Back to Journals » Drug Design, Development and Therapy » Volume 8
Potent tumor targeting drug release system comprising MMP-2 specific peptide fragment with self-assembling characteristics
Authors Hua D, Kong W, Zheng X, Zhou Z, Yu B, Li Y, Wang Y, Yang X, Liu C, Tang L, Li Y, Gong M
Received 6 May 2014
Accepted for publication 28 June 2014
Published 14 October 2014 Volume 2014:8 Pages 1839—1849
DOI https://doi.org/10.2147/DDDT.S67305
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 5
Dan Hua,1 Weiling Kong,2 Xuemin Zheng,2 Zhixing Zhou,2 Bing Yu,2 Yazhou Li,2 Yuli Wang,2 Xue Yang,2 Changxiao Liu,2 Lida Tang,2 Ying Li,3 Min Gong2
1School of Pharmacy, Tianjin Medical University, 2Tianjin Institute of Pharmaceutical Research, State Key Laboratory of Drug Delivery and Pharmacokinetics, 3Tianjin Neurological Institute, Tianjin Medical University General Hospital, Tianjin, People’s Republic of China
Abstract: Self-assembling peptides are capable of forming a complex containing a cavity where cytotoxic agents can be wrapped in a self-assembling manner. These complexes are beneficial for improving the pharmacological properties and pharmacokinetics of cytotoxic agents, such as doxorubicin and paclitaxel. In the present study, this self-assembling feature was successfully integrated into a hexapeptide with matrix metalloproteinase (MMP)-2 specific targeting activity, producing a supramolecule possessing controlled drug release characteristics. The MMP-2 specific substrate fragment, PVGLIG, makes this supramolecule disassociate in the presence of MMP-2, and this system is considered to be a powerful tool for the treatment of tumors with high expression of MMP-2 or tumor metastasis. Our findings show that this modified self-assembling peptide with the PVGLIG fragment was able to significantly enhance specificity against HT1080 cells, a tumor cell line with high expression of MMP-2. In addition, residence time of the complex in blood was prolonged since paclitaxel was wrapped into the supramolecule. Our results suggest that the modified MMP-2 specific substrate, SAMTA7, could act as a controlled and sustained drug carrier for treatment of tumors with high expression of MMP-2 and for tumor metastasis.
Keywords: matrix metalloproteinase, self-assembly, drug targeting system, paclitaxel
Introduction
Levels of membrane-type matrix metalloproteinases (MMPs), which are capable of degrading constituents of the extracellular matrix and the cellular membrane,1 are abnormally elevated in tumor cells, leading to metastases by invasion of the extracellular matrix at sites distant from the primary tumor.2–4 Gelatinases are overexpressed in metastatic tumor cells when compared with their nonmetastatic counterparts, and abnormal expression of gelatinases by endothelial cells plays a key role in angiogenesis.5 It was found that gelatinases are also produced by nonmalignant cells present in tumor cells, such as mesenchymal cells and tumor-infiltrating macrophages.6 Gelatinases and other proteinases are critical for migration of endothelial cells and extensive tissue remodeling,6–9 which are necessary for formation of new capillaries and angiogenesis.10 Because gelatinases and other MMPs are potential targets for therapeutic intervention in cancer, there has been much interest in developing synthetic MMP inhibitors, especially for MMP-2 and MMP-9. It was previously reported that MMP-2 (gelatinase A, 72 kDa) and MMP-9 (gelatinase B, 92 kDa) play a critical role in tumor progression, angiogenesis, and metastasis. These enzymes help tumor cells to survive and grow by breaking down the extracellular matrix, releasing growth factors to stimulate cell proliferation and angiogenesis factors to promote blood vessel formation.8,9
In experimental animals, generic MMP-2 or MMP-9 inhibitors prevented tumor dissemination and formation of metastases via their antiangiogenic properties.11–15 MMPs are generally inhibited by compounds containing reactive zinc-chelating groups, such as thiol or hydroxamate.16,17 In recent years, several MMP inhibitors have been developed and investigated at the preclinical stage, and selected candidates are being evaluated in Phase I–III studies.18–22 Unfortunately, no inhibitors specific for MMP family members have been described to date.
The specific protease activity of MMPs provides an alternative strategy for controlled drug release. The MMP-activated drug targeting system is designed for enzymatically metabolized prodrugs, in which therapeutic drugs are covalently bound to MMP substrate peptides.23–25 The prodrug is then activated upon cleavage of the peptide by MMP. There are many such prodrugs developed for the treatment of cancer. For example, a complex of doxorubicin and a peptide specific for MMP was shown to preferentially release leucine-modified doxorubicin and free doxorubicin in a targeted fashion in MMP-expressing xenografts.25 Similarly, Robert Langer’s group has developed a targeted prodrug comprising a conjugate of MMP-specific peptide and methotrexate for tumor-targeted delivery of chemotherapeutics.26,27 The presence of the MMP-specific peptide allows drug release upon MMP-2, an important tumor-associated gelatinase.26–28 It was found that this prodrug specifically inhibited growth of MMP-overexpressing tumor cells, but not RT-112 cells, a slower growing model that does not overexpress MMP.
However, clinical development of MMP-specific peptide and cytotoxic drug conjugates has been greatly limited by their poor stability. A hypothesis was then proposed that peptides with self-assembling features might be beneficial to enhance the stability of these conjugates. In this study, a novel MMP-related controlled drug release system was designed. The MMP-specific peptide was characterized by self-assembling features through integration of active sequences for MMP cleavage. The modified peptides were able to form supramolecules containing an anticancer drug in a self-assembling manner. In a previous study, we showed that this self-assembling peptide successfully produced a supramolecule containing a cavity into which the drug molecule could be loaded.29 Stabilization of a drug such as doxorubicin was thereby improved significantly. It was considered that this self-assembling feature in combination with the targeting properties of MMP-2 might result in a novel drug release system that has both specificity and prolonged stability. If so, use of this tool could avoid the complicated conjugation and purification procedure required to form a peptide-drug complex. It was presumed that a “bifunctional” drug release carrier could be designed, ie, the formation of a complex could improve the stability of the wrapped drug and the MMP-2 protein could “switch on” to release the drug by specific proteolysis. Our data demonstrated that the supramolecule formed by self-assembling the MMP substrate peptide and paclitaxel was stable, and that the drug could be released upon specific cleavage by MMP-2. Here we similarly used a heptapeptide as an MMP-2 specific substrate peptide fragment.26,27 After modification of this fragment with a self-assembling feature, paclitaxel was loaded in for formation of a drug-containing supramolecule. The results of an in vitro assay suggested that the candidate drug (SAMTA7) exerted an antiproliferative effect specific to an HT1080 cell line overexpressing MMP-2 when compared with HEp2 cells that do not express MMP. In addition, SAMTA7 had marked antiproliferative activity in mice bearing HT1080 tumors. Along with a prolonged blood residence time in experimental animals treated with SAMTA7, SAMTA7 was a potent sustained and controlled drug release carrier.
In summary, use of peptides in drug targeting has been a rapidly expanding area in the controlled drug release field. Although limited by poor stabilization, integration of the self-assembling feature into targeting peptides offers a new pathway for development of a sustained and targeted drug carrier system for clinical applications.
Materials and methods
Materials
All the peptides used in this study were purchased from Sangon Biotech Co., Ltd., (Shanghai, People’s Republic of China) and purified by high-performance liquid chromatography (HPLC). Purity was >95%, as identified by mass spectrometry (MS). The freeze-dried peptide was weighed and dissolved in saline to make 10 mg/mL stock solutions for further analysis. Paclitaxel was obtained from Demochem Medical Tech Co., Ltd., (Shanghai, People’s Republic of China). All other chemicals were obtained from Sigma-Aldrich (St Louis, MO, USA), unless otherwise indicated.
Cell culture
HT1080 and HEp2 cell lines were cultured in Dulbecco’s Modified Eagle’s Medium (Gibco; Invitrogen, Carlsbad, CA, USA) and Roswell Park Memorial Institute 1640 medium (Gibco) supplemented with 10% fetal bovine serum (Gibco) at 37°C in a 5% CO2 atmosphere.
Analysis of paclitaxel and self-assembled MMP substrate peptide mixture by HPLC
Paclitaxel and various peptides were mixed at several different molar ratios and incubated at 4°C overnight in order to investigate whether the modified peptides had the ability to self-assemble. Varying amounts of peptides were used, while the concentration of paclitaxel was kept at 10 μM. The final molar ratios of paclitaxel to peptides were 1:1, 1:5, and 1:10, respectively. Mixtures (20 μL) containing 10% dimethyl sulfoxide were analyzed using a Surveyor HPLC system with a C18 analytical column (Thermo Scientific, Waltham, MA, USA). The column was eluted at a flow rate of 1 mL per minute in gradient mode with a mixture of mobile phase A (H2O +20% acetonitrile +0.1% trifluoroacetic acid) and mobile phase B (acetonitrile +0.1% trifluoroacetic acid). Mobile phase A was eluted for 10 minutes and mobile phase B was then increased from 20% to 100% over 60 minutes. HPLC analyses were performed at ambient temperature and the ultraviolet detection wavelength was set at 214 nm. Next, 20 μL aliquots of paclitaxel solution (10 μM) and SAMTA7 stock solution (1 mg/mL) were injected into the C18 HPLC column as the controls.
MMP-2 proteolysis assays
Proteolysis assays were performed to determine whether the complexes formed by the modified peptides and paclitaxel were still cleavable by the MMP-2 enzyme. Various complexes of peptides and paclitaxel at a molar ratio of 1:10 (peptide to paclitaxel, the amount of paclitaxel being 100 μg) were incubated with and without MMP-2 (5 μg) in phosphate-buffered saline (pH 7.0) containing 100 μM ZnSO4 at 37°C for 48 hours. HPLC-MS was used to monitor the amount of free paclitaxel released from the mixture.
HPLC-MS measurements
Three Thermo Finnigan quadrupole MS devices (TSQ7000) with Xcalibur® and Lcquan® software were equipped with an electrospray ion source and a diverter valve. The chromatographic system consisted of an HPLC model 616 pump and a 616S controller from Waters Corporation (Milford, MA, USA) and a Series 200 autosampler from Perkin-Elmer (Norwalk, CT, USA). An Oasis HLB chromatography column (30 mm particle size, 1×50 mm) was purchased from Waters. Electrospray ionization was performed in the positive ion mode. The heated capillary was set at 245°C and the spray voltage at 4.5 kV. Nitrogen was used as the sheath and auxiliary gas, and set at 90 lb per square inch and 25 arbitrary units, respectively. The argon collision gas pressure was set to 2.5 mTorr. The plasma samples or cell lysis solutions (50 μL) were chromatographed at ambient temperature on the Oasis HLB column. Solvent A was acetonitrile and solvent B was 5 mM sodium trifluoroacetate in 0.1% aqueous formic acid. The analyses were chromatographed at a flow rate of 0.8 mL per minute with the following linear gradient: 0 minute, 5% A; 0.5 minutes, 5% A; 5.5 minutes, 82% A. The gradient was followed by a 1.0 mL per minute rinse for 1 minute using 100% methanol and 30 seconds using 95% acetonitrile, and then re-equilibration of the column to the initial conditions. The flow was diverted to the MS device for quantitative analysis.
MTT assay
The cytotoxicity of free paclitaxel and that of the SAMTA7-paclitaxel complex were determined using the 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyl tetrazolium bromide (MTT) assay. HT1080, HEp2, MCF7, and U-87MG cells (1×105 cells/100 μL per well) were cultured in 96-well plates at 37°C in 5% CO2. Aqueous solutions of free paclitaxel containing a final concentration of 2% dimethyl sulfoxide or the complex of SAMTA7 and paclitaxel (molar ratio at 1:6) were dissolved in culture medium at final paclitaxel concentrations of 0.1, 0.5, 1, 5, 10, and 100 μg/mL. After incubation for 24, 48, and 72 hours, the MTT solution (in 2 mg/mL phosphate-buffered saline) was added, the plates were incubated for 4 hours, and the cells were lysed with 50% N,N-dimethylformamide containing 20% sodium dodecyl sulfate (pH 4.5). Absorbance at 570 nm was measured for each well using a SpectraMax M5 (Molecular Devices, Sunnyvale, CA USA). The absorbance of control cells was taken as 100% viability, and the values of the treated cells were calculated as a percentage of the control.
Scratch assay
HT1080 and HEp2 cells were seeded in 24-well plates and cultured overnight to form a confluent monolayer. After being scratched with a sterile P200 pipette tip, the cells were rinsed gently with phosphate-buffered saline to remove the detached cells. Subsequently, