Back to Journals » OncoTargets and Therapy » Volume 7
Potential implications of GRP58 expression and susceptibility of cervical cancer to cisplatin and thymoquinone-based therapy
Authors Hafiza WA, Latifah SY
Received 24 February 2014
Accepted for publication 6 April 2014
Published 7 August 2014 Volume 2014:7 Pages 1375—1387
DOI https://doi.org/10.2147/OTT.S62928
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Wan Abd Ghani Wan Nor Hafiza,1,2 Saiful Yazan Latifah1,3
1Department of Biomedical Science, Faculty of Medicine and Health Sciences, Universiti Putra Malaysia, Selangor, 2College of Medical Laboratory Technology, Institute for Medical Research, Ministry of Health, Jalan Pahang, Kuala Lumpur, 3Laboratory of Molecular Biomedicine, Institute of Bioscience, Universiti Putra Malaysia, Selangor, Malaysia
Abstract: A new therapeutic approach of looking at the expression of glucose-regulated protein (GRP) 58 as an indication of cisplatin sensitivity may eradicate fruitless treatment and side effects in patients with cervical cancer. Thymoquinone, the bioactive compound in Nigella sativa, has been reported to have an antiproliferative effect on cervical cancer cells. This study compared the cytotoxic effects of cisplatin, a drug commonly used in the treatment of cervical cancer, and thymoquinone in cervical cancer (HeLa and SiHa) cell lines by 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide assay, and measured GRP58 expression in the cells by quantitative real-time polymerase chain reaction and Western blotting. Cisplatin had higher antiproliferative activity towards the cervical cancer cell lines than thymoquinone in a dose-dependent and time-dependent manner. However, cisplatin was more toxic to normal 3T3 and Vero cell lines than thymoquinone. The half maximal inhibitory concentration (IC50) of cisplatin in HeLa and SiHa cells at 72 hours was 13.3±2.52 µM and 19.5±2.12 µM, respectively. Meanwhile, the IC50 of thymoquinone in HeLa and SiHa cells was 29.57±5.81 µM and 23.41±1.51 µM, respectively (P<0.05). A significant correlation was found between the cytotoxicity of cisplatin and expression of GRP58, but this relationship was not significant for thymoquinone. Therefore, the response of cervical cancer cells to cisplatin can be predicted on the basis of GRP58 expression.
Keywords: glucose-regulated protein 58, cervical cancer, cisplatin, thymoquinone
Introduction
Currently, cisplatin alone or in combination with other drugs, such as 5-fluorouracil,1 etoposide,2 vinorelbine,3 topotecan,4,5 paclitaxel,6 gemcitabine, and irinotecan, is used together with radiotherapy as a standard regimen in the management of advanced cervical cancer.7,8 However, despite their effectiveness, concerns remain regarding the side effects of these chemotherapeutic drugs.4,9–11 Prediction of chemosensitivity before giving the treatment to those individuals who are most likely to benefit from cisplatin is particularly challenging, and it is important to avoid or minimize any harmful effects and unnecessary cost.
In recent years, there has been increasing research interest in the use of natural products to treat cervical cancers, in particular herbal extracts, because they are less likely to have side effects. A number of in vitro studies have demonstrated the potential mechanisms of action of natural compounds in inducing cell death or apoptosis in cervical cancer cells. Thymoquinone is an active constituent of the plant Nigella sativa, and has a potential role in the treatment of cancer. Previous researchers have reported that thymoquinone reduces the proliferation of lung cancer, breast cancer, colon cancer, melanoma,12,13 liver cancer,14 neuroblastoma,15 and oral cancer cells.16
One of the protein molecules that may be used as a potential marker in predicting the response of cervical cancer cells towards cisplatin is thiol-disulfide oxidoreductase, ie, glucose-regulated protein 58 kDa (GRP58/Erp57/ER60/PDI/ERp60/ERp61/P58/Q2/HIP-70). A recent study showed that GRP58, an endoplasmic reticulum stress (ER)-responsive protein that is activated in response to glucose deprivation modulates the invasiveness of cervical cancer.17 Upregulation of GRP58 in cancers of the breast, uterus, lung, ovary, and stomach has been reported previously.18 However, downregulation of GRP58 expression has also been documented in gastric cancer,19 esophageal cancer,20 cervical carcinoma, and renal cell carcinoma.21,22 One study reported that downregulation of GRP58 significantly enhanced the neurotoxicity of prions, an ER stress inducer of neuronal cells.23 Several have shown a relationship between GRP58 and inhibition of proliferation of cancer cells. For instance, knockdown of GRP58 was associated with inhibition of proliferation of breast cancer and melanoma cells.24,25 In contrast, downregulation of GRP58 was associated with a poor prognosis in early-stage cervical cancer.26
Functionally, GRP58 forms complexes with calnexin and calreticulin, which are chaperones that act together with newly synthesized glycoproteins in the ER.27,28 GRP58 is also involved in nuclear localization and interacts with specific DNA sequences in the HeLa cervical cancer cell line. The level of GRP58 expression may then be influenced by the cytotoxicity of cisplatin since the mechanism of action involves targeting of DNA to form different types of cisplatin-DNA adducts.29 There is also some evidence showing that the antitumor activity of cisplatin in cancer cells occurs via nucleus-independent activation of caspase-12 and upregulation of GRP78, the hallmark molecule of ER stress.30
In this study, we investigated the relationship between GRP58 expression levels in HeLa and SiHa (squamous carcinoma) cells and their response to cisplatin. Thymoquinone was also assessed as a candidate agent in the management of cervical cancer. The work is considered significant in the discovery of the potential role of GRP58 a as predictor of the cytotoxicity of cisplatin and that of thymoquinone. Our findings suggest that GRP58 is a potential prognostic marker in cervical cancer and has direct implications for drug therapy and clinical usage.
Materials and methods
Cell culture
Cervical adenocarcinoma (HeLa, ATCC-CCL-2; passage 10), squamous cell carcinoma (SiHa, ATCC-HTB-35, passage 6), 3T3 (passage 15), and Vero (passage 4) cell lines were purchased from the American Type Culture Collection (Rockville, MD, USA). HeLa, 3T3, and Vero cells were grown in Roswell Park Memorial Institute-1640 medium (PAA Laboratories, Linz, Austria) and the SiHa cells in Minimum Essential Medium Eagle with Earle’s (EMEM) (Sigma, St Louis, MO, USA). Both types of culture medium were supplemented with 10% (volume/volume [v/v]) fetal bovine serum and 1% penicillin-streptomycin (100 IU/mL of penicillin and 100 μg/mL of streptomycin, PAA Laboratories). The cells were allowed to grow at 37°C in a 5% CO2 atmosphere.
Determination of cytotoxicity of cisplatin and thymoquinone
Cytotoxicity was assessed using the MTT assay.31 The cells (0.7×105 mL−1 for HeLa, 3T3, and Vero cells, and 1×105 mL−1 for SiHa cells) were treated with different concentrations of cisplatin and thymoquinone (3.0–200 μM) in a 96-well plate for 24, 48, and 72 hours. Untreated controls were also included. Following incubation, 20 μL of 5 mg/mL MTT was added to each well and kept at 37°C for 3 hours. Next, 100 μL of dimethyl sulfoxide was added to each well to solubilize the blue crystal precipitate (formazan product). The absorbance at 570 nm and the reference wavelength of 630 nm was measured using an enzyme linked immunosorbent assay reader (Opsys MR®; Dynex Technologies, Chantilly, VA, USA). Each experiment was performed in triplicate and the average absorbance values were calculated. The IC50 (concentration of a drug that achieves a 50% maximal inhibitory response in cells compared with a control) was obtained from the best fit standard curve of percentage cell viability on the ordinate against the extract concentration on the abscissa:
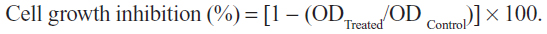
RNA extraction and complementary DNA preparation
HeLa and SiHa cells were treated with cisplatin (3 μM and 6 μM) and thymoquinone (6 μM and 12 μM) for 48 hours to achieve an apoptotic index between 15% and 30%. The concentrations were chosen based on studies published by Ng et al32 in 2011 using the Annexin V/fluorescein isothiocyanate assay. RNA was extracted using a Total RNA Extraction Kit (Mini) from Real Biotech Corporation (Banqiao City, Taiwan) according to the manufacturer’s instructions. The concentration and integrity of RNA were measured using a nanodrop system (NanoPhotometer™; Implen, Munich, Germany; OD260/OD280 nm absorption ratio >1.90). The aliquoted RNA was stored at −80°C. Complementary DNA templates were synthesized from the extracted RNAs in a volume of 6 μL, which was added to the reaction mix of 5× iScript Transcription Supermix (Bio-Rad Laboratories, Hercules, CA, USA) containing oligo (dT), random primers, buffer, RNase inhibitor, MgCl2, and dNTP mix in a final volume of 20 μL.
Quantitative real-time polymerase chain reaction
Highly purified salt-free primers were designed by Next Gene Scientific Sdn Bhd (Puchong, Malaysia) and synthesized by AITbiotech Pte Ltd (Science Park 1, Singapore). The descriptions of the primers used for amplification of GRP58, HPRT, and β-actin genes are shown in Table 1.
 | Table 1 Primers for amplification of GRP58, HPRT, and β-actin genes |
Polymerase chain reaction (PCR) products were synthesized using IQ™ SYBR® Green Supermix (Bio-Rad). Real-time PCR reactions were performed in a total volume of 25 μL using the CFX 96 Real-Time PCR Detection System (Bio-Rad). Quantitative real-time PCR (RT-qPCR) conditions for all PCRs were optimized in a gradient cycler (Mastercycler Gradient, Bio-Rad) with regard to various annealing temperatures (50°C–60°C) (Table 2). RT-qPCR amplification products were separated by 1% agarose gel electrophoresis (Fisher Scientific, Loughborough, UK). Optimized results were transferred to the following LightCycler PCR protocol.
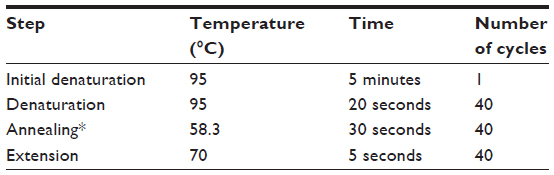 | Table 2 Polymerase chain reaction protocol for GRP58, HPRT, and β-actin |
Upon completion of amplification, the specificity of the amplified products was confirmed using melting curve analysis whereby the PCR products were incubated by increasing the incubation temperature from 70°C to 90°C with an increase of 0.5°C per second. All reactions were performed in triplicate, and a mixture without a complementary DNA template (NTC) was used as the negative control. Data acquisition and analysis were performed using CFX Manager version 2.0.
Western blot analysis
Western blotting was performed using a modification of the method described previously.33 The Bradford assay was used to quantify the protein level.34 Total protein was resolved by sodium dodecyl sulfate polyacrylamide gel electrophoresis. Samples (20 μg/well) with a rainbow protein molecular weight marker (Fermentas, Loughborough, UK) were run through bis-acrylamide stacking gel (0.5 M Tris [pH 6.8]) at 50 V for 20 minutes, followed by 100 V for one hour through a 10% (weight/volume [w/v]) bis-acrylamide resolving gel (1.5 M Tris [pH 8.8] using a 1× running buffer [95 mM Tris-Base, 960 mM glycine, and 1% of 10% sodium dodecyl sulfate, pH 8.6]). The proteins on the gel were transferred to a polyvinylidene fluoride membrane (Bio-Rad) in the 1× transfer buffer (transfer buffer 10×; 190 mM Tris-Base, 1.92 M glycine, 100% ethanol, and water) at 300 mA for 1.5 hours. The membrane was blocked with phosphate-buffered saline/Tween-20 (0.5%) and 5% milk (blocking agent) for one hour. Subsequently, the membrane was incubated with the primary antibody, mouse anti-GRP58 (Abcam Corporation, Cambridge, MA, USA) and anti-β-actin (Santa Cruz Biotechnology Inc., Santa Cruz, CA, USA) at a dilution of 1:2,000 overnight at 4°C. These steps were followed by incubation with the secondary antibody, rabbit anti-mouse immunoglobulin G (Santa Cruz Biotechnology Inc.), for one hour at 25°C on a rocker (dilution 1:4,000). All immunoblots were visualized by enhanced chemiluminescence plus Western blotting detection reagents (Abcam Corporation, Cambridge, MA, USA). Densitometric quantification of autoradiograms was performed using ImageJ software (version 1.41; National Institutes of Health, Bethesda, MD, USA).
Statistical analysis
The statistical analysis was performed using Pearson’s correlation, General Linear Model (univariate), Duncan’s multiple range test, and Dunnett’s test using Statistical Package of Social Sciences for Windows version 21.0 software (IBM Corporation, Armonk, NY, USA). All data are presented as the mean ± standard deviation. P<0.05 was considered to indicate a statistically significant difference.
Results and discussion
Cytotoxicity of cisplatin and thymoquinone
Our results indicate differences in the sensitivity of HeLa and SiHa cells to cisplatin and thymoquinone. Cisplatin showed more antiproliferative activity and cytotoxicity than thymoquinone in the cervical cancer cell lines in a dose-dependent and time-dependent manner. However, cisplatin was more toxic than thymoquinone in the normal 3T3 and Vero cell lines (Figure 1 and Table 3). At 72 hours, the values for cisplatin and thymoquinone were 9.65±0.08 μM and 17.38±2.94 μM, respectively. Any of the cell types used as a control is of human origin, and in particular 3T3 cells are mouse fibroblast particularly sensitive to most of the known cytotoxics. 3T3 cells are recommended by the US National Institute of Environmental Health Sciences to access in vitro basal cytotoxicity for acute oral toxicity testing. Vero cells are homologous to human body cells. These cells have been banked, are well characterized, and are approved by the World Health Organization for production of human vaccines.
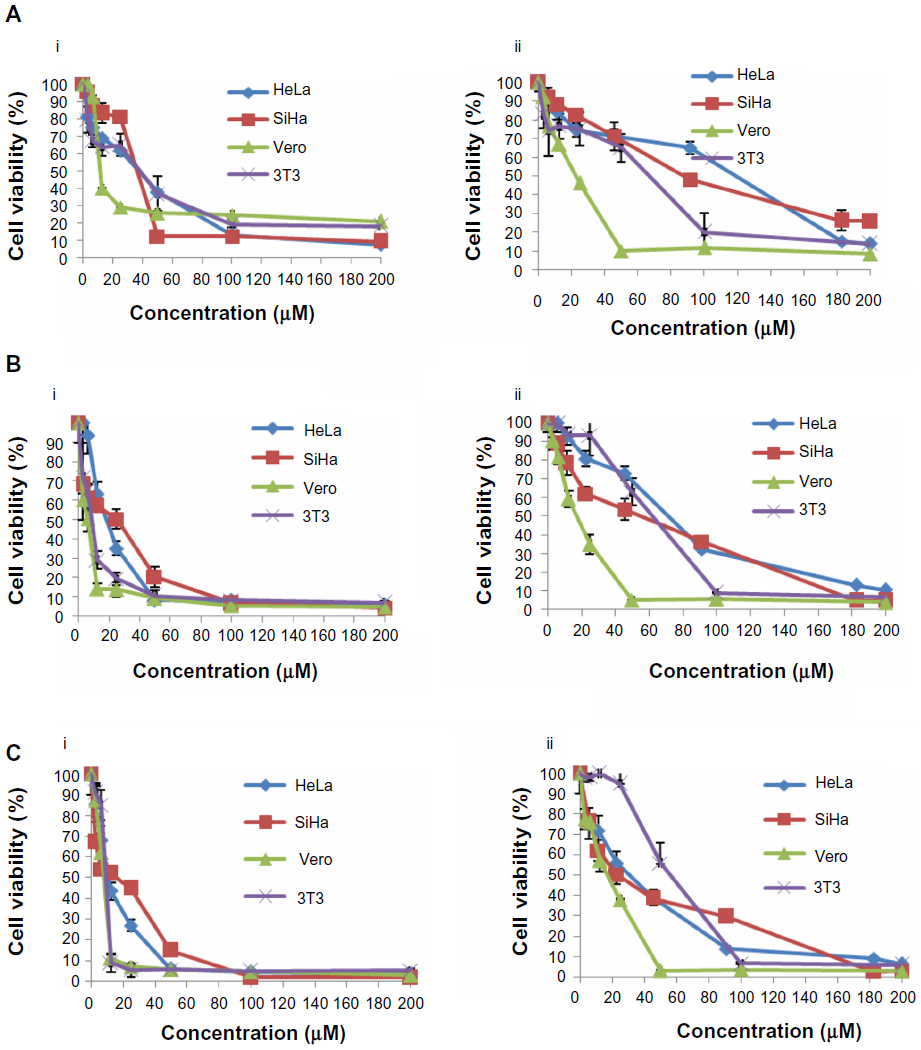 | Figure 1 Dose-response curves for HeLa, SiHa, Vero, and 3T3 cells following treatment with cisplatin (i) and thymoquinone (ii) at 24 hours (A), 48 hours (B), and 72 hours (C). The cells (0.7×105 mL−1 for HeLa, 3T3, and Vero cells, and 1×105 mL-1 for SiHa cells) were treated with different concentrations of cisplatin and thymoquinone and subjected to MTT assay. |
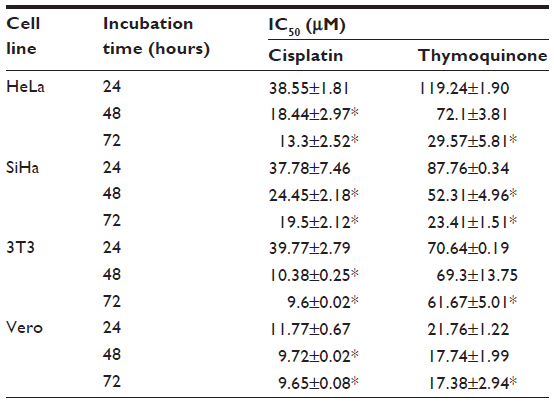 | Table 3 Cytotoxicity of cisplatin and thymoquinone towards various human cell lines represented as IC50 value determined by MTT assay |
Expression of GRP58 mRNA following treatment with cisplatin and thymoquinone
Intensive research has been conducted for the past 30 years, and several mechanisms accounting for the cisplatin-resistant tumor cell phenotype have been reported. However, no correlation between GRP58 expression and response to cisplatin in advanced cervical cancer has been reported. High GRP58 expression has been reported to be associated with resistance to anticancer drugs in patients with oral squamous cell carcinoma35 and those with ovarian cancer.36 In a model of cochlear damage, exposure to cisplatin resulted in a decreased level of GRP58 expression.37
In the present study, we investigated whether GRP58 expression could be a predictive marker of susceptibility to cervical cancer treatment using cisplatin and thymoquinone. The cells were treated with both drugs to achieve an apoptotic index of 15%–30%, which is optimal for studying the early molecular events in the ER stress-induced pathway. RT-qPCR was initially used to measure the transcription level of GRP58 mRNA. In this experiment, the specificity of qRT-PCR products was documented using high resolution gel electrophoresis and resulted in a single product of the desired length (GRP58, 161 bp; HPRT, 185 bp; β-actin 250 bp). In addition, a LightCycler melting curve analysis was performed, which resulted in a single product-specific annealing temperature as follows: GRP58 and β-actin, 60°C; HPRT, 58.3°C (Figure 2A). No primer-dimers were generated during the 40 real-time PCR amplification cycles. RT-qPCR efficiencies were calculated from the given slopes using LightCycler software. The corresponding real-time PCR efficiency (E) of one cycle in the exponential phase was calculated according to the equation: E=10[−1/slope]. The transcripts showed high real-time PCR efficiency rates, ie, 110.3% for GRP58, 105.9% for β-actin, and 108.9% for HPRT, in the investigated range of 2.18–70 ng complementary DNA input (n=3) with high linearity (Pearson correlation coefficient R2>0.99, Figure 2B–D).
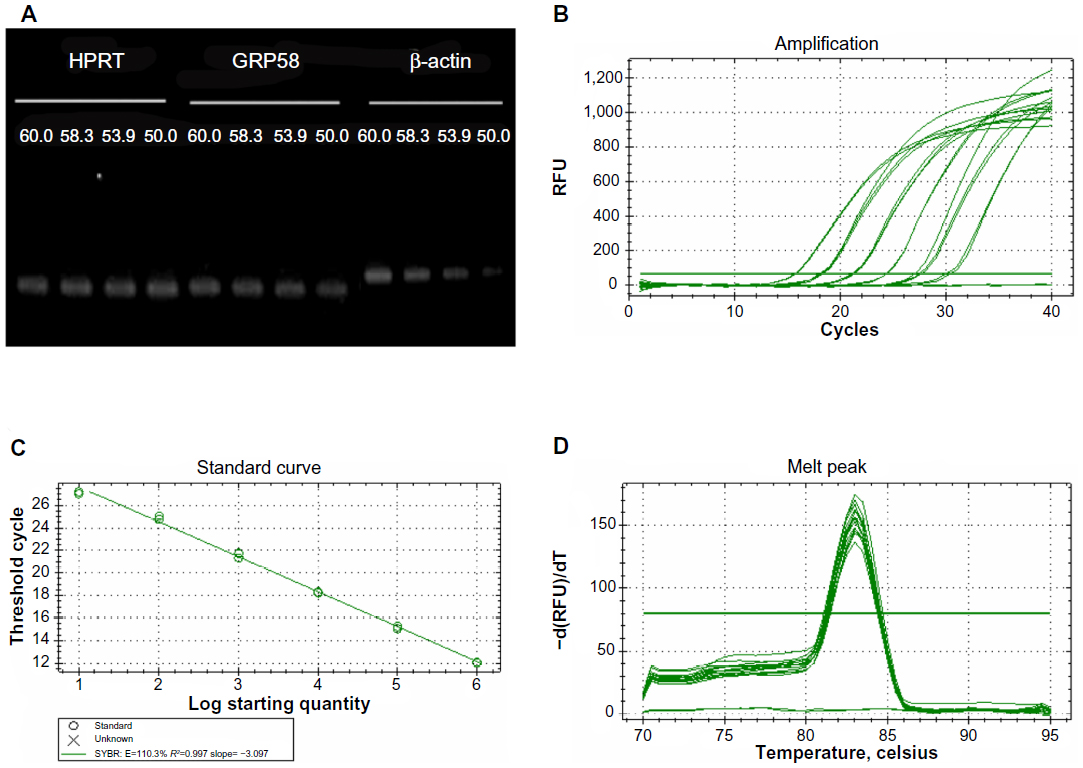 | Figure 2 Optimization of quantitative real-time polymerase chain reaction. |
Gene analysis data showed that GRP58 was expressed in both untreated (control) cell lines, with a higher level in HeLa cells than in SiHa cells. RNA expression levels were analyzed based on the cycle threshold value. The lower the cycle threshold value, the higher the expression of GRP58 (Figure 3C). The results show that the GRP58 level differed between the cell line types, as reported by previous researchers who noted that patients with adenocarcinoma had higher levels of GRP58 than those with squamous cell carcinoma.17 This difference is due to the different types of metabolism used by the two types of cells. Adenocarcinoma cells rely mainly on aerobic glycolysis for production of ATP, whereas under hypoxic squamous carcinoma cells conditions undergo mitochondrial oxidation with anaerobic glycolysis.38 GRP58 is activated in response to glucose deprivation, so the physiological changes in HeLa and SiHa cells may contribute to the observed difference in GRP58 levels; however, given that these two cell lines were grown under the same conditions, ie, with no differences in the hypoxic microenvironment or metabolism, the reasons for the difference in GRP58 levels remain unclear. Otherwise, differences in GRP58 expression are most likely due to differences in the human papillomavirus (HPV) genotypes and oncoproteins present in HeLa and SiHa cells. The results of epidemiologic research support the experimental data showing involvement of HPV in cervical cancer.39 Each HeLa cell contains 10–50 copies of HPV-18, while each SiHa cells contains one copy of HPV-16.40–42 These high-risk types of HPV encode the E6 and E7 oncogenes. E6 interacts with the tumor suppressor protein p53 through a ubiquitin-dependent proteasome pathway, while E7 encodes viral oncoproteins that target retinoblastoma protein.43,44 These E6 and E7 proteins affect cell cycle control by facilitating stable maintenance of episomes and stimulating differentiated cells to proliferate.45 GRP58 might be involved in either the E6-dependent or E7-dependent pathway. For example, adeno-associated virus transfection has been shown to moderately reduce the growth rate of tumors arising from SiHa cells but not that of tumors arising from HeLa cells. This differential suppression exerted by adeno-associated virus may be due to differences in HPV genotype.46 In another study, delivery of monoclonal antibodies that bind to HPV-16 E6 and neutralize its biological activity in vitro could restore p53 function in SiHa cells but not in HeLa cells. Proliferation of SiHa cells was markedly diminished, but no apoptosis was detectable in HeLa cells.47 On that basis, involvement of GRP58 in the cell death process may vary from cell type to cell type (eg, glandular versus squamous), HPV copy number, expression of oncoproteins, or other variables that remain to be elucidated.
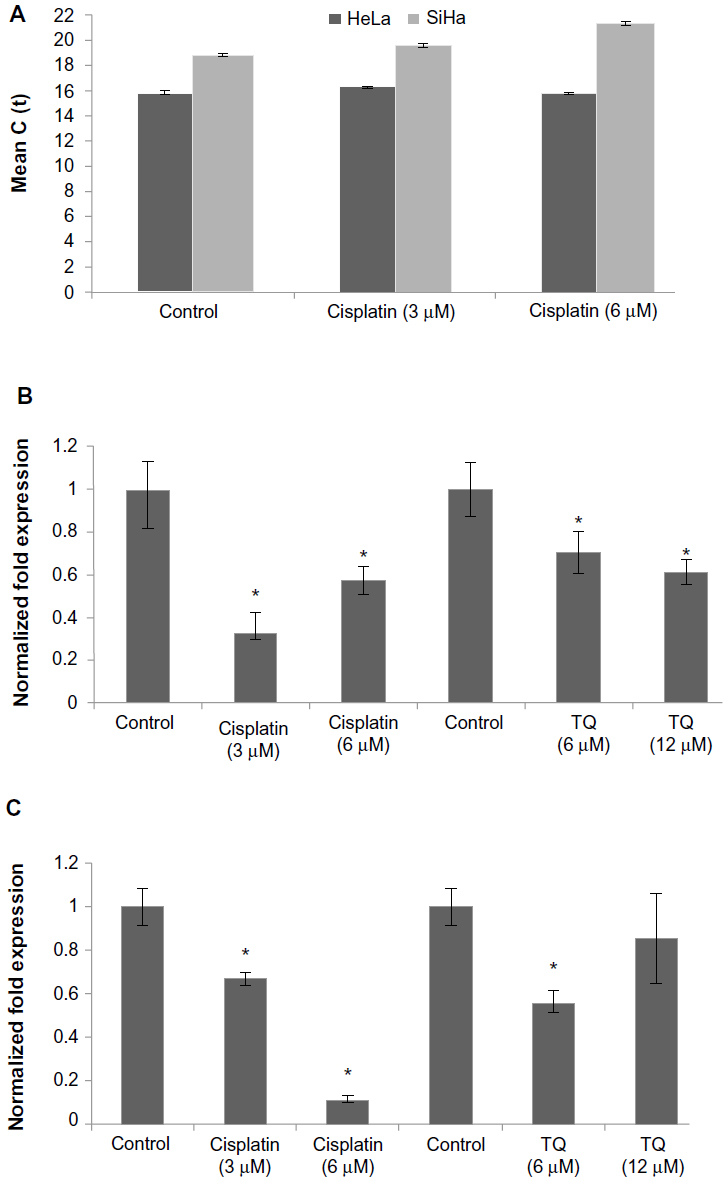 | Figure 3 mRNA level of GRP58 in HeLa and SiHa cells following treatment with cisplatin and thymoquinone for 48 hours as measured by quantitative real-time polymerase chain reaction. |
Figure 3 presents the data for GRP58 expression after treatment with cisplatin and thymoquinone. GRP58 was significantly downregulated following exposure to both agents in a dose-dependent manner. The GRP58 expression level was lower in SiHa cells than in HeLa cells at a cisplatin concentration of 6 μM (P<0.05). However, GRP58 expression decreased significantly in SiHa cells treated with thymoquinone at 12 μM.
Expression of GRP58 protein following treatment with cisplatin and thymoquinone
The RT-qPCR data were further validated using Western blot analysis using β-actin as a control to ensure equal protein loading (Figure 4). Untreated (control) HeLa cells showed a higher level of GRP58 expression than did control SiHa cells (Figure 4A and B), which was verified by quantification of the intensity of the protein bands by densitometry analysis. Downregulation of GRP58 was observed following treatment with cisplatin and thymoquinone. At 24 hours, the GRP58 level was reduced by 8-fold in HeLa cells after treatment with cisplatin 3 μM when compared with the control cells, and in SiHa cells, there was a 1.5-fold decrease. For thymoquinone 12 μM, the GRP58 level was reduced by 1.3-fold after 24 hours. The GRP58 expression level at 48 hours was significantly lower than that at 24 hours following treatment with cisplatin and thymoquinone (P<0.05). GRP58 expression was downregulated more efficiently after treatment with cisplatin than after treatment with thymoquinone in both cell lines (Figure 5). Although GRP58 mRNA expression was not downregulated by thymoquinone 12 μM in SiHa cells, protein expression was reduced. Expression levels of GRP58 might not necessarily be associated with protein levels because not all mRNA templates detected in RT-qPCR analysis are fully translated to functional proteins. At the higher thymoquinone concentration of 12 μM, GRP58 mRNA and protein expression levels were both reduced. Treatment of the cells with increasing amounts of thymoquinone decreased the stability of GRP58 mRNA, resulting in a decreased level of expression. However, there was an indication that a post-translational mechanism to increase GRP58 protein stability activity in the thymoquinone-treated cells existed at a lower concentration (6 μM). The mechanism by which expression of GRP58 is regulated at the translation level is poorly understood.
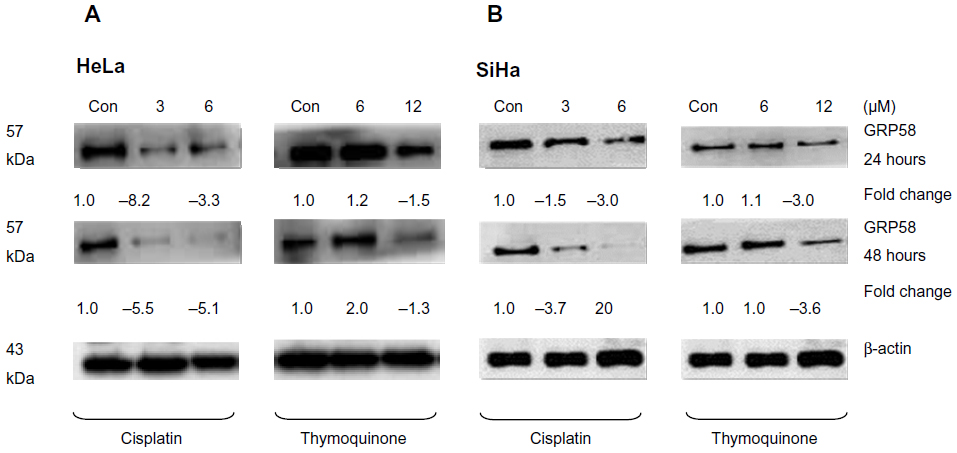 | Figure 4 GRP58 expression in human cervical cancer cell lines treated with cisplatin and thymoquinone as determined by Western blot analysis. |
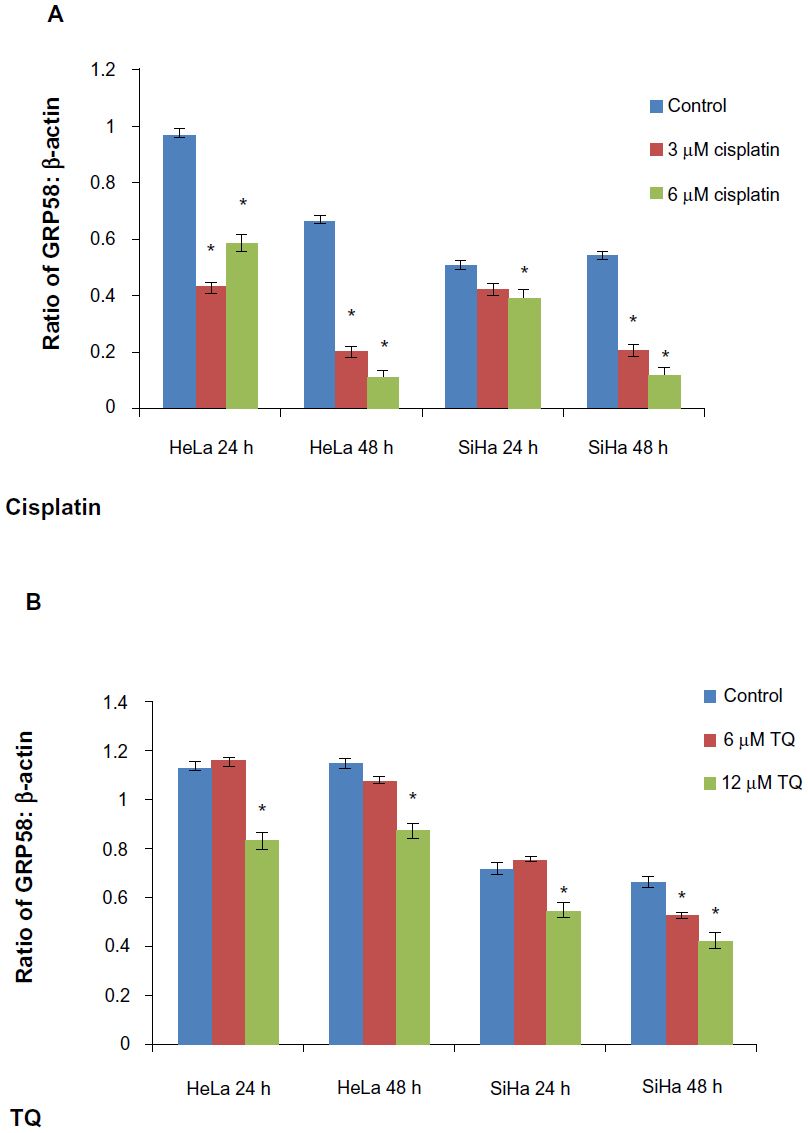 | Figure 5 GRP58 levels in HeLa and SiHa cell lines following treatment with cisplatin (A) and thymoquinone (B). |
Correlation between GRP58 expression and sensitivity to cisplatin and thymoquinone
GRP58, an ER resident protein, could be a target for development of novel chemotherapeutic strategies. We predicted that the response (sensitivity) to cisplatin and thymoquinone would be increased by downregulation of GRP58. To confirm this hypothesis, correlation analysis between GRP58 expression and the cytotoxicity (based on IC50 values) of cisplatin and thymoquinone was performed. Sensitivity towards cisplatin was found to be significantly correlated with GRP58 levels in the two cell lines (P<0.05). However, the correlation was not significant for thymoquinone (P>0.05, Figure 6).
