated by the MTT assay. (B–D) Mel-loaded NISVs inhibited the growth of S. aureus ATCC 25923, MRSA ATCC 43300 and VISA-87. Bacterial cells were treated with empty NISVs or Mel-loaded NISVs at the indicated dilutions for 24 hours. These dilutions were diluted from the stock that approximately equaled 690.10±38.58 μg/mL. Bacterial growth was determined by CFU counting. Results represent the mean ± SEM from three independent experiments performed in triplicate. *p<0.05, significant compared to control; #p<0.05, significant compared to empty NISVs. |
Antibacterial Activity of Mel-Loaded NISVs
To determine the efficacy of bacterial growth inhibition by Mel-loaded NISVs, bacterial strains including S. aureus ATCC 25923, MRSA ATCC 43300 and VISA-87 were incubated with Mel-loaded NISVs for 24 hours and the inhibition of bacterial growth was evaluated by CFU counting. The results showed that Mel-loaded NISVs significantly (p<0.05) inhibited bacterial growth in a dose-dependent manner (Figure 8B–D). Mel-loaded NISVs were found to be effective against VISA-87, whereas oxacillin did not inhibit the bacterial growth. Although treatment with empty NISV demonstrated a toxicity at high concentrations (50×, 10× and 2× dilutions), dilutions in the range 100× to 10× showed non-cytotoxicity to the cell lines related to skin tissue. Thus, this result indicates that Mel-loaded NISVs have a potent antibacterial activity on skin pathogenic bacteria and could be applied as a treatment for skin infections.
Ex Vivo Studies
Differentiation of Epidermis and Dermis Layers of Porcine Ear Skin
Porcine ear skin paraffin sections were histologically investigated and stained with PAS, which specifically binds to carbohydrates and carbohydrate-rich macromolecules, glycoproteins and basement membranes. On average, the epidermis of the porcine ear skin was 20 μm thick (Figure 9A). The obtained data provided useful information for further experiments.
 |
Figure 9 Histological images of porcine ear skin. (A) Differentiation of the epidermis and dermis compartment was carried out by PAS staining. The basement membrane region in the porcine ear skin is illustrated, and the epidermis and dermis layers of the skin section are indicated. The scale bar represents 20 μm. (B) Ex vivo penetration study at 2 and 4 hours post-application of Mel-loaded NISVs in a porcine skin model. The FITC-labeled Mel (green) was formulated and NISV formulation was labeled with NR (red). The overlay fluorescence images demonstrated the penetration of Mel-loaded NISVs into the skin (yellow). The cellular nuclei were counterstained by DAPI (blue). The scale bar represents 100 μm. (C) Orthogonal imaging analysis was performed to confirm the localization of Mel-loaded NISVs in the skin tissue (yellow). The depth of Mel-loaded NISV permeation reached up to 820 μm after 4 hours’ incubation. |
Skin Permeation of Mel-Loaded NISVs
CLSM studies were conducted to evaluate the extent of penetration and the transdermal potential of the permeated system, as indicated by an increase in the depth of penetration. The fluorescence images of the epidermis and dermis layers of porcine skin after treatment with Mel-loaded NISVs are shown in Figure 9B. The images show the penetration ability of Mel-loaded NISVs through the epidermis layer after 2 hours of treatment, extending to the deeper layers after the incubation time had increased to 4 hours. The Mel-loaded NISVs showed a great penetration ability up to 820 μm of depth, as demonstrated by the fluorescence intensity in the dermis layer. The orthogonal image analysis by z-stack scanning also confirmed the permeation of Mel via the niosomal nanocarrier (Figure 9C).
These results confirm that the niosomal nanocarrier system provided the ability to improve drug distribution in the stratum corneum (SC) and suggest that the Mel-loaded NISVs had an effective ability to permeate the epidermis and dermis.
Reduction of Bacterial Skin Infection by Mel-Loaded NISVs
To examine the effect of Mel-loaded NISVs on inhibition of bacterial skin infections, porcine ear models were used. In this study, inhibition of skin infections was studied on undamaged and burn wound skin. The skin samples were infected with FITC-labeled S. aureus ATCC 25923, followed by treatment with NR-labeled, Mel-loaded NISVs. Twenty hours after infection, the bacteria had colonized the models, predominantly at the surface of the undamaged skin or the wound bed, and disseminated into deeper skin layers of the wound burn skin. A progressive infection was demonstrated in the wounded skin, as evidenced by the fluorescence intensity of FITC-tagged S. aureus deeper in the model, whereas the fluorescence intensity appeared mainly at the surface and declined in the deeper layers of the undamaged skin (Figure 10A). This confirmed the fact that bacterial skin pathogens mainly infect the skin and soft tissue through breaches in the skin and via hair follicle routes.
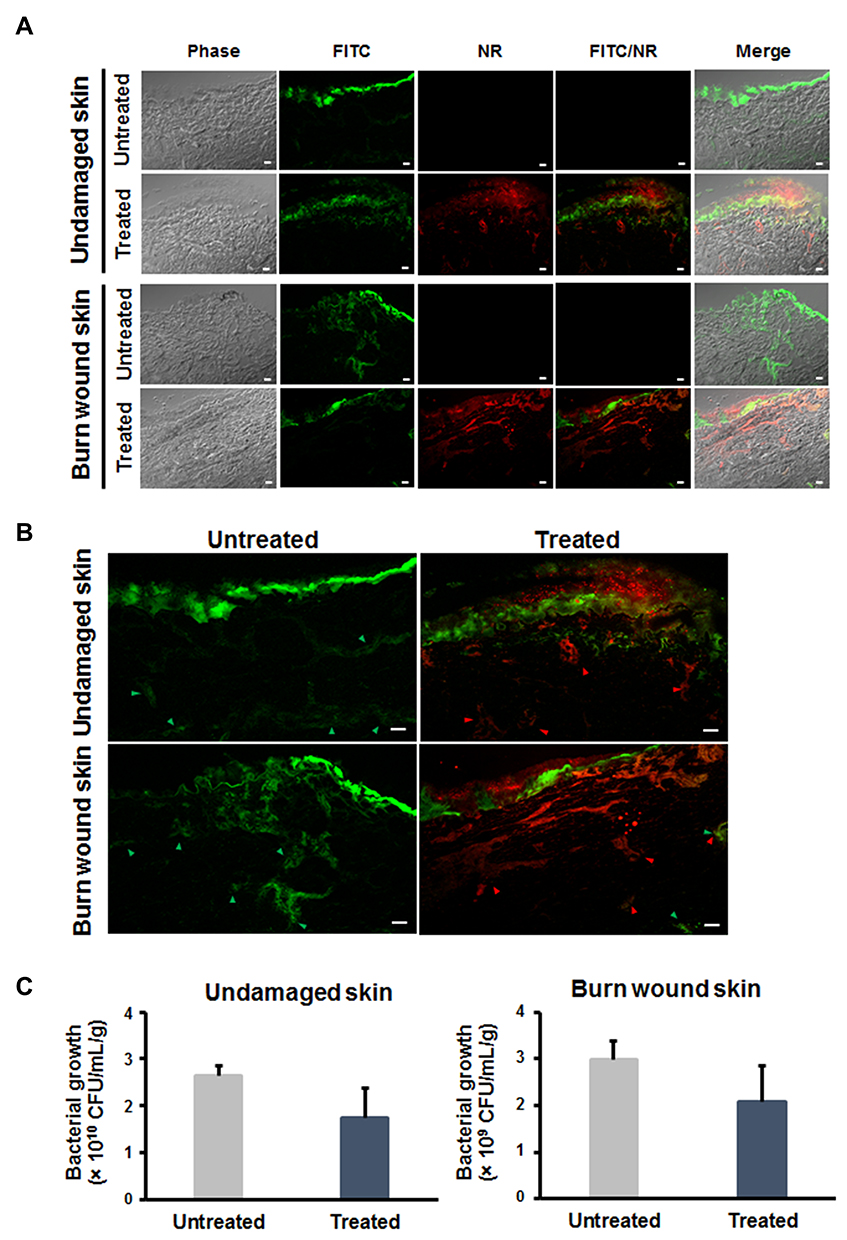 |
Figure 10 Inhibitory effect of Mel-loaded NISVs on S. aureus-infected skin models. (A) Undamaged and wound skin models were infected with FITC-labeled S. aureus (green) for 20 hours, followed by 4 hours’ treatment with Mel-loaded NISVs (red). The results were observed by CLSM. The scale bar represents 50 μm. Spreading of S. aureus infection (green arrowheads) in (B) undamaged and wound skin sections. Reduction of bacterial growth was found in Mel-loaded NISV treatment (red arrowheads). (C) Bacterial quantification of undamaged and wound skin infection. The scale bar represents 50 μm. The number of bacteria in the treated skin sample was determined by CFU count compared to the untreated skin (n=3). |
Administration of Mel-loaded NISVs on the infected skin sections for 4 hours showed a diminishing spread of infection. As illustrated in Figure 10B, the CLSM images showed a reduced amount of FITC-labeled S. aureus in the deeper layers of both undamaged and wound skin models. In particular, in areas that showed NR-labeled Mel-loaded NISVs, FITC-labeled S. aureus was not present. These results clearly show that S. aureus infection was limited after the application of Mel-loaded NISVs.
Quantitative Assessment of Bacterial Growth
Approximately 108 bacterial cells were inoculated onto the skin samples for 20 hours, followed by 4 hours’ treatment with Mel-loaded NISVs. The average CFU count rose to approximately 2.64×1010 and 3×109 CFU/mL/g tissue in undamaged and burn wound skin tissues, respectively, without the treatment of Mel-loaded NISVs. The level of bacterial growth in Mel-loaded NISV treated skin decreased to approximately 1.75×1010 CFU/mL/g tissue for undamaged samples and 2.09×109 CFU/mL/g tissue for burn wound skin. The growth inhibition of the treated sections was 33.70% and 30.33% for undamaged and wound skin samples, respectively (Figure 10C). Although the bacterial growth rate was not remarkably reduced, these results strongly implicated the influence of Mel-loaded NISVs on the inhibition of skin infection.
Discussion
The rapid emergence of bacterial resistance among S. aureus isolates, not only MRSA but also resistance to vancomycin (both VISA and VRSA), is causing serious problems and public health concerns.8,9 To provide an alternative strategy to combat infections caused by these pathogens, the exploration of effective antimicrobial substances is a definite requirement. Previous studies have demonstrated the antimicrobial activities of Mel against many microorganisms,15,16 including S. aureus and MRSA.17,48 However, its activity on VISA has not been investigated. In the current study, VISA were identified from MRSA clinical isolates and subsequently treated with Mel. The results showed that Mel had potent antimicrobial activity against all tested bacterial pathogens, especially the strains that were less susceptible to antibiotics.
