Back to Journals » Diabetes, Metabolic Syndrome and Obesity » Volume 16
Potential Roles of mtDNA Mutations in PCOS-IR: A Review
Authors Dong XC ![]() , Liu C, Zhuo GC, Ding Y
, Liu C, Zhuo GC, Ding Y ![]()
Received 25 November 2022
Accepted for publication 9 January 2023
Published 25 January 2023 Volume 2023:16 Pages 139—149
DOI https://doi.org/10.2147/DMSO.S393960
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Juei-Tang Cheng
Xiao-Chao Dong,1 Chang Liu,1 Guang-Chao Zhuo,2 Yu Ding2
1Department of Gynecology and Obstetrics, Hangzhou First People’s Hospital, Zhejiang University School of Medicine, Hangzhou, People’s Republic of China; 2Central Laboratory, Hangzhou First People’s Hospital, Zhejiang University School of Medicine, Hangzhou, People’s Republic of China
Correspondence: Yu Ding, Central Laboratory, Hangzhou First People’s Hospital, Zhejiang University School of Medicine, 261 Huansha Road, Hangzhou, People’s Republic of China, Tel/Fax +86-571-5600-5600, Email [email protected]
Abstract: Polycystic ovary syndrome (PCOS) is the most common heterogeneous endocrine disease that affecting females in reproductive age. Insulin resistance (IR), an important molecular basis for PCOS, accounts for at least 75% of women carrying this syndrome. Although there have been many studies on PCOS-IR, the detailed mechanisms are not fully understood. As essential hub for energy generation, mitochondria are critical to insulin secretion and normal function, whereas mutations in mitochondrial DNA (mtDNA) result in mitochondrial dysfunctions contributing to the pathophysiology of PCOS-IR via the regulation of balance of oxidative stress (OS), energy deficiency, or hormone metabolism. In the current review, we summarize the clinical and molecular features of PCOS-IR and discuss molecular mechanisms related to mtDNA mutations.
Keywords: PCOS-IR, mitochondrial dysfunction, mtDNA mutations
Introduction
Polycystic ovary syndrome (PCOS) is the most common endocrine disease occurring during reproductive years. It was a kind of endocrine and metabolic disorders that results in obesity, irregularity of menstruation, OS, hyperinsulinemia, hyperandrogenism, infertility, and sterility.1,2 First identified in 1935, PCOS was also recognized as Stein–Leventhal syndrome.3 Diagnosis of PCOS in adults can be made when at least two of three criteria are met: impairment of ovarian function, clinical and/or biochemical hyperandrogenism, and polycystic ovaries.4,5 Despite significant progress in diagnostic criteria for PCOS, the syndrome is still underdiagnosed or misunderstood by many practitioners.6
In the early stage, PCOS is often complicated with infertility and adverse pregnancy outcomes, while in the long term, the incidence of endometrial cancer, type 2 diabetes mellitus (T2DM), and cardiovascular diseases gradually increase, seriously harming women’s physical and mental health. Based on the National Institutes of Health’s diagnostic criteria, the geographical prevalence of this disease is 8.7%, 17.8%, and 12% based on the definition proposed by the Androgen Excess and PCOS Society.7 In a 2019 study, the prevalence of PCOS in Chinese women of reproductive age was 5.6%, which was consistent with other studies,8 while its incidence in Indian women was 9.13% according to a recent study.9 Interestingly, PCOS seems to be more frequent in black women (8.0%) than white women (4.8%), with an incidence of 6.6%.10 The prevalence of oligoanovulation and hyperandrogenemia is 56.6% and 60% among women with PCOS, respectively.11
Multiple morbidities are linked to PCOS, such as infertility, impairment of glucose tolerance, T2DM, coronary heart disease, depression, gynecological oncology, and nonalcoholic fatty-liver disease.12 Despite these well-characterized phenotypes, the pathogenesis of PCOS remains unclear. Increasing evidence suggests that genetic, epigenetic, and environmental factors contribute to PCOS progression.13 However, it was generally accepted that IR and hyperandrogenism play key roles in its etiology.14
The Role of IR in PCOS
Insulin is the master regulator of glucose metabolism. This hormone works under the condition of glucose uptake by insulin-sensitive tissue (muscle, liver, and adipose).15,16 IR is caused by defects in insulin signaling, reducing the ability of insulin to stimulate glucose utilization, and can thus lead to high insulin levels (hyperinsulinemia). It has been suggested that >75% of patients have associated IR.17
At the molecular level, IR and hyperinsulinemia may stimulate P450c17α and influence the activity of 17-hydroxylase and 17,20-lyase.18 Subsequently, these biochemical processes promote the secretion of androgen, increase free-androgen levels, and inhibit insulin signal transduction and translocation of glucose transporter 4, which affects glucose and lipid metabolism.19 Androgens can produce IR by directly affecting insulin action in skeletal muscle and adipose tissue, changing adipokine secretion and increasing visceral adiposity. Moreover, insulin and IGF120 synergize with luteinizing hormone (LH).21 Hyperinsulinemia enhances LH binding and androgen-producing response to LH.22 Hyperinsulinemia also reduces hepatic sex hormone–binding globulin,23–25 increasing free-testosterone levels in the blood and thus contributing to PCOS phenotypes (Figures 1 and 2).