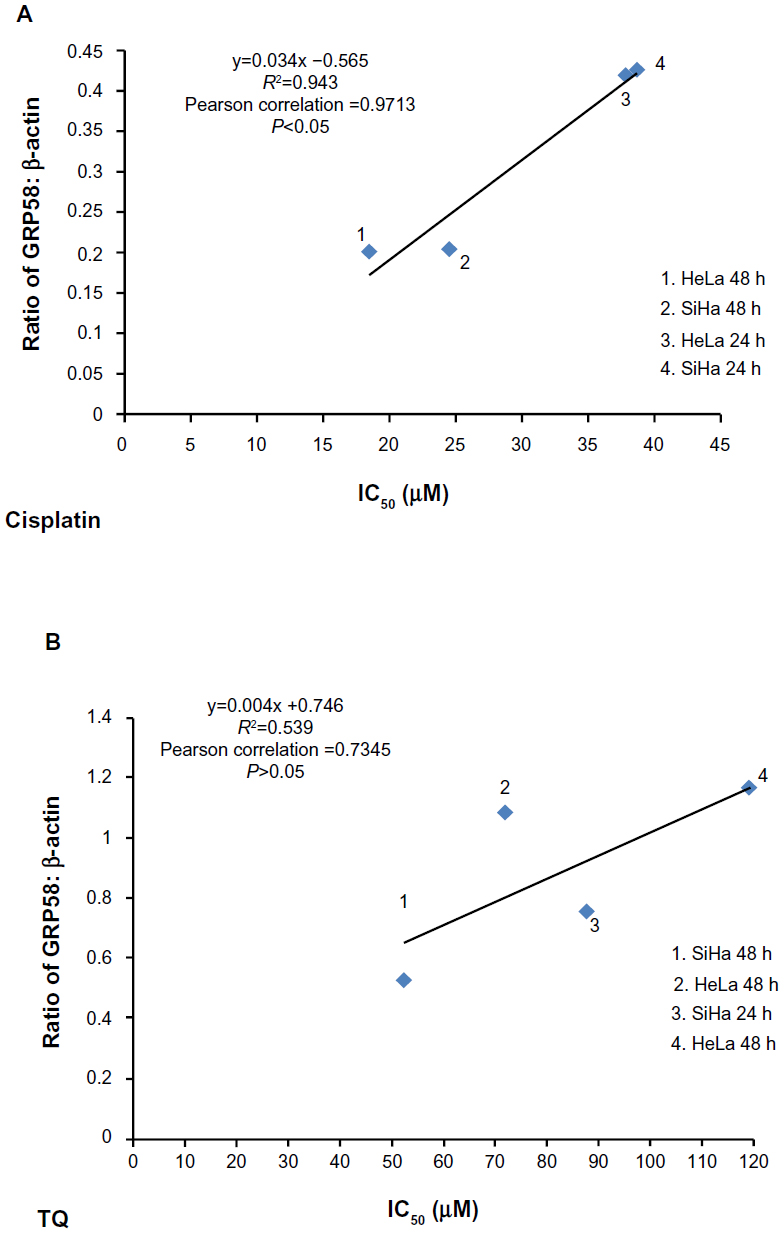 | Figure 6 Correlation between density values of the ratio of GRP58 to β-actin and cytotoxicity (IC50 values) in HeLa and SiHa cells following treatment with cisplatin (A) and thymoquinone (B) using Pearson’s correlation method for at least three independent experiments. P<0.05 was considered to be statistically significant. |
So far, there is yet any study on the involvement of GRP58 in cisplain-induced cytotoxicity. We postulated that GRP58 plays a role in the ER stress-induced apoptosis signaling pathway by cisplatin in the cervical cancer cells of which previously, nucleus independent activation of caspase-12 and upregulation of GRP78 have been involved.30 Knockdown of GRP58 has been shown to lead to ER stress-induced apoptosis in breast cancer cells.25 The current study indicates that the antitumor effect of cisplatin could be increased by downregulating GRP58 expression.
There are several pathways that have been directly associated with ER stress-induced apoptosis, all of which require activation by GRP78. It has previously been demonstrated that knockdown of GRP58 increases the level of GRP78.48 Considering that GRP78 is a key player in ER stress-induced apoptosis, these findings may support the involvement of GRP58 in ER stress-induced pathways. The first pathway involves induction of transcription factor CCAAT/enhancer-binding protein homologous protein (CHOP)/growth and DNA damage-inducible transcription factor (GADD153). The second pathway involves activation of the c-Jun N-terminal kinase (JNK) pathway by ER transmembrane protein kinase type I, IRE1 (inositol requiring 1), and PERK (PKR-like ER kinase).49 The third pathway involves cleavage of caspase-12. CHOP/GADD153 modulates the level of PUMA (p53 upregulated modulator of apoptosis), an important regulator in p53-mediated apoptosis. Overexpression of PUMA is accompanied by increased expression of BAX, release of cytochrome c, and reduction in the mitochondrial membrane potential.50
Studies have shown that this protein can interact with antiapoptotic Bcl-2 family members, resulting in activation of caspase-9,51 which subsequently activates caspase-3, leading to apoptosis. Thus, sensitivity of cervical cancer cells towards cisplatin might be associated with activation of the ER-induced apoptosis signaling pathway and the GRP58 level. Further, GRP58 modulates STAT3 (signal transducer and activator of transduction 3) and regulates mTOR1 (mammalian target of rapamycin 1) signaling.52,53 Activated STAT3 and mTOR1 promote development of cancer cells by preventing apoptosis.54,55 Downregulation of GRP58 may inhibit proliferation of cancer cells. Based on our findings, the proposed mechanism of induction of apoptosis by cisplatin in HeLa and SiHa cells is shown in Figure 7. This phenomenon could be of therapeutic benefit in the treatment of cervical cancer.
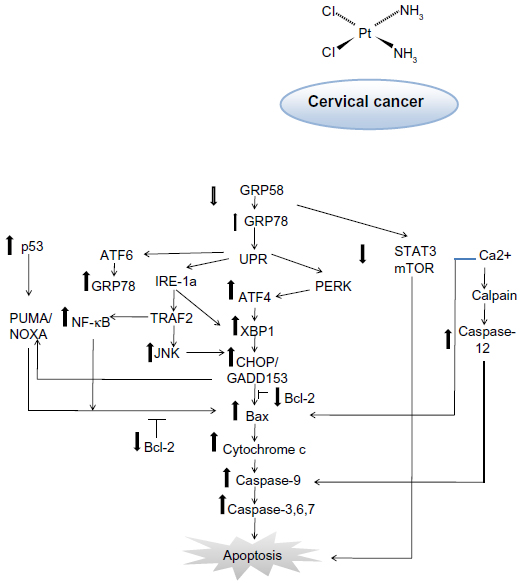 | Figure 7 Schematic diagram of proposed mechanism of action for GRP58 in cisplatin-induced apoptosis. The pathways involve the endoplasmic reticulum stress-apoptotic-dependent pathway. |
On the other hand, involvement of GRP58 in the cytotoxicity of thymoquinone was not identified in this analysis. Indeed, no involvement of any ER stress proteins in thymoquinone-induced apoptotic pathways has been documented to date. However, treatment of SiHa cells with thymoquinone has been shown to cause upregulation of p53 and downregulation of Bcl-2.32 Thus, it is postulated that thymoquinone may also induce apoptosis via pathways similar to those mentioned earlier, whereby GADD153/CHOP modulates PUMA to undergo apoptosis via p53-mediated pathway under conditions of ER stress.50 However, a previous study demonstrated that p53 was not involved in upregulation of PUMA in ER stressed-induced apoptosis.56 In other types of cancer, treatment with thymoquinone caused activation of the STAT3,57 NF-κB,58,59 JNK, and mitogen-associated protein kinase (MAPK) pathways.60,61 These pathways are linked to regulation of apoptosis with ER stress-induced pathway. However, STAT3, NF-κB, JNK, and MAPK are molecules that could be having proapoptotic or antiapoptotic effects. For instance, in gastric cancer, induction of ER stress protects cells from apoptosis, especially via MAPK pathways.61 This may explain the negative correlation between GRP58 expression level and cytotoxicity of thymoquinone in cervical cancer cells.
Conclusion
In summary, the GRP58 expression level depending on the type of cell and type of drug. In this study, there was a correlation between expression of GRP58 protein and the cytotoxicity of cisplatin in HeLa and SiHa cells. However, no significant correlation was found for thymoquinone. This indicates that the GRP58 expression level might have potential implications for the susceptibility of cervical cancer cells to cisplatin and thymoquinone-based therapy. A further mechanistic study using small interfering RNA is needed for confirmation.
Acknowledgments
The technical assistance of Dr Tan Sheau Wei with RT-qPCR is greatly appreciated. This study was funded by the Research University Grant Scheme of Universiti Putra Malaysia (04-01-09-0714RU) and by the Fundamental Research Grant Scheme (04-04-10-884FR).
Disclosure
The authors report no conflict of interest in this work.
References
Jurado M, Martinez-Monge R, Garcia-Foncillas J, et al. Pilot study of concurrent cisplatin, 5-fluorouracil, and external beam radiotherapy prior to radical surgery ± intraoperative electron beam radiotherapy in locally advanced cervical cancer. Gynecol Oncol. 1999;74(1):30–37. | |
Bae JH, Lee SJ, Lee A, et al. Neoadjuvant cisplatin and etoposide followed by radical hysterectomy for stage 1B-2B cervical cancer. Gynecol Oncol. 2008;111(3):444–448. | |
Pignata S, Silvestro G, Ferrari E, et al. Phase II study of cisplatin and vinorelbine as first-line chemotherapy in patients with carcinoma of the uterine cervix. J Clin Oncol. 1999;17(3):756–760. | |
Rose PG, Sill MW, McMeekin DS, et al. A phase I study of concurrent weekly topotecan and cisplatin chemotherapy with whole pelvic radiation therapy in locally advanced cervical cancer: a gynecologic oncology group study. Gynecol Oncol. 2012;125(1):158–162. | |
Manci N, Marchetti C, Di Tucci C, et al. A prospective phase II study of topotecan (Hycamtin(R)) and cisplatin as neoadjuvant chemotherapy in locally advanced cervical cancer. Gynecol Oncol. 2011;122(2):285–290. | |
Moore DH, Blessing JA, McQuellon RP, et al. Phase III study of cisplatin with or without paclitaxel in stage IVB, recurrent, or persistent squamous cell carcinoma of the cervix: a gynecologic oncology group study. J Clin Oncol. 2004;22(15):3113–3119. | |
Duenas-González A, Orlando M, Zhou Y, Quinlivan M, Barraclough H. Efficacy in high burden locally advanced cervical cancer with concurrent gemcitabine and cisplatin chemoradiotherapy plus adjuvant gemcitabine and cisplatin: prognostic and predictive factors and the impact of disease stage on outcomes from a prospective randomized phase III trial. Gynecol Oncol. 2012;126(3):334–340. | |
Sugiyama T, Yakushiji M, Noda K, et al. Phase II study of irinotecan and cisplatin as first-line chemotherapy in advanced or recurrent cervical cancer. Oncology. 2000;58(1):31–37. | |
Oike T, Ohno T, Noda SE, et al. Comparison of hematological toxicities between innovator and generic cisplatin formulations in cervical cancer patients treated with concurrent chemoradiotherapy. J Radiat Res. 2012;14:14. | |
Long HJ, Bundy BN, Grendys EC, et al. Randomized Phase III trial of cisplatin with or without topotecan in carcinoma of the uterine cervix: a Gynecologic Oncology Group Study. J Clin Oncol. 2005;23(21):4626–4633. | |
Kong TW, Chang SJ, Paek J, et al. Comparison of concurrent chemoradiation therapy with weekly cisplatin versus monthly fluorouracil plus cisplatin in FIGO stage IIB-IVA cervical cancer. J Gynecol Oncol. 2012;23(4):235–241. | |
Attoub S, Sperandio O, Raza H, et al. Thymoquinone as an anticancer agent: evidence from inhibition of cancer cells viability and invasion in vitro and tumor growth in vivo. Fundam Clin Pharmacol. 2013;27(5):557–569. | |
Woo CC, Loo SY, Gee V, et al. Anticancer activity of thymoquinone in breast cancer cells: possible involvement of PPAR-gamma pathway. Biochem Pharmacol. 2011;82(5):464–475. | |
Raghunandhakumar S, Paramasivam A, Senthilraja S, et al. Thymoquinone inhibits cell proliferation through regulation of G1/S phase cell cycle transition in N-nitrosodiethylamine-induced experimental rat hepatocellular carcinoma. Toxicol Lett. 2013;3(13):01290–01293. | |
Paramasivam A, Sambantham S, Shabnam J, et al. Anti-cancer effects of thymoquinone in mouse neuroblastoma (Neuro-2a) cells through caspase-3 activation with down-regulation of XIAP. Toxicol Lett. 2012;213(2):151–159. | |
Abdelfadil E, Cheng YH, Bau DT, et al. Thymoquinone induces apoptosis in oral cancer cells through p38beta inhibition. Am J Chin Med. 2013;41(3):683–696. | |
Liao CJ, Wu TI, Huang YH, et al. Glucose-regulated protein 58 modulates cell invasiveness and serves as a prognostic marker for cervical cancer. Cancer Sci. 2011;102(12):2255–2263. | |
Chay D, Cho H, Lim BJ, et al. ER-60 (PDIA3) is highly expressed in a newly established serous ovarian cancer cell line, YDOV-139. Int J Oncol. 2010;37(2):399–412. | |
Leys CM, Nomura S, LaFleur BJ, et al. Expression and prognostic significance of prothymosin-alpha and ERp57 in human gastric cancer. Surgery. 2007;141(1):41–50. | |
Ayshamgul H, Ma H, Ilyar S, Zhang LW, Abulizi A. Association of defective HLA-I expression with antigen processing machinery and their association with clinicopathological characteristics in Kazak patients with esophageal cancer. Chin Med J (Engl). 2011;124(3):341–346. | |
Mehta AM, Jordanova ES, Kenter GG, Ferrone S, Fleuren GJ. Association of antigen processing machinery and HLA class I defects with clinicopathological outcome in cervical carcinoma. Cancer Immunol Immunother. 2008;57(2):197–206. | |
Tanaka T, Kuramitsu Y, Fujimoto M, Naito S, Oka M, Nakamura K. Downregulation of two isoforms of ubiquitin carboxyl-terminal hydrolase isozyme L1 correlates with high metastatic potentials of human SN12C renal cell carcinoma cell clones. Electrophoresis. 2008;29(12):2651–2659. | |
Hetz C, Russelakis-Carneiro M, Walchli S, et al. The disulfide isomerase Grp58 is a protective factor against prion neurotoxicity. J Neurosci. 2005;25(11):2793–2802. | |
Corazzari M, Lovat PE, Armstrong JL, et al. Targeting homeostatic mechanisms of endoplasmic reticulum stress to increase susceptibility of cancer cells to fenretinide-induced apoptosis: the role of stress proteins ERdj5 and ERp57. Br J Cancer. 2007;96(7):1062–1071. | |
Lwin ZM, Yip GW, Chew FT, Bay BH. Downregulation of ER60 protease inhibits cellular proliferation by inducing G1/S arrest in breast cancer cells in vitro. Anat Rec (Hoboken). 2012;295(3):410–416. | |
Chung H, Cho H, Perry C, et al. Downregulation of ERp57 expression is associated with poor prognosis in early-stage cervical cancer. Biomarkers. 2013;18(7):573–579. | |
Jessop CE, Tavender TJ, Watkins RH, Chambers JE, Bulleid NJ. Substrate specificity of the oxidoreductase ERp57 is determined primarily by its interaction with calnexin and calreticulin. J Biol Chem. 2009;284(4):2194–2202. | |
Mayer M, Frey S, Koivunen P, Myllyharju J, Buchner J. Influence of the oxidoreductase ER57 on the folding of an antibody fab fragment. J Mol Biol. 2004;341(4):1077–1084. | |
Chichiarelli S, Ferraro A, Altieri F, et al. The stress protein ERp57/GRP58 binds specific DNA sequences in HeLa cells. J Cell Physiol. 2007;210(2):343–351. | |
Mandic A, Hansson J, Linder S, Shoshan MC. Cisplatin induces endoplasmic reticulum stress and nucleus-independent apoptotic signaling. J Biol Chem. 2003;278(11):9100–9106. | |
Mosmann T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J Immunol Methods. 1983;65(1–2):55–63. | |
Ng WK, Yazan LS, Ismail M. Thymoquinone from Nigella sativa was more potent than cisplatin in eliminating of SiHa cells via apoptosis with down-regulation of Bcl-2 protein. Toxicol In Vitro. 2011;25(7):1392–1398. | |
Rahman HS, Rasedee A, Abdul AB, et al. Zerumbone-loaded nanostructured lipid carrier induces G2/M cell cycle arrest and apoptosis via mitochondrial pathway in a human lymphoblastic leukemia cell line. Int J Nanomedicine. 2014;9:527–538. | |
Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. | |
Nakatani K, Nakamura M, Uzawa K, et al. Establishment and gene analysis of a cisplatin-resistant cell line, Sa-3R, derived from oral squamous cell carcinoma. Oncol Rep. 2005;13(4):709–714. | |
Bernardini M, Lee CH, Beheshti B, et al. High-resolution mapping of genomic imbalance and identification of gene expression profiles associated with differential chemotherapy response in serous epithelial ovarian cancer. Neoplasia. 2005;7(6):603–613. | |
Coling DE, Ding D, Young R, et al. Proteomic analysis of cisplatin-induced cochlear damage: methods and early changes in protein expression. Hear Res. 2007;226(1–2):140–156. | |
Meijer TW, Schuurbiers OC, Kaanders JH, et al. Differences in metabolism between adeno- and squamous cell non-small cell lung carcinomas: spatial distribution and prognostic value of GLUT1 and MCT4. Lung Cancer. 2012;76(3):316–323. | |
Clifford GM, Rana RK, Franceschi S, Smith JS, Gough G, Pimenta JM. Human papillomavirus genotype distribution in low-grade cervical lesions: comparison by geographic region and with cervical cancer. Cancer Epidemiol Biomarkers Prev. 2005;14(5):1157–1164. | |
Schwarz E, Freese UK, Gissmann L, et al. Structure and transcription of human papillomavirus sequences in cervical carcinoma cells. Nature. 1985;314(6006):111–114. | |
Meissner JD. Nucleotide sequences and further characterization of human papillomavirus DNA present in the CaSki, SiHa and HeLa cervical carcinoma cell lines. J Gen Virol. 1999;80 Pt 7:1725–1733. | |
Baker CC, Phelps WC, Lindgren V, Braun MJ, Gonda MA, Howley PM. Structural and transcriptional analysis of human papillomavirus type 16 sequences in cervical carcinoma cell lines. J Virol. 1987;61(4):962–971. | |
Brimer N, Vande Pol SB. Papillomavirus E6 PDZ interactions can be replaced by repression of p53 to promote episomal human papillomavirus genome maintenance. J Virol. 2014;88(5):3027–3030. | |
Zhang B, Chen W, Roman A. The E7 proteins of low- and high-risk human papillomaviruses share the ability to target the pRB family member p130 for degradation. Proc Natl Acad Sci U S A. 2006;103(2):437–442. | |
Longworth MS, Laimins LA. Pathogenesis of human papillomaviruses in differentiating epithelia. Microbiol Mol Biol Rev. 2004;68(2):362–372. | |
Su PF, Wu FY. Differential suppression of the tumorigenicity of HeLa and SiHa cells by adeno-associated virus. Br J Cancer. 1996;73(12):1533–1537. | |
Courtete J, Sibler AP, Zeder-Lutz G, et al. Suppression of cervical carcinoma cell growth by intracytoplasmic codelivery of anti-oncoprotein E6 antibody and small interfering RNA. Mol Cancer Ther. 2007;6(6):1728–1735. | |
Xu D, Perez RE, Rezaiekhaligh MH, Bourdi M, Truog WE. Knockdown of ERp57 increases BiP/GRP78 induction and protects against hyperoxia and tunicamycin-induced apoptosis. Am J Physiol Lung Cell Mol Physiol. 2009;297(1):L44–L51. | |
Urano F, Wang X, Bertolotti A, et al. Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science. 2000;287(5453):664–666. | |
Liu FT, Newland AC, Jia L. Bax conformational change is a crucial step for PUMA-mediated apoptosis in human leukemia. Biochem Biophys Res Commun. 2003;310(3):956–962. | |
Nakano K, Vousden KH. PUMA, a novel proapoptotic gene, is induced by p53. Mol Cell. 2001;7(3):683–694. | |
Guo GG, Patel K, Kumar V, et al. Association of the chaperone glucose-regulated protein 58 (GRP58/ER-60/ERp57) with Stat3 in cytosol and plasma membrane complexes. J Interferon Cytokine Res. 2002;22(5):555–563. | |
Ramirez-Rangel I, Bracho-Valdes I, Vazquez-Macias A, Carretero-Ortega J, Reyes-Cruz G, Vazquez-Prado J. Regulation of mTORC1 complex assembly and signaling by GRp58/ERp57. Mol Cell Biol. 2011;31(8):1657–1671. | |
Serra V, Markman B, Scaltriti M, et al. NVP-BEZ235, a dual PI3K/mTOR inhibitor, prevents PI3K signaling and inhibits the growth of cancer cells with activating PI3K mutations. Cancer Res. 2008;68(19):8022–8030. | |
Catlett-Falcone R, Landowski TH, Oshiro MM, et al. Constitutive activation of Stat3 signaling confers resistance to apoptosis in human U266 myeloma cells. Immunity. 1999;10(1):105–115. | |
Zou CG, Cao XZ, Zhao YS, et al. The molecular mechanism of endoplasmic reticulum stress-induced apoptosis in PC-12 neuronal cells: the protective effect of insulin-like growth factor I. Endocrinology. 2009;150(1):277–285. | |
Li F, Rajendran P, Sethi G. Thymoquinone inhibits proliferation, induces apoptosis and chemosensitizes human multiple myeloma cells through suppression of signal transducer and activator of transcription 3 activation pathway. Br J Pharmacol. 2010;161(3):541–554. | |
Sethi G, Ahn KS, Aggarwal BB. Targeting nuclear factor-κB activation pathway by thymoquinone: role in suppression of antiapoptotic gene products and enhancement of apoptosis. Mol Cancer Res. 2008;6(6):1059–1070. | |
Sakalar C, Yuruk M, Kaya T, Aytekin M, Kuk S, Canatan H. Pronounced transcriptional regulation of apoptotic and TNF-NF-kappa-B signaling genes during the course of thymoquinone mediated apoptosis in HeLa cells. Mol Cell Biochem. 2013;14:14. | |
Wada T, Penninger JM. Mitogen-activated protein kinases in apoptosis regulation. Oncogene. 2004;23(16):2838–2849. | |
Feng R, Zhai WL, Yang HY, Jin H, Zhang QX. Induction of ER stress protects gastric cancer cells against apoptosis induced by cisplatin and doxorubicin through activation of p38 MAPK. Biochem Biophys Res Commun. 2011;406(2):299–304. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.