The bacterial killing kinetics of Mel was assessed and showed a rapid elimination of tested bacteria, including S. aureus ATCC 25923, MRSA ATCC 43300 and VISA clinical isolates, with 99% reduction at 1×MIC within 1 hour and having completely killed the bacteria after 6 hours’ incubation. This rapid elimination by Mel represented a unique advantage of this peptide over conventional antibiotic drugs. This may lead to better treatment outcomes, especially in resistant strains. Understanding the mechanism of the antimicrobial activity of Mel on the bacteria is essential for furthering its development as a potential therapeutic option. Observation of Mel localization after treatment of the bacterial cells by CLSM, with z-stack scanning, demonstrated the accumulation of Mel at the bacterial membrane level (Figure 4). This supports previous findings indicating the membrane-targeting property of Mel.49 Several studies have described that antimicrobial peptides (AMPs) such as magainin and lacticin Q, including Mel, are commonly known for their non-receptor-mediated membrane lytic bactericidal activity, through transmembrane insertion from a surface-binding state when above a threshold peptide concentration. Thus, bilayer-spanning pores are created in the cell membranes,48–50 resulting in the loss of membrane integrity. This occurrence was confirmed by PI staining, as the membrane-impermeant dye passes through and intercalates with bacterial DNA. Noticeably, the RFI of PI was increased significantly (p<0.05) in Mel-treated VISA (Figure 5B). This finding might be explained by Gram-positive bacterial cell walls having a large amount of anionic polysaccharides (up to 60%) attached to the cytoplasmic membrane or the peptidoglycan layer,51,52 resulting in a negatively charged cell surface.53 Since a thickened cell wall and increased peptidoglycan multilayer has been highlighted as a characteristic phenotype commonly found in clinical VISA,8,52,54 this probably resulted in a more negative charge. Moreover, it is known that the mechanism of action of AMPs relies on the electrostatic interaction of their cationic nature with the negatively charged lipoteichoic acid (LTA) or teichoic acid (TA) of Gram-positive bacteria.12,14,55 Hence, the greater susceptibility of VISA to Mel was possibly induced by the anionic-rich membrane. As a lower rate of peptidoglycan cross-linking is also a common phenotypic change observed in VISA,6,56,57 this may affect the rearrangement and compactness of the cell-wall structure. Consequently, PI easily entered the cell wall and cytoplasmic membrane after structural destabilization by Mel. The results clearly demonstrated the fast permeabilization characteristics of Mel and indicated the anti-VISA activity of Mel. These effects could be described by many possible lethal mechanisms of bacteria. First, the orientation of a membrane-bound Mel is probably altered, leading to the Mel-induced transmembrane channels based on the toroidal pore model.18 This results in the loss of membrane integrity, leading to the leakage of intracellular molecules into the extracellular environment. Second, it was able to induce enzyme secretion, aggregation of membrane proteins and even lipid extraction from the membrane.13,14,50 Besides destruction of membrane, interaction with essential targets may induce cell stress and disrupt the intracellular mechanism of bacteria, including apoptosis-like processes activated by DNA damage.58
Although Mel exhibits potent biological properties, the application of Mel is limited by its toxicity and undesirable effects.22 Besides the unique characteristics of Mel, application of Mel in a topical dosage form remains a problem because of the barrier nature of the skin. The diffusional barrier nature of the skin restricts the entry of most drugs, resulting in a low penetration rate of hydrophilic substances.24,26,39,59 Several physical and chemical advanced techniques have been introduced to enhance percutaneous drug penetration.26,33,34 Among these, lipid-based formulations, such as liposomes and niosomes, have been extensively investigated and proved to be suitable to improve skin penetration.26 Unfortunately, the use of a liposome formulation is not suitable for Mel delivery because the phospholipid composition of the vesicle can be disrupted by Mel, leading to drug leakage.30,31 Hence, the niosomal vesicle approach, which is based mainly on the use of surfactant solutions, is the most promising solution. Niosomes, which are analogs of liposomes, are a self-assembly of non-ionic surfactants and lipids such as cholesterol, making them capable of entrapping both hydrophilic and lipophilic substances.33,39 Because the materials used to prepare niosomes are non-ionic surfactants, their vesicular structure shows better stability.60,61 In addition, non-ionic surfactants have no charged groups in their hydrophilic heads,33,36 and this characteristic may be favorable for the encapsulation of cationic peptides. Previously, a niosome system has been employed to entrap proteins and peptides.34,37–39 Also, it was demonstrated that co-encapsulation of recombinant lysostaphin and LL-37, a cationic antimicrobial peptide, was successfully entrapped by a niosomal system, and this formulation also had an anti-staphylococcal effect.38 Furthermore, Dabbagh Moghaddam et al demonstrated successful loading of Mel based on a niosome formulation, and its therapeutic effect for breast cancer through an apoptosis induction mechanism.32
It has been reported that a combination of Span and Tween can lead to the production of niosomes with highly stable membranes,38,62 and the addition of cholesterol could increase the rigidity and enable the formation of less leaky niosomes.63,64 In the current research, niosomal formulations were prepared using different ratios of surfactants (Tween 80 and Span 60) and cholesterol, followed by loading of Mel. In this formulation, Tween 80 has a large hydrophilic head group with high hydrophilic–lipophilic balance (HLB) (HLB=15), which cannot form vesicles without cholesterol.65 In contrast, a non-ionic surfactant with a single alkyl tail, such as Span 60, can form vesicular structures since it has a relatively large hydrophobic moiety (HLB=4.7) and low water solubility. The presence of proper amounts of cholesterol provided a suitable molecular geometry and hydrophobicity for bilayer vesicle formation and rendered the integrity of the vesicles.38,65,66 Thus, a mixture of Tween 80, Span 60 and cholesterol in this formulation could provide an equilibrium of chemical compositions for vesicle structure. Similarly, the work performed by Dabbagh Moghaddam et al demonstrated the success of Mel loaded within NISVs incorporating Tween 60 and Span 60 as a surfactant portion.32 Although there are differences in using Tween 80 (HLB=15) in our formulation instead Tween 60 (HLB=14.9), the HLB values of these surfactants are approximate. This may not distinctly affect their physicochemical characteristics. The results obtained with both formulations were similar in particle size, but different in the PDI and % EE. The difference in PDI, a parameter indicating the dispersion and homogeneity of the population of particles,67 may possibly be caused by several factors. For instance, the differences in the conditions used for the hydration (time and speed of evaporator) and sonication (time, power and amplitude) processes may affect the homogeneity of the particle population. Likewise, ZP values other than −30 mV to +30 mV are generally considered, since a small ZP value indicates the tendency of the particles aggregate.68 Regarding the difference in % EE, the concentration of Mel used in our formulation was smaller than in the other study. We used approximately 100–200 μg/mL of Mel loaded with the lipid phase, corresponding to a ratio of 0.5:10, while the other work used 1 mg/mL. This may haveresulted in the higher encapsulation capacity of our formulation.
The impact of particle size on the application of nanocarrier systems was explained earlier.67 It should be noted that transdermal delivery systems need a diameter of less than 300 nm to permeate into the deeper skin layers.67 In the present study, all of the formulations obtained from the mixture of surfactants and cholesterol were able to form vesicles of different sizes. The mean particle size of all formulations of empty NISVs was 100–180 nm, while that of Mel-loaded NISVs was 120–200 nm for formulations F1–F3 and 200–450 nm for F4. The particle size of each formulation was found to be different owing to variation in the composition of the formulation, and the larger size of Mel-loaded NISVs was caused by the inclusion of the Mel. In particular, in the F4 formulation, the amount of both Tween 80 and cholesterol was reduced, which may have resulted in the lower rigidity of the vesicle membrane when incorporated with Mel, resulting in the larger size of particles, which tended to aggregate easily. Considering the PDI, all formulations in this study showed a PDI in the range of 0.3–0.4. DLS analysis also showed a monomodal distribution, indicating a relatively homogeneously sized vesicle population.
The presence of charge on the surface of the vesicles produced a repulsive force between the vesicles. This made the vesicles stable, devoid of agglomeration and able to settle more quickly, and provided an evenly distributed suspension.68 In this study, the ZP of empty NISVs was negative, ranging from −7.7 to −9.0 mV. When Mel was added to the system, the ZP was −7.1 to −11.5 mV, which was not markedly altered. This may be due to the lower ratio of Mel:lipid phase, which did not affect the net charge of the niosomes. From this result, it can be inferred that Mel may be located in the aqueous core of the formulation or adsorbed on the membrane and/or the surface of the particles. However, because the charge values are not high enough to strongly induce repulsion, poor stability of the particles and aggregation when stored for a period of time may occur. To improve the stability, some modifications, such as the addition of charge-inducing agents, may be needed.
The EE of Mel in the niosomes was more than 90% for all formulations, as summarized in Table 3. The efficiency of drug entrapment depends on several factors, including the ratio of surfactant, the structure of the surfactant and the method of preparation. In addition, the EE could be dependent on the length of the surfactant, with long chains producing high entrapment.59,69 Moreover, it is possible that positively charged antimicrobial peptides lead to greater entrapment in anionic nanovesicles,70 leading to the high percentage of EE. Thus, it may be concluded that the high EE of Mel observed in this study resulted from the interaction of positively charged Mel (Supplementary data 2; Figure S2) with anionic niosomes. Similar results have been reported in other studies. For instance, the encapsulation of lysostaphin and LL-37 into negatively charged anionic niosomes (Span 60, Tween 60 and cholesterol) showed a high EE, up to 90%.38 This can also be explained by the fact that amphiphilic molecules bearing both highly hydrophilic groups and highly hydrophobic groups can form rigid membranes,65 and the addition of cholesterol could improve the stability of the niosomes by stabilization of the bilayers, prevention of drug leakage and restraining the permeation of molecules inside the aqueous cores.66 Therefore, the mixture of Tween 80 and Span 60 with cholesterol produced suitable hydrophobic and high hydrophilic properties, providing a system that was excellent for the entrapment of Mel.
From these results, formulation F2 was selected as the optimal niosomal formulation and its bioactivity was investigated in further experiments. The supporting information in terms of the physical morphology and chemical structure of Mel-loaded NISVs was obtained using SEM and FT-IR spectroscopy, respectively (Figure 6A and B). The SEM images of empty NISVs and Mel-loaded NISVs confirmed the nanometer size of niosomes and the average size of Mel-loaded NISVs, which was slightly larger than the empty NISVs. This was concordant with previous results analyzed by DLS. It was clear from the FT-IR spectra that the functional groups corresponding to Mel and empty NISVs were observed in Mel-loaded NISVs, even though the absorption signals of Mel-loaded NISVs slightly shifted from the characteristic signals of Mel. This is presumably due to the interaction between the amide group of Mel and the NISVs. This result indicated that Mel was successfully loaded into the NISVs.
The rate of drug release from drug delivery systems is critical and should be studied to achieve an optimal system with desirable release characteristics. In this study, release of Mel from NISVs appeared in a sustained manner. The initial burst release from niosomes occurred in the first 2 and 4 hours at pH 7.4 and 5.5, respectively, which may be due to the peptides available on the surface and inside the niosomal structure. Subsequently, the intense hydrophobic interactions between the lipid nanostructure and peptide, part of which remains inside the lipid core, leads to a reduction in release, which reaches a plateau. Similar release patterns were found in a previous study, which showed less cumulative drug release at a lower pH value.66 The release of molecules across a bilayer membrane depends on the composition and fluidity of the bilayer membrane.64,71 In addition, electrostatic interaction between drug and surfactants is an important factor72 that may cause a lower and slower release rate of Mel compared to neutral pH.
The ability of the vesicular carrier to aid Mel internalization in the cells was assessed in A375 cells. The images obtained for each marker displayed their location inside the cell, as illustrated in Figure 5A and B. When cells were incubated with Mel-loaded NISVs for 4 hours, Mel-loaded NISVs were already internalized and accumulated in the cytoplasm. Meanwhile, Mel was released from NISVs and distributed throughout the whole cytoplasm and nucleus. This event was also found in previous work.73 In agreement with other studies, our findings demonstrate that fusion of Mel-loaded NISVs with the cell membrane may be the mechanism responsible for the initial and rapid internalization that enabled cytoplasm saturation. Furthermore, many studies have also suggested that several entry mechanisms exist to explain vesicle uptake in a variety of cell types.73–75
Since Mel-loaded NISVs are aimed at being applied as topical dosage forms, cytotoxicity assays were carried out on NIH-3T3 and A375 cell lines. Cytotoxicity of empty NISVs was not observed on the tested cell lines in the dilution range from 100× to 10×, while at a higher dosage cell viability significantly (p<0.05) decreased (Figure 8A). This result reveals the low toxicity of NISV formulations in an in vitro model. The antibacterial activity of Mel-loaded NISVs was examined against S. aureus ATCC 25923, MRSA ATCC 43300 and VISA-87. The susceptibility study showed that Mel-loaded NISVs could effectively inhibit the growth of these bacterial strains (Figure 8B–D). It is noteworthy that the antimicrobial ability of Mel-loaded NISVs was more effective on VISA strains at a lower dose. This inhibitory effect on bacterial cells may be due to the interaction of the outer membrane of the bacteria with subsequent fusion of niosomes, which facilitates intracellular drug delivery through the membrane bilayer matrix (hydrophobic) pathway or the self-promoted route, usually used by cationic substances.38,76 Moreover, two main mechanisms of the enhanced antimicrobial activities of liposomal drug delivery, which are applicable to the niosome system, have been suggested, namely, the reduction of electrostatic repulsion of liposomal antibiotics from bacterial membranes and protection of the drug from bacterial enzymes.76,77
From these findings, it could be inferred that NISV provided a proper system for the entrapment of Mel, with a more stable structure than conventional liposomes, resulting in the prevention of leakage of Mel. Importantly, the adverse effects of Mel involving lytic activity on human erythrocytes and peripheral blood lymphocytes could be minimized. This statement can be supported by a previous study, which mentioned the absence of toxicity and hemolytic effects in Mel-loaded NISVs compared to free Mel.32 Thus, NISV has a potential ability to improve the limitations of Mel application. Meanwhile, using Mel-loaded NISV enhanced the effective activity to reduce the growth of pathogens, especially drug-resistant strains, with a small dosage which reduced the adverse effects caused by Mel.
In this work, we intended to prepare Mel-loaded NISVs for transdermal delivery. Surfactants could act as percutaneous permeation enhancers through adsorption at interfaces, interaction with biological membranes and alteration of the barrier function of the SC, as a result of reversible lipid modification.59,78 It has been reported that the intercellular lipid barrier in the SC can be dramatically changed to be more permeable when treated with niosomes.36,59 Therefore, the interaction between the skin and niosomes may be conducive to the improvement of transdermal drug delivery.
A study in an ex vivo skin model was conducted on porcine ear skin, which has similar histological characteristics to human skin, presenting similar SC thickness and hair follicle density.44,79 As shown in Figure 7, the Mel-loaded NISVs were mainly detected in the epidermis after 2 hours’ incubation, and increased penetration into deeper skin layers was seen when incubation time was longer, up to 4 hours. This result may be explained by the effect of the vesicle size and chemical structure of the surfactants, which reversibly altered the permeability of the SC, induced by phospholipids.59 Besides, cholesterol is present in the cell membrane and relatively enriched in the skin, which may enhance absorption through the skin surface and fusion with the SC, and promote drug release from the niosomes.80 Thereby, the diffusion of Mel-loaded NISVs into the hydrophilic dermal layer was promoted.
The inhibitory effect of Mel-loaded NISVs on a S. aureus skin infection was investigated in undamaged and wound burn models. Colonization by bacteria was seen predominantly on the surface and epidermis layer of the skin after infection, while the dissemination of the pathogen into deeper layers was evidenced in the wound skin model. This can be explained by the fact that bacterial skin infections usually occur through a breach of skin and via hair follicle routes. Thus, invasive infection in this study was observed in the burn wound model, which was ruptured and lost its barrier ability. When the infected skin was treated with Mel-loaded NISVs, the number of bacterial cells on the skin surface decreased. Meanwhile, in the deeper layer, the presence of bacteria was not found in the same area as the Mel-loaded NISV distribution. This phenomenon was demonstrated in both undamaged and damaged skin models, as shown in Figure 10. In addition, the rate of bacterial growth was inhibited, by approximately 30%, upon treatment with Mel-loaded NISVs in both undamaged and wound skin models. This was an indication of the influence of Mel-loaded NISVs on the inhibition of skin infections with a single treatment. This was a proof-of-concept study, which has provided useful pilot data for the improvement of drug regimens or treatment procedures in further investigations. To avoid deactivation of the entrapped drug, development of the formulation with regard to drug stability and skin proteases is required,81 and its pharmacokinetics also need to be explored.
Conclusion
In this study, the inhibitory effect of Mel against VISA was investigated for the first time. Surprisingly, compared with S. aureus and MRSA, VISA was more susceptible to Mel. Moreover, we also successfully formulated Mel-loaded NISVs with favorable physicochemical properties and demonstrated the sustained release of Mel. In addition, the assessment of biological properties indicated the low cytotoxicity and biocompatibility of the formulation. Mel-loaded NISVs were able to permeate the bilayer cell membrane and were distributed throughout the intracellular space. Taken together, these results show that Mel-loaded NISVs exhibited an effective activity against antibiotic-susceptible and antibiotic-resistant S. aureus. This formulation also provided an excellent permeation ability and was able to restrict dissemination of bacterial infection in ex vivo porcine skin models. This current observation implies that Mel-loaded NISVs have the potential to be further developed for skin treatment and may be a candidate for topical application.
Acknowledgments
This research was funded by the Research and Researchers for Industries (RRi) from the Thailand Science Research and Innovation (TSRI) [grant number PhD59I0055], the National Research Council of Thailand (NRCT), and funding for Teaching Assistant and Research Assistant (TA/RA) Scholarships from The Graduate School, Chiang Mai University. This research work was also partially supported by Chiang Mai University.
We thank Prof. Dr Watchara Kasinrerk from the Biomedical Technology Research Center, National Center for Genetic Engineering and Biotechnology, National Sciences and Technology Development Agency at the Faculty of Associated Medical Sciences, Chiang Mai University, Chiang Mai, Thailand, for assisting us with the fluorescence microscopy. We thank Ms Supattara Suwanpairoj, application specialist, Rushmore Precision Co., Ltd, Bangkok, Thailand, for supporting confocal laser scanning microscopy (LSM 800 with Airyscan), and Mr Supakorn Sirinutsomboon and Ms Sudarat Iemtod, product specialist, Scientific Instrument Division, Hollywood International Co., Ltd, Bangkok, Thailand, for supporting 3D holographic microscopy and 3D imaging analysis.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Ibrahim F, Khan T, Pujalte GG. Bacterial skin infections. Prim Care. 2015;42(4):485–499. doi:10.1016/j.pop.2015.08.001
2. Cardona AF, Wilson SE. Skin and soft-tissue infections: a critical review and the role of telavancin in their treatment. Clin Infect Dis. 2015;61(suppl 2):69–78. doi:10.1093/cid/civ528
3. Baldwin HE, Bhatia ND, Friedman A, et al. The role of cutaneous microbiota harmony in maintaining a functional skin barrier. J Drugs Dermatol. 2017;16:12–18.
4. Lacey KA, Geoghegan JA, McLoughlin RM. The role of Staphylococcus aureus virulence factors in skin infection and their potential as vaccine antigens. Pathogens. 2016;5(1):1–17. doi:10.3390/pathogens5010022
5. Russo A, Concia E, Cristini F, et al. Current and future trends in antibiotic therapy of acute bacterial skin and skin-structure infections. Clin Microbiol Infect. 2016;22:27–36. doi:10.1016/S1198-743X(16)30095-7
6. Dhanda Dhanda G, Sarkar P, Samaddar S, et al. Battle against vancomycin-resistant bacteria: recent developments in chemical strategies. J Med Chem. 2019;62(7):3184–3205. doi:10.1021/acs.jmedchem.8b01093
7. Craft KM, Nguyen JM, Berg LJ, et al. Methicillin-resistant Staphylococcus aureus (MRSA): antibiotic-resistance and the biofilm phenotype. Medchemcomm. 2019;10(8):1231–1241. doi:10.1039/C9MD00044E
8. Guo Y, Song G, Sun M, et al. Prevalence and therapies of antibiotic-resistance in Staphylococcus aureus. Front Cell Infect Microbiol. 2020;10:1–11. doi:10.3389/fcimb.2020.00107
9. Rengaraj R, Mariappan S, Sekar U, et al. Detection of vancomycin resistance among Enterococcus faecalis and Staphylococcus aureus. J Clin Diagn Res. 2016;10:4–6.
10. Aufschnaiter A, Kohler V, Khalifa S, et al. Apitoxin and its components against cancer, neurodegeneration and rheumatoid arthritis: limitations and possibilities. Toxins. 2020;12:1–15.
11. Somwongin S, Chantawannakul P, Chaiyana W. Antioxidant activity and irritation property of venoms from Apis species. Toxicon. 2018;145:32–39. doi:10.1016/j.toxicon.2018.02.049
12. Miyazaki Y, Okazaki S, Shinoda W. Free energy analysis of membrane pore formation process in the presence of multiple melittin peptides. Biochim Biophys Acta Biomembr. 2019;1861(7):1409–1419. doi:10.1016/j.bbamem.2019.03.002
13. Gajski G, Domijan AM, Zegura B, et al. Melittin induced cytogenetic damage, oxidative stress and changes in gene expression in human peripheral blood lymphocytes. Toxicon. 2016;110:56–67. doi:10.1016/j.toxicon.2015.12.005
14. Therrien A, Fournier A, Lafleur M. Role of the cationic c-terminal segment of melittin on membrane fragmentation. J Phys Chem B. 2016;120(17):3993–4002. doi:10.1021/acs.jpcb.5b11705
15. Dosler S, Karaaslan E, Gerceker A, et al. Antibacterial and anti-biofilm activities of melittin and colistin, alone and in combination with antibiotics against Gram-negative bacteria. J Chemother. 2016;28(2):95–103. doi:10.1179/1973947815Y.0000000004
16. Leandro LF, Mendes CA, Casemiro LA, et al. Antimicrobial activity of apitoxin, melittin and phospholipase A2 of honey bee (Apis mellifera) venom against oral pathogens. An Acad Bras Cienc. 2015;87(1):147–155. doi:10.1590/0001-3765201520130511
17. Choi JH, Jang AY, Lin S, et al. Melittin, a honeybee venom-derived antimicrobial peptide, may target methicillin-resistant Staphylococcus aureus. Mol Med Rep. 2015;12(5):6483–6490. doi:10.3892/mmr.2015.4275
18. Hong J, Lu X, Deng Z, et al. How melittin inserts into cell membrane: conformational changes, inter-peptide cooperation, and disturbance on the membrane. Molecules. 2019;24(9):1–17. doi:10.3390/molecules24091775
19. Jozefiak A, Engberg RM. Insect proteins as a potential source of antimicrobial peptides in livestock production. A review. J Anim Feed Sci. 2017;26(2):87–99. doi:10.22358/jafs/69998/2017
20. Nguyen MHL, DiPasquale M, Rickeard BW, et al. Time-resolved SANS reveals pore-forming peptides cause rapid lipid reorganization. New J Chem. 2020;45(1):447–456. doi:10.1039/D0NJ04717A
21. Pucca MB, Cerni AF, Oliveira IS, et al. Bee updated: current knowledge on bee venom and bee envenoming therapy. Front Immunol. 2019;10:1–15.
22. Lee G, Bae H. Anti-inflammatory applications of melittin, a major component of bee venom: detailed mechanism of action and adverse effects. Molecules. 2016;21(5):1–10. doi:10.3390/molecules21050616
23. Chamundeeswari M, Jeslin J, Verma ML. Nanocarriers for drug delivery applications. Environ Chem Lett. 2019;17(2):849–865. doi:10.1007/s10311-018-00841-1
24. Alkilani AZ, McCrudden MT, Donnelly RF. Transdermal drug delivery: innovative pharmaceutical developments based on disruption of the barrier properties of the stratum corneum. Pharmaceutics. 2015;7:438–470.
25. Schoellhammer CM, Blankschtein D, Langer R. Skin permeabilization for transdermal drug delivery: recent advances and future prospects. Expert Opin Drug Deliv. 2014;11(3):393–407. doi:10.1517/17425247.2014.875528
26. Jijie R, Barras A, Boukherroub R, et al. Nanomaterials for transdermal drug delivery: beyond the state of the art of liposomal structures. J Mater Chem B. 2017;5:8653–8675.
27. Yang L, Cui F, Shi K, et al. Design of high payload PLGA nanoparticles containing melittin/sodium dodecyl sulfate complex by the hydrophobic ion-pairing technique. Drug Dev Ind Pharm. 2009;35(8):959–968. doi:10.1080/03639040902718039
28. Zetterberg MM, Reijmar K, Pranting M, et al. PEG-stabilized lipid disks as carriers for amphiphilic antimicrobial peptides. J Control Release. 2011;156(3):323–328. doi:10.1016/j.jconrel.2011.08.029
29. Mao J, Liu S, Ai M, et al. A novel melittin nano-liposome exerted excellent anti-hepatocellular carcinoma efficacy with better biological safety. J Hematol Oncol. 2017;10(1):1–4. doi:10.1186/s13045-017-0442-y
30. Tian JL, Ke X, Chen Z, et al. Melittin liposomes surface modified with poloxamer 188: in vitro characterization and in vivo evaluation. Pharmazie. 2011;65:362–367.
31. Li Y, Ruan S, Wang Z, et al. Hyaluronic acid coating reduces the leakage of melittin encapsulated in liposomes and increases targeted delivery to melanoma cells. Pharmaceutics. 2021;13(8):1–14. doi:10.3390/pharmaceutics13081235
32. Dabbagh Moghaddam F, Akbarzadeh I, Marzbankia E, et al. Delivery of melittin-loaded niosomes for breast cancer treatment: an in vitro and in vivo evaluation of anti-cancer effect. Cancer Nano. 2021;12(1):1–35. doi:10.1186/s12645-021-00085-9
33. Moghassemi S, Hadjizadeh A. Nano-niosomes as nanoscale drug delivery systems: an illustrated review. J Control Release. 2014;185:22–36. doi:10.1016/j.jconrel.2014.04.015
34. Ge X, Wei M, He S, et al. Advances of non-ionic surfactant vesicles (niosomes) and their application in drug delivery. Pharmaceutics. 2019;11(2):1–16. doi:10.3390/pharmaceutics11020055
35. Seleci DA, Seleci M, Walter J, et al. Niosomes as nanoparticular drug carriers: fundamentals and recent applications. J Nanomater. 2016;2016:1–13. doi:10.1155/2016/7372306
36. Kumar GP, Rajeshwarrao P. Nonionic surfactant vesicular systems for effective drug delivery-An overview. Acta Pharm Sin B. 2011;1(4):208–219. doi:10.1016/j.apsb.2011.09.002
37. Dufes C, Gaillard F, Uchegbu IF, et al. Glucose-targeted niosomes deliver vasoactive intestinal peptide (VIP) to the brain. Int J Pharm. 2004;285(1–2):77–85. doi:10.1016/j.ijpharm.2004.07.020
38. Sadeghi Bakhshandeh H, Ahangari Cohan R, Ahangari Cohan R, et al. Synergistic anti-Staphylococcal activity of niosomal recombinant lysostaphin-LL-37. Int J Nanomedicine. 2019;14:9777–9792. doi:10.2147/IJN.S230269
39. Muzzalupo R, Tavano L. Niosomal drug delivery for transdermal targeting recent advances. Dovepress. 2015;4:23–33.
40. Lu X, Jin X, Zhu J. Flow cytometry and electron microscopy study of Staphylococcus aureus and Escherichia coli treated with Mdc-Hly. Microsc Microanal. 2015;21(2):351–357. doi:10.1017/S1431927615000203
41. Obeid MA, Gebril AM, Tate RJ, et al. Comparison of the physical characteristics of monodisperse non-ionic surfactant vesicles (NISV) prepared using different manufacturing methods. Int J Pharm. 2017;521(1–2):54–60. doi:10.1016/j.ijpharm.2017.02.007
42. Akbarzadeh I, Yaraki MT, Bourbour M, et al. Optimized doxycycline-loaded niosomal formulation for treatment of infection-associated prostate cancer: an in-vitro investigation. J Drug Deliv Sci Technol. 2020;57:1–12.
43. Jantakee K, Prapan A, Chaiwaree S, et al. Fabrication and characterization of human serum albumin particles loaded with non-sericin extract obtained from silk cocoon as a carrier system for hydrophobic substances. Polymers. 2021;13(3):1–17. doi:10.3390/polym13030334
44. Alves DR, Booth AP, Scavone P, et al. Development of a high-throughput ex-vivo burn wound model using porcine skin, and its application to evaluate new approaches to control wound infection. Front Cell Infect Microbiol. 2018;8:1–15. doi:10.3389/fcimb.2018.00196
45. Al Kindi A, Alkahtani AM, Nalubega M, et al. Staphylococcus aureus internalized by skin keratinocytes evade antibiotic killing. Front Microbiol. 2019;10:1–10. doi:10.3389/fmicb.2019.02242
46. Peng HT, Huang H, Shek PN. PEGylation of melittin: structural characterization and hemostatic effects. J Bioact Compat Polym. 2010;25(1):75–97. doi:10.1177/0883911509354230
47. Rehman MU, Rasul A, Khan MI, et al. Development of niosomal formulations loaded with cyclosporine A and evaluation of its compatibility. Trop J Pharm Res. 2018;17(8):1457–1464. doi:10.4314/tjpr.v17i8.1
48. Han SM, Kim JM, Hong IP, et al. Antibacterial activity and antibiotic-enhancing effects of honeybee venom against methicillin-resistant Staphylococcus aureus. Molecules. 2016;21(1):1–9. doi:10.3390/molecules21010079
49. Le CF, Fang CM, Sekaran SD. Intracellular targeting mechanisms by antimicrobial peptides. Antimicrob Agents Chemother. 2017;61(4):1–16. doi:10.1128/AAC.02340-16
50. Lu X, Liu J, Gou L, et al. Designing melittin-graphene hybrid complexes for enhanced antibacterial activity. Adv Healthc Mater. 2019;1801521:1–10.
51. Maria-Neto S, de Almeida KC, Macedo ML, et al. Understanding bacterial resistance to antimicrobial peptides: from the surface to deep inside. Biochim Biophys Acta. 2015;1848(11):3078–3088. doi:10.1016/j.bbamem.2015.02.017
52. Howden BP, Davies JK, Johnson PD, et al. Reduced vancomycin susceptibility in Staphylococcus aureus, including vancomycin-intermediate and heterogeneous vancomycin-intermediate strains: resistance mechanisms, laboratory detection, and clinical implications. Clin Microbiol Rev. 2010;23:99–139.
53. Gottenbos B, Grijpma DW, van der Mei HC, et al. Antimicrobial effects of positively charged surfaces on adhering Gram-positive and Gram-negative bacteria. J Antimicrob Chemother. 2001;48(1):7–13. doi:10.1093/jac/48.1.7
54. Nannini E, Murray BE, Arias CA. Resistance or decreased susceptibility to glycopeptides, daptomycin, and linezolid in methicillin-resistant Staphylococcus aureus. Curr Opin Pharmacol. 2010;10(5):516–521. doi:10.1016/j.coph.2010.06.006
55. Yasir M, Dutta D, Willcox MDP. Mode of action of the antimicrobial peptide Mel4 is independent of Staphylococcus aureus cell membrane permeability. PLoS One. 2019;14(7):1–22. doi:10.1371/journal.pone.0215703
56. Sieradzki K, Tomasz A. Alterations of cell wall structure and MetabolismAccompany reduced susceptibility to vancomycin in an IsogenicSeries of clinical isolates of Staphylococcus aureus. J Bacteriol. 2003;185(24):7103–7110. doi:10.1128/JB.185.24.7103-7110.2003
57. Hu Q, Peng H, Rao X. Molecular events for promotion of vancomycin resistance in vancomycin intermediate Staphylococcus aureus. Front Microbiol. 2016;7:1–18. doi:10.3389/fmicb.2016.01601
58. Bayles KW. Bacterial programmed cell death: making sense of a paradox. Nat Rev Microbiol. 2014;12(1):63–69. doi:10.1038/nrmicro3136
59. Muzzalupo R, Tavano L, Cassano R, et al. A new approach for the evaluation of niosomes as effective transdermal drug delivery systems. Eur J Pharm Biopharm. 2011;79(1):28–35. doi:10.1016/j.ejpb.2011.01.020
60. Yeo PL, Lim CL, Chye SM, et al. Niosomes: a review of their structure, properties, methods of preparation, and medical applications. Asian Biomed. 2017;11(4):301–314. doi:10.1515/abm-2018-0002
61. Bartelds R, Nematollahi MH, Pols T, et al. Niosomes, an alternative for liposomal delivery. PLoS One. 2018;13(4):e0194179. doi:10.1371/journal.pone.0194179
62. Liu T, Guo R. Investigation of PEG 6000/Tween 80/Span 80/H2O niosome microstructure. Colloid Polym Sci. 2007;285(6):711–713. doi:10.1007/s00396-006-1627-z
63. Ertekin ZC, Bayindir ZS, Yuksel N. Stability studies on piroxicam encapsulated niosomes. Curr Drug Deliv. 2015;12(2):192–199. doi:10.2174/1567201811666140723115852
64. Ruckmani K, Sankar V. Formulation and optimization of zidovudine niosomes. AAPS PharmSciTech. 2010;11:1119–1127.
65. Junyaprasert VB, Singhsa P, Suksiriworapong J, et al. Physicochemical properties and skin permeation of Span 60/Tween 60 niosomes of ellagic acid. Int J Pharm. 2012;423(2):303–311. doi:10.1016/j.ijpharm.2011.11.032
66. Jin Y, Wen J, Garg S, et al. Development of a novel niosomal system for oral delivery of Ginkgo biloba extract. Int J Nanomedicine. 2013;8:421–430. doi:10.2147/IJN.S37984
67. Danaei M, Dehghankhold M, Ataei S, et al. Impact of particle size and polydispersity index on the clinical applications of lipidic nanocarrier systems. Pharmaceutics. 2018;10(2):1–7. doi:10.3390/pharmaceutics10020057
68. Joseph E, Singhvi G. Multifunctional nanocrystals for cancer therapy: a potential nanocarrier. In: Grumezescu AM, editor. Nanomaterials for Drug Delivery and Therapy. William and Andrew: Applied Science Publisher; 2019:91–116.
69. Abdelbary G, El-Gendy N. Niosome-encapsulated gentamicin for ophthalmic controlled delivery. AAPS PharmSciTech. 2008;9(3):740–747. doi:10.1208/s12249-008-9105-1
70. Colas JC, Shi W, Rao VS, et al. Microscopical investigations of nisin-loaded nanoliposomes prepared by Mozafari method and their bacterial targeting. Micron. 2007;38:841–847.
71. Muller RH, Mader K, Gohla S. Solid lipid nanoparticles (SLN) for controlled drug delivery - a review of the state of the art. Eur J Pharm Biopharm. 2000;50(1):161–177. doi:10.1016/S0939-6411(00)00087-4
72. Tavano L, Muzzalupo R, Picci N, et al. Co-encapsulation of antioxidants into niosomal carriers: gastrointestinal release studies for nutraceutical applications. Colloids Surf B Biointerfaces. 2014;114:82–88. doi:10.1016/j.colsurfb.2013.09.058
73. Manca ML, Manconi M, Nacher A, et al. Development of novel diolein-niosomes for cutaneous delivery of tretinoin: influence of formulation and in vitro assessment. Int J Pharm. 2014;477(1–2):176–186. doi:10.1016/j.ijpharm.2014.10.031
74. Papakostas D, Rancan F, Sterry W, et al. Nanoparticles in dermatology. Arch Dermatol Res. 2011;303(8):533–550. doi:10.1007/s00403-011-1163-7
75. Rejman J, Oberle V, Zuhorn IS, et al. Size-dependent internalization of particles via the pathways of clathrin- and caveolae-mediated endocytosis. Biochem J. 2004;377(1):159–169. doi:10.1042/bj20031253
76. Satish J, Amusa AS, Gopalakrishna P. In vitro activities of fluoroquinolones entrapped in non-ionic surfactant vesicles against ciprofloxacin-resistant bacteria strains. IJPSDR. 2012;1:1–11.
77. Mugabe C, Halwani M, Azghani AO, et al. Mechanism of enhanced activity of liposome-entrapped aminoglycosides against resistant strains of Pseudomonas aeruginosa. Antimicrob Agents Chemother. 2006;50(6):2016–2022. doi:10.1128/AAC.01547-05
78. Choi MJ, Maibach HI. Liposomes and niosomes as topical drug delivery systems. Skin Pharmacol Physiol. 2005;18(5):209–219. doi:10.1159/000086666
79. Praca FSG, Medina WSG, Eloy JO, et al. Evaluation of critical parameters for in vitro skin permeation and penetration studies using animal skin models. Eur J Pharm Sci. 2018;111:121–132. doi:10.1016/j.ejps.2017.09.034
80. Zhang Y, Zhang K, Wu Z, et al. Evaluation of transdermal salidroside delivery using niosomes via in vitro cellular uptake. Int J Pharm. 2015;478(1):138–146. doi:10.1016/j.ijpharm.2014.11.018
81. Dubey S, Perozzo R, Scapozza L, et al. Using protease inhibitors to improve protein stability in the presence of skin: a case study on the stability of insulin like growth factor 1. Eur J Pharm Biopharm. 2021;158:379–438. doi:10.1016/j.ejpb.2020.12.009
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.