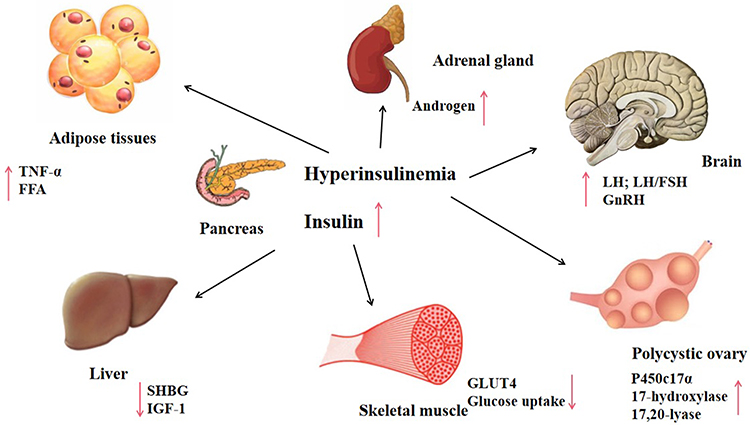 |
Figure 1 Influence of hyperinsulinemia on various human organs. |
 |
Figure 2 Summarized scheme of the pathophysiology of PCOS. Abbreviations: GnRH, gonadotropin-releasing hormone; LH, luteinizing hormone; FSH, follicule-stimulating hormone; SHBG, sex hormone –binding globulin. |
Mitochondrial Structure and Function
Mitochondria are very important organelles consisting an of outer membrane, intermembrane space, and inner membrane that surround the matrix. Structurally, the inner membrane is tightlyfolded and is the major site for electron-transport chain (ETC) (complexes I–IV), which are essential for oxygen consumption in mammalian cells.26 Among these, complex I — nicotinamide adenine dinucleotide Q (NADH-Q) oxidoreductase, comprises enzymes consisting of iron sulfur and flavin mononucleotide.27 Complex II, also known as succinate dehydrogenase (SDH), contains four nuclear encoded subunits: SDHA, SDHB, SDHC, and SDHD. Interestingly, complex II has a dual role, ie, in the ETC. and the tricarboxylic acid cycle, linking the two essential energy-producing processes of the cell.28,29 Complex I and II oxidize NADH and flavin adenine dinucleotide 2, respectively, transferring the resulting electrons to ubiquinol, which carries electrons to complex III. Complex III shunts the electrons across the intermembrane space to cytochrome C, which brings electrons to complex IV.30,31 Complex IV then uses the electrons to reduce oxygen to water. There are many enzymes located within the mitochondrial matrix that are critical for metabolic pathways, including tricarboxylic acid cycle or β-oxidation.
Mitochondria are also important for the maintenance of cellular energy homeostasis. They are often called the powerhouses of the cell because of their significant role in the supplementation of ATP via oxidative phosphorylation (OxPhos). In contrast, mitochondria also generate reactive oxygen species (ROS) through ETC complexes, and excess ROS production will induce OS and cause mitochondrial dysfunctions.32 Mitochondria contain their own genetic material, mtDNA, which encodes seven genes of the ETC complex I: one for ETC complex III, three for ETC complex IV, and two for ETC complex V.33 The rest of the mitochondrial proteins are encoded by nuclear genes (Figure 3).34
 |
Figure 3 Genetic map of human mitochondrial genome, which has a 16,569 bp sequence. Red boxes indicated PCOS-IR–associated mtDNA mutations. |
Homoplasmy and Heteroplasmy
mtDNA has a very high sequence-evolution rate, in part because it is exposed to ROS. mtDNA mutations include point mutations, deletions, and insertions. mtDNA mutations can be either homoplasmic or heteroplasmic when just one or more than two variants exist, respectively.35,36 The heteroplasmic level of a certain mtDNA mutation is critical in clinical phenotypes.36
Heteroplasmic mtDNA mutations are frequently associated with human pathologies because they cause more severe mitochondrial dysfunction than homoplasmic mtDNA mutations. Under normal conditions, mtDNA can “repair” mitochondrial dysfunction. When it comes to a certain heteroplasmic level, nevertheless, such compensation will be insufficient and lead to clinical expression of disease (Figure 4).37,38
 |
Figure 4 Heteroplasmy and the threshold effect. |
OS and PCOS-IR
Because mitochondria are the major sites for ROS generation, overproduction of ROS will lead to OS and consequent imunbalance between the oxidant and antioxidant systems.39 This imbalance may be caused by several metabolic activities, including obesity, hyperinsulinemia, and dyslipidemia.40 The predominant ROS in the mitochondria are superoxide anions (O2−), which are produced by the leakage of electrons from the ETC, which can then react with O2.41
Endometrial IR is linked to hyperandrogenemia, obesity, and inflammation and strongly associated with OS, resulting in an upregulation of OS caused by excessive ROS and pregnancy impairment.42 OS can lead to IR through the impairment of insulin signaling and causing adipokine dysregulation.43 OS also regulates some classical signaling pathways, such as NFκB and JNK, which in turn phosphorylate insulin-receptor substrate proteins and lead to their degradation.44 Overproduction of ROS also suppresses GLUT4 translocation in cells via affecting insulin signaling.45
PCOS-IR–Associated mtDNA Mutations
ND1 T3394C Mutation
The ND1 T3394C (p.Y30H) mutation changes an amino acid (AA) that is extremely conserved in >90% of mammalian mtDNAs and believed to be associated with PCOS-IR.46,47 Functional analysis has revealed that this mutation affects the stability of ND1 mRNA, as well as complex I assembly and activity, decreases ATP levels and mitochondrial membrane potential (MMP), and enhances ROS production.47,48
ND2 C5178A Mutation
The m.C5178A mutation causes the alternation of leucine to methionine at position 237 of the corresponding AA, which occurrs within the ND2 gene in complex I and is associated with PCOS-IR,46 longevity,49 and acute myocardial infarction.50 Markedly decreased ATP, MMP, superoxide dismutaseand significantly increased ROS, malondialdehyde, and 8-hydroxydeoxyguanosine have been identified in polymononuclear leukocytes derived from subjects harboring this mutation, suggesting that the m.C5178A mutation may cause OS and result in mitochondrial dysfunction.51
ND5 T12338C and T12811C Mutations
We previously identified homoplasmic m.T12338C (p.M1T) together with tRNASer(UCN) C7492T mutation in a patient with PCOS-IR.52 At the molecular level, m.T12338C altered well-conserved methionine with threonine; therefore, the ND5 mRNA was expected to be shortened by two AAs.53 Using cybrid cell models, the m.T12338C mutation decreased the stability of the ND5 polypeptide, affecting the assembly and activity of respiratory chain complexes.54 Therefore, m.T12338C causes a mitochondrial dysfunction that plays a key role in PCOS-IR.
The m.T12811C (p.Y159H) mutation occurs at extremely conserved residues in ND5, which is essential for the functions of complex I.55 The alternation of tyrosine to histidine is believed to affect the structure and function of the transmembrane region of the ND5 protein.56 Since ND5 plays a a putative role in maintening the functions of complex I, the m.T12811C mutation may affect the ND5 polypeptide and influence ETC activities.57
D-loop Mutations
The D-loop region is where mtDNA replication and transcription occur, and is important for transcription of both heavy and light strands.58 A recent case–control study by Deng et al suggested that variants m.G207A, m.16036GGins, and m.16049Gins may decrease the risk of PCOS in a Chinese population.59 The m.G207A substitution was located at the heavy strand, which is critical for mtDNA replication, suggesting that m.G207A may affect the binding affinity and influence the replication of mtDNA.60 While m.16036GGins and m.16049Gins both occurred at hypervariable region 1, notably they were found to reduce the risk of endometriosis,61 highlighting the importance of these mutations in maintaining mitochondrial functions.
4977-bp Deletion
The 4977-bp deletion is one of the most common deletions of mtDNA, spanning approximately a third of the entire mitochondrial genome (nucleotides 8470–13,447), and is regarded as a pathogenic deletion in PCOS.62,63 The 4977-bp deletion removes five tRNAs and seven genes encoding respiratory chain complexes that are important for normal OxPhos functions. The 4977-bp deletion results in an impairment of protein synthesis and reduces ATP and mtDNA copy number.64
tRNALeu(UUR) Mutations
We previously described a Chinese pedigree with PCOS-IR that harbored a heteroplasmic tRNALeu(UUR) A3302G mutation.65 The proband’s mother and grandmother were diagnosed with T2DM. The m.A3302G mutation occurred at two nucleotides from the 5’ end of tRNALeu(UUR), which is evolutionarily conserved from various species (Figure 5A). As such, it can be anticipated that m.A3302G mutation may influence 5’ end processing.66 Biochemical analysis has revealed that this mutation leads to severe complex I and IV deficiencies. A marked decreased in the stability of tRNALeu(UUR) was identified in cybrids with this mutation. In addition, the m.A3302G mutation led to abnormal processing of RNA19, an unprocessed RNA intermediate comprising mt-16S rRNA, mt-tRNALeu(UUR), and mtND1.67,68 Therefore, the m.A3302G mutation can cause mitochondrial dysfunctions involved in PCOS-IR.
 |
Figure 5 (A-G) Cloverleaf structure of mt-tRNA genes, arrows indicate the positions of PCOS-IR related mutations. |
Another PCOS-IR–associated mutation is m.C3275T in tRNALeu(UUR), which has been reported in another family with PCOS and metabolic syndrome.69 The m.C3275T mutation was located in a well-conserved position in the variable region of tRNALeu(UUR) (Figure 5A). A recent study revealed that the m.C3275T mutation was a risk factor of Leber’s hereditary optic neuropathy.70 Intriguingly, m.C3275T disrupted Watson–Crick base-pairing (28A–46C) and may have caused a failure in tRNALeu(UUR) metabolism.
tRNAGln Mutations
By mutational screening for mt-tRNA genes in 80 patients with PCOS-IR and 50 healthy controls, we identified a set of tRNA mutations.71 Among these, m.T4363C mutation occurred at the anticodon stem of tRNAGln (conventional position 38). A mutation at position 38 needed to be modified and played important roles in tRNA structure and function (Figure 5B).72 Bioinformatic analysis indicated that the m.T4363C mutation caused the thermodynamic change of tRNAGln69. The m.T4363C mutation has been identified in a Chinese pedigree with hypertension, but detailed molecular mechanisms remain unexplored.73 While the m.T4395C mutation occurs at the sixth base of mt-tRNAGln-accept arm, adjacent to the 5’ end of tRNAGln (Figure 5B), interestingly the mutant 4395C formed a novel (6C–64G) base-pairing that was associated with essential hypertension.74 The secondary structure altered by the m.T4395C mutation may influence tRNA function and impair mitochondrial translation.
tRNACys Mutation
The homoplasmic m.G5821A mutation resides at the acceptor arm of tRNACys gene (position 6). This mutation has been reported to be associated with cardiomyopathy,75 and is also a risk factor in clinical expression of deafness-related m.A1555G mutation.76 At the molecular level, m.G5821A abolishes conserved base-pairing (6G–67C); therefore, it may lead to failure of tRNA metabolism via the alternation of its structure (Figure 5C).77
tRNASer(UCN) Mutation
The m.C7492T mutation in homoplasmy is located at the anticodon stem of tRNASer(UCN) (position 26). Notably, the cytosine at that position is conserved from bacteria to human mitochondria, emphasizing the importance of m.C7492T mutation to tRNA function (Figure 5D).78 A heteroplasmic mutation (m.T4295C) occurring at the same conventional position in tRNAIle has been reported to cause chronic progressive external ophthalmoplegia.79 Therefore, the m.C7492T mutation may have the same impact on tRNA function.
tRNAAsp Mutation
Adenine-to-guanine alternation at position 7543 affects a well-conserved adenosine in the anticodon stem of tRNAAsp, which may influence the posttranscriptional modification of this tRNA (Figure 5E).80 Yeast genes harboring C-to-T transition at position 28 are transcribed and can be further processed to form the maturation of 4S-size tRNAAsp, whereas mutant tRNA may not be charged with radiolabeled aspartate.81 Subjects with m.A7543G mutation have shown partial cytochrome C oxidase deficiency, suggesting the potential pathogenicity of this mutation in mitochondrial dysfunction.82
tRNALys Mutation
The homoplasmic A8343G mutation affects the first adenine in the TψC loop of tRNALys (position 54). The nucleotide at that position is often modified, thus playing an important role in the structure and function of tRNA.83 This mutation may affect tRNA aminoacylation ability and binding affinity with mitochondrial elongation factor Tu, which is critical for mitochondrial protein synthesis (Figure 5F).84,85 Therefore, the m.A8343G mutation is pathogenic in PCOS-IR.
tRNAGlu Mutation
The well-known m.A14693G mutation occurs at a conserved position of the TΨC loop of tRNAGlu (Figure 5G). The nucleotide at position 54 (m.A14693G) of tRNAGlu is often modified, thus having an impact on tRNA functions.86 It has been proposed that the m.A14693G mutation can cause failure in tRNAGlu metabolism and impair mitochondrial protein synthesis.87
Mechanism of PCOS-IR–Associated mtDNA Mutations
Mutations in mtDNA have structural and functional consequences, such as affecting OxPhos complexes and influencing mitochondrial protein synthesis. Mmost of these mtDNA mutations occurred with mt-tRNA genes (Figure 5 and Table 1). mt-tRNA mutations may destabilize tRNA tertiary structure, alter RNA processing, and lead to defects in nucleotide modification. Subsequently, these mutations lead to failures in tRNA metabolism. These mitochondrial protein-synthesis defects resulte in decreased in ATP production in granulosa cells or pancreatic cells, thus contributing to PCOS clinical phenotypes.
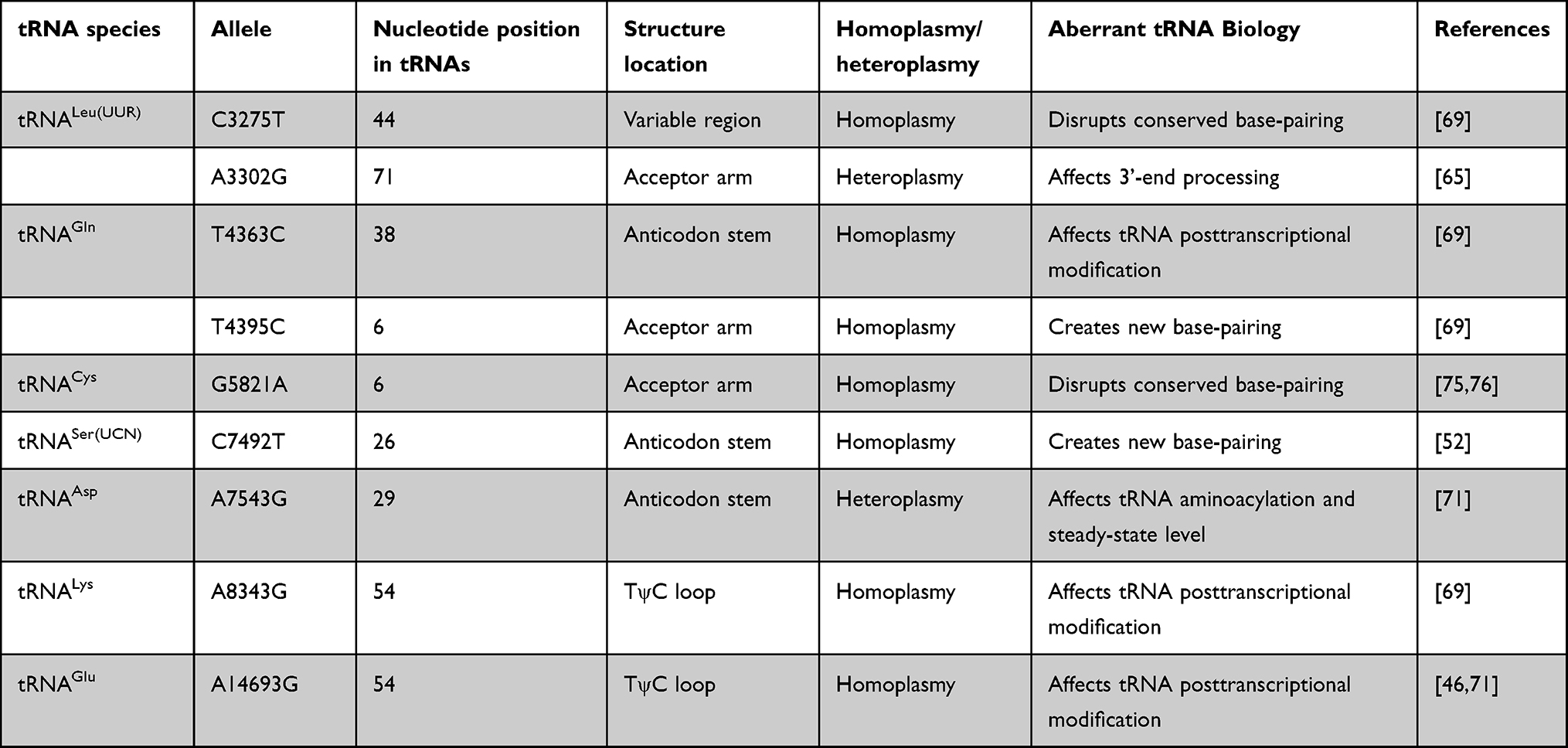 |
Table 1 Summary of PCOS-IR–associated Mt-tRNA mutations |
Conclusion
Mutations in mtDNA important contributors to PCOS-IR. Genetic variants in mitochondrial genomes can perturb OxPhos and are thought to contribute to the clinical pathology of PCOS. mtDNA damage is believed to increase OS and create a proinflammatory state, which could accelerate the progression of PCOS.88 Therefore, mtDNA may offer a viable alternative target for genetic studies tackling this complex but common disease and attempting to explain the discrepancies in clinical phenotype and progression of PCOS.
Abbreviations
PCOS, polycystic ovary syndrome; IR, insulin resistance; mtDNA, mitochondrial DNA; ROS, reactive oxygen species; OS, oxidative stress; T2DM, type 2 diabetes mellitus; LH, luteinizing hormone; ATP, adenosine triphosphate; OxPhos, oxidative phosphorylation; nDNA, nuclear DNA; ETC, electron transport chain; NADH-Q, nicotinamide adenine dinucleotide Q; SDH, succinate dehydrogenase; AA, amino acid; MMP, mitochondrial membrane potential.
Acknowledgments
This work was supported by grants from the Health Commission of Zhejiang Province (2021RC022), Hangzhou Bureau of Science and Technology (20201203B210), and Hangzhou Municipal Health Commission (ZD20220010).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Swaroop A, Jaipuriar AS, Gupta SK, et al. Efficacy of a novel fenugreek seed extract (Trigonella foenum-graecum, Furocyst) in polycystic ovary syndrome (PCOS). Int J Med Sci. 2015;12(10):825–831. doi:10.7150/ijms.13024
2. Teede H, Deeks A, Moran L. Polycystic ovary syndrome: a complex condition with psychological, reproductive and metabolic manifestations that impacts on health across the lifespan. BMC Med. 2010;8:41. doi:10.1186/1741-7015-8-41
3. Azziz R, Carmina E, Dewailly D, et al.; Androgen Excess Society. Positions statement: criteria for defining polycystic ovary syndrome as a predominantly hyperandrogenic syndrome: an Androgen Excess Society guideline. J Clin Endocrinol Metab. 2006;91(11):4237–4245. doi:10.1210/jc.2006-0178
4. Rotterdam ESHRE/ASRM-Sponsored PCOS Consensus Workshop Group. Revised 2003 consensus on diagnostic criteria and long-term health risks related to polycystic ovary syndrome. Fertil Steril. 2004;81(1):19–25. doi:10.1016/j.fertnstert.2003.10.004
5. Conway G, Dewailly D, Diamanti-Kandarakis E, et al.; ESE PCOS Special Interest Group. The polycystic ovary syndrome: a position statement from the European Society of Endocrinology. Eur J Endocrinol. 2014;171(4):P1–29. doi:10.1530/EJE-14-0253
6. Dokras A, Saini S, Gibson-Helm M, et al. Gaps in knowledge among physicians regarding diagnostic criteria and management of polycystic ovary syndrome. Fertil Steril. 2017;107(6):1380–1386.e1. doi:10.1016/j.fertnstert.2017.04.011
7. Bozdag G, Mumusoglu S, Zengin D, et al. The prevalence and phenotypic features of polycystic ovary syndrome: a systematic review and meta-analysis. Hum Reprod. 2016;31(12):2841–2855. doi:10.1093/humrep/dew218
8. Khan MJ, Ullah A, Basit S. Genetic basis of polycystic ovary syndrome (PCOS): current perspectives. Appl Clin Genet. 2019;12:249–260. doi:10.2147/TACG.S200341
9. Nidhi R, Padmalatha V, Nagarathna R, et al. Prevalence of polycystic ovarian syndrome in Indian adolescents. J Pediatr Adolesc Gynecol. 2011;24(4):223–227. doi:10.1016/j.jpag.2011.03.002
10. McGowan MP. Polycystic ovary syndrome: a common endocrine disorder and risk factor for vascular disease. Curr Treat Options Cardiovasc Med. 2011;13(4):289–301. doi:10.1007/s11936-011-0130-0
11. Lizneva D, Suturina L, Walker W, et al. Criteria, prevalence, and phenotypes of polycystic ovary syndrome. Fertil Steril. 2016;106(1):6–15. doi:10.1016/j.fertnstert.2016.05.003
12. Zhang C, Ma J, Wang W, et al. Lysyl oxidase blockade ameliorates anovulation in polycystic ovary syndrome. Hum Reprod. 2018;33(11):2096–2106. doi:10.1093/humrep/dey292
13. Bednarska S, Siejka A. The pathogenesis and treatment of polycystic ovary syndrome: what’s new? Adv Clin Exp Med. 2017;26(2):359–367. doi:10.17219/acem/59380
14. Teede HJ, Hutchison SK, Zoungas S. The management of insulin resistance in polycystic ovary syndrome. Trends Endocrinol Metab. 2007;18(7):273–279. doi:10.1016/j.tem.2007.08.001
15. Mastrototaro L, Roden M. Insulin resistance and insulin sensitizing agents. Metabolism. 2021;125:154892. doi:10.1016/j.metabol.2021.154892
16. Vecchio I, Tornali C, Bragazzi NL, et al. The discovery of insulin: an important milestone in the history of medicine. Front Endocrinol. 2018;9:613. doi:10.3389/fendo.2018.00613
17. Tsatsoulis A, Mantzaris MD, Bellou S, et al. Insulin resistance: an adaptive mechanism becomes maladaptive in the current environment-an evolutionary perspective. Metabolism. 2013;62(5):622–633. doi:10.1016/j.metabol.2012.11.004
18. Bannigida DM, Nayak BS, Vijayaraghavan R. Insulin resistance and oxidative marker in women with PCOS. Arch Physiol Biochem. 2020;126(2):183–186. doi:10.1080/13813455.2018.1499120
19. He FF, Li YM. Role of gut microbiota in the development of insulin resistance and the mechanism underlying polycystic ovary syndrome: a review. J Ovarian Res. 2020;13(1):73. doi:10.1186/s13048-020-00670-3
20. Docea AO, Vassilopoulou L, Fragou D, et al. CYP polymorphisms and pathological conditions related to chronic exposure to organochlorine pesticides. Toxicol Rep. 2017;4:335–341. doi:10.1016/j.toxrep.2017.05.007
21. Rothenberg SS, Beverley R, Barnard E, et al. Polycystic ovary syndrome in adolescents. Best Pract Res Clin Obstet Gynaecol. 2018;48:103–114. doi:10.1016/j.bpobgyn.2017.08.008
22. Rosenfield RL, Ehrmann DA. The pathogenesis of polycystic ovary syndrome (PCOS): the hypothesis of PCOS as functional ovarian hyperandrogenism revisited. Endocr Rev. 2016;37(5):467–520. doi:10.1210/er.2015-1104
23. Cassar S, Misso ML, Hopkins WG, et al. Insulin resistance in polycystic ovary syndrome: a systematic review and meta-analysis of euglycaemic-hyperinsulinaemic clamp studies. Hum Reprod. 2016;31(11):2619–2631. doi:10.1093/humrep/dew243
24. Condorelli RA, Calogero AE, Di Mauro M, et al. PCOS and diabetes mellitus: from insulin resistance to altered beta pancreatic function, a link in evolution. Gynecol Endocrinol. 2017;33(9):665–667. doi:10.1080/09513590.2017.1342240
25. Pandurevic S, Macut D, Fanelli F, et al. Biomediators in polycystic ovary syndrome and cardiovascular risk. Biomolecules. 2021;11(9):1350. doi:10.3390/biom11091350
26. Hernansanz-Agustín P, Enríquez JA. Inner mitochondrial membrane sensitivity to Na+ reveals partially segmented functional CoQ pools. J Vis Exp. 2022;2022(185):1. doi:10.3791/63729
27. Kang PT, Chen CL, Lin P, et al. Mitochondrial complex I in the post-ischemic heart: reperfusion-mediated oxidative injury and protein cysteine sulfonation. J Mol Cell Cardiol. 2018;121:190–204. doi:10.1016/j.yjmcc.2018.07.244
28. Ackrell BA. Progress in understanding structure-function relationships in respiratory chain complex II. FEBS Lett. 2000;466(1):1–5. doi:10.1016/s0014-5793(99)01749-4
29. Cecchini G. Function and structure of complex II of the respiratory chain. Annu Rev Biochem. 2003;72:77–109. doi:10.1146/annurev.biochem.72.121801.161700
30. Klimova T, Chandel NS. Mitochondrial complex III regulates hypoxic activation of HIF. Cell Death Differ. 2008;15(4):660–666. doi:10.1038/sj.cdd.4402307
31. Chandel NS. Mitochondrial complex III: an essential component of universal oxygen sensing machinery? Respir Physiol Neurobiol. 2010;174(3):175–181. doi:10.1016/j.resp.2010.08.004
32. Starkov AA. The role of mitochondria in reactive oxygen species metabolism and signaling. Ann N Y Acad Sci. 2008;1147:37–52. doi:10.1196/annals.1427.015
33. Wallace DC. A mitochondrial bioenergetic etiology of disease. J Clin Invest. 2013;123(4):1405–1412. doi:10.1172/JCI61398
34. Wallace DC. A mitochondrial paradigm of metabolic and degenerative diseases, aging, and cancer: a Dawn for evolutionary medicine. Annu Rev Genet. 2005;39:359–407. doi:10.1146/annurev.genet.39.110304.095751
35. Kang E, Wu J, Gutierrez NM, et al. Mitochondrial replacement in human oocytes carrying pathogenic mitochondrial DNA mutations. Nature. 2016;540(7632):270–275. doi:10.1038/nature20592
36. Grandhi S, Bosworth C, Maddox W, et al. Heteroplasmic shifts in tumor mitochondrial genomes reveal tissue-specific signals of relaxed and positive selection. Hum Mol Genet. 2017;26(15):2912–2922. doi:10.1093/hmg/ddx172
37. Stewart JB, Chinnery PF. Extreme heterogeneity of human mitochondrial DNA from organelles to populations. Nat Rev Genet. 2021;22(2):106–118. doi:10.1038/s41576-020-00284-x
38. Rossignol R, Faustin B, Rocher C, et al. Mitochondrial threshold effects. Biochem J. 2003;370(Pt 3):751–762. doi:10.1042/BJ20021594
39. Luo L, Gu F, Jie H, et al. Early miscarriage rate in lean polycystic ovary syndrome women after euploid embryo transfer - a matched-pair study. Reprod Biomed Online. 2017;35(5):576–582. doi:10.1016/j.rbmo.2017.07.010
40. Macut D, Bjekić-Macut J, Savić-Radojević A. Dyslipidemia and oxidative stress in PCOS. Front Horm Res. 2013;40:51–63. doi:10.1159/000341683
41. Chenna S, Koopman WJH, Prehn JHM, et al. Mechanisms and mathematical modeling of ROS production by the mitochondrial electron transport chain. Am J Physiol Cell Physiol. 2022;323(1):C69–C83. doi:10.1152/ajpcell.00455.2021
42. Shan H, Luo R, Guo X, et al. Abnormal endometrial receptivity and oxidative stress in polycystic ovary syndrome. Front Pharmacol. 2022;13:904942. doi:10.3389/fphar.2022.904942
43. Houstis N, Rosen ED, Lander ES. Reactive oxygen species have a causal role in multiple forms of insulin resistance. Nature. 2006;440(7086):944–948. doi:10.1038/nature04634
44. Evans JL, Maddux BA, Goldfine ID. The molecular basis for oxidative stress-induced insulin resistance. Antioxid Redox Signal. 2005;7(7–8):1040–1052. doi:10.1089/ars.2005.7.1040
45. Ma J, Nakagawa Y, Kojima I, et al. Prolonged insulin stimulation down-regulates GLUT4 through oxidative stress-mediated retromer inhibition by a protein kinase CK2-dependent mechanism in 3T3-L1 adipocytes. J Biol Chem. 2014;289(1):133–142. doi:10.1074/jbc.M113.533240
46. Zhuo G, Ding Y, Feng G, et al. Analysis of mitochondrial DNA sequence variants in patients with polycystic ovary syndrome. Arch Gynecol Obstet. 2012;286(3):653–659. doi:10.1007/s00404-012-2358-7
47. Zhu J, Vinothkumar KR, Hirst J. Structure of mammalian respiratory complex I. Nature. 2016;536(7616):354–358. doi:10.1038/nature19095
48. Ji Y, Zhang J, Yu J, et al. Contribution of mitochondrial ND1 3394T>C mutation to the phenotypic manifestation of Leber’s hereditary optic neuropathy. Hum Mol Genet. 2019;28(9):1515–1529. doi:10.1093/hmg/ddy450
49. Kokaze A, Ishikawa M, Matsunaga N, et al. Longevity-associated mitochondrial DNA 5178 A/C polymorphism and blood pressure in the Japanese population. J Hum Hypertens. 2004;18(1):41–45. doi:10.1038/sj.jhh.1001632
50. Mukae S, Aoki S, Itoh S, et al. Mitochondrial 5178A/C genotype is associated with acute myocardial infarction. Circ J. 2003;67(1):16–20. doi:10.1253/circj.67.16
51. Jiang Z, Teng L, Zhang S, et al. Mitochondrial ND1 T4216C and ND2 C5178A mutations are associated with maternally transmitted diabetes mellitus. Mitochondrial DNA a DNA Mapp Seq Anal. 2021;32(2):59–65. doi:10.1080/24701394.2020.1856101
52. Ding Y, Zhuo G, Zhang C, et al. Point mutation in mitochondrial tRNA gene is associated with polycystic ovary syndrome and insulin resistance. Mol Med Rep. 2016;13(4):3169–3172. doi:10.3892/mmr.2016.4916
53. Jiang Z, Cai X, Kong J, et al. Maternally transmitted diabetes mellitus may be associated with mitochondrial ND5 T12338C and tRNAAla T5587C variants. Ir J Med Sci. 2022;191(6):2625–2633. doi:10.1007/s11845-021-02911-w
54. Zhang J, Ji Y, Lu Y, et al. Leber’s hereditary optic neuropathy (LHON)-associated ND5 12338T > C mutation altered the assembly and function of complex I, apoptosis and mitophagy. Hum Mol Genet. 2018;27(11):1999–2011. doi:10.1093/hmg/ddy107
55. Cai W, Fu Q, Zhou X, et al. Mitochondrial variants may influence the phenotypic manifestation of Leber’s hereditary optic neuropathy-associated ND4 G11778A mutation. J Genet Genomics. 2008;35(11):649–655. doi:10.1016/S1673-8527(08)60086-7
56. Bi R, Zhang AM, Jia X, et al. Complete mitochondrial DNA genome sequence variation of Chinese families with mutation m.3635G>A and Leber hereditary optic neuropathy. Mol Vis. 2012;18:3087–3094.
57. Huoponen K, Lamminen T, Juvonen V, et al. The spectrum of mitochondrial DNA mutations in families with Leber hereditary optic neuroretinopathy. Hum Genet. 1993;92(4):379–384. doi:10.1007/BF01247339
58. Nicholls TJ, Minczuk M. In D-loop: 40 years of mitochondrial 7S DNA. Exp Gerontol. 2014;56:175–181. doi:10.1016/j.exger.2014.03.027
59. Deng X, Ji D, Li X, et al. Polymorphisms and haplotype of mitochondrial DNA D-loop region are associated with polycystic ovary syndrome in a Chinese population. Mitochondrion. 2021;57:173–181. doi:10.1016/j.mito.2020.12.006
60. Máximo V, Soares P, Lima J, et al. Mitochondrial DNA somatic mutations (point mutations and large deletions) and mitochondrial DNA variants in human thyroid pathology: a study with emphasis on Hürthle cell tumors. Am J Pathol. 2002;160(5):1857–1865. doi:10.1016/S0002-9440(10)61132-7
61. Li X, Ji D, Marley JL, et al. Association between mitochondrial DNA D-loop region polymorphisms and endometriosis in a Chinese population. J Assist Reprod Genet. 2020;37(9):2171–2179. doi:10.1007/s10815-020-01853-z
62. Yusoff AAM, Abdullah WSW, Khair SZNM, et al. A comprehensive overview of mitochondrial DNA 4977-bp deletion in cancer studies. Oncol Rev. 2019;13(1):409. doi:10.4081/oncol.2019.409
63. Ye M, Hu B, Shi W, et al. Mitochondrial DNA 4977 bp deletion in peripheral blood is associated with polycystic ovary syndrome. Front Endocrinol. 2021;12:675581. doi:10.3389/fendo.2021.675581
64. Wei YH, Lee CF, Lee HC, et al. Increases of mitochondrial mass and mitochondrial genome in association with enhanced oxidative stress in human cells harboring 4977 BP-deleted mitochondrial DNA. Ann N Y Acad Sci. 2001;928:97–112. doi:10.1111/j.1749-6632.2001.tb05640.x
65. Ding Y, Zhuo G, Zhang C. The mitochondrial tRNALeu(UUR) A3302G mutation may be associated with insulin resistance in woman with polycystic ovary syndrome. Reprod Sci. 2016;23(2):228–233. doi:10.1177/1933719115602777
66. Hutchison WM, Thyagarajan D, Poulton J, et al. Clinical and molecular features of encephalomyopathy due to the A3302G mutation in the mitochondrial tRNA(Leu(UUR)) gene. Arch Neurol. 2005;62(12):1920–1923. doi:10.1001/archneur.62.12.1920
67. Maniura-Weber K, Helm M, Engemann K, et al. Molecular dysfunction associated with the human mitochondrial 3302A>G mutation in the MTTL1 (mt-tRNALeu(UUR)) gene. Nucleic Acids Res. 2006;34:6404–6415. doi:10.1093/nar/gkl727
68. Bindoff LA, Howell N, Poulton J, et al. Abnormal RNA processing associated with a novel tRNA mutation in mitochondrial DNA. A potential disease mechanism. J Biol Chem. 1993;268(26):19559–19564. doi:10.1016/S0021-9258(19)36552-4
69. Ding Y, Xia BH, Zhang CJ, et al. Mitochondrial tRNALeu(UUR) C3275T, tRNAGln T4363C and tRNALys A8343G mutations may be associated with PCOS and metabolic syndrome. Gene. 2018;642:299–306. doi:10.1016/j.gene.2017.11.049
70. Garcia-Lozano JR, Aguilera I, Bautista J, et al. A new mitochondrial DNA mutation in the tRNA leucine 1 gene (C3275A) in a patient with Leber’s hereditary optic neuropathy. Hum Mutat. 2000;15(1):120–121. doi:10.1002/(SICI)1098-1004(200001)15:1<120::AID-HUMU33>3.0.CO;2-8
71. Ding Y, Xia BH, Zhang CJ, et al. Mutations in mitochondrial tRNA genes may be related to insulin resistance in women with polycystic ovary syndrome. Am J Transl Res. 2017;9(6):2984–2996.
72. Suzuki T, Nagao A, Suzuki T. Human mitochondrial tRNAs: biogenesis, function, structural aspects, and diseases. Annu Rev Genet. 2011;45:299–329. doi:10.1146/annurev-genet-110410-132531
73. Wang L, Dong Z, Lin W, et al. Molecular characterization of a pedigree carrying the hypertension‑associated mitochondrial tRNAGln T4363C mutation. Mol Med Rep. 2017;16(5):6029–6033. doi:10.3892/mmr.2017.7371
74. Zhu H-Y, Wang S-W, Liu L, et al. Genetic variants in mitochondrial tRNA genes are associated with essential hypertension in a Chinese Han population. Clin Chim Acta. 2009;410(1–2):64–69. doi:10.1016/j.cca.2009.09.023
75. Ozawa T, Tanaka M, Sugiyama S, et al. Patients with idiopathic cardiomyopathy belong to the same mitochondrial DNA gene family of Parkinson’s disease and mitochondrial encephalomyopathy. Biochem Biophys Res Commun. 1991;177(1):518–525. doi:10.1016/0006-291x(91)92014-b
76. Lu J, Qian Y, Li Z, et al. Mitochondrial haplotypes may modulate the phenotypic manifestation of the deafness-associated 12S rRNA 1555A>G mutation. Mitochondrion. 2010;10(1):69–81. doi:10.1016/j.mito.2009.09.007
77. Sprinzl M, Horn C, Brown M, et al. Compilation of tRNA sequences and sequences of tRNA genes. Nucleic Acids Res. 1998;26(1):148–153. doi:10.1093/nar/26.1.148
78. Peng W, Zhong Y, Zhao X, et al. Low penetrance of hearing loss in two Chinese families carrying the mitochondrial tRNASer(UCN) mutations. Mol Med Rep. 2020;22(1):77–86. doi:10.3892/mmr.2020.11100
79. Silvestri G, Servidei S, Rana M, et al. A novel mitochondrial DNA point mutation in the tRNA(Ile) gene is associated with progressive external ophtalmoplegia. Biochem Biophys Res Commun. 1996;220(3):623–627. doi:10.1006/bbrc.1996.0453
80. Suzuki T, Suzuki T, Wada T, et al. Taurine as a constituent of mitochondrial tRNAs: new insights into the functions of taurine and human mitochondrial diseases. EMBO J. 2002;21(23):6581–6589. doi:10.1093/emboj/cdf656
81. Najarian D, Shu HH, Martin NC. Sequence and expression of four mutant aspartic acid tRNA genes from the mitochondria of Saccharomyces cerevisiae. Nucleic Acids Res. 1986;14(24):9561–9578. doi:10.1093/nar/14.24.9561
82. Shtilbans A, El-Schahawi M, Malkin E, et al. A novel mutation in the mitochondrial DNA transfer ribonucleic acidAsp gene in a child with myoclonic epilepsy and psychomotor regression. J Child Neurol. 1999;14(9):610–613. doi:10.1177/088307389901400910
83. Delaunay S, Pascual G, Feng B, et al. Mitochondrial RNA modifications shape metabolic plasticity in metastasis. Nature. 2022;607(7919):593–603. doi:10.1038/s41586-022-04898-5
84. Rinaldi T, Lande R, Bolotin-Fukuhara M, et al. Additional copies of the mitochondrial Ef-Tu and aspartyl-tRNA synthetase genes can compensate for a mutation affecting the maturation of the mitochondrial tRNAAsp. Curr Genet. 1997;31(6):494–496. doi:10.1007/s002940050235
85. Belostotsky R, Frishberg Y, Entelis N. Human mitochondrial tRNA quality control in health and disease: a channelling mechanism? RNA Biol. 2012;9(1):33–39. doi:10.4161/rna.9.1.18009
86. Ding Y, Li Y, You J, et al. Mitochondrial tRNA(Glu) A14693G variant may modulate the phenotypic manifestation of deafness-associated 12S rRNA A1555G mutation in a Han Chinese family. J Genet Genomics. 2009;36(4):241–250. doi:10.1016/S1673-8527(08)60111-3
87. Wiedemann N, Pfanner N. Mitochondrial machineries for protein import and assembly. Annu Rev Biochem. 2017;86:685–714. doi:10.1146/annurev-biochem-060815-014352
88. Boyapati RK, Dorward DA, Tamborska A, et al. Mitochondrial DNA is a pro-inflammatory damage-associated molecular pattern released during active IBD. Inflamm Bowel Dis. 2018;24(10):2113–2122. doi:10.1093/ibd/izy095
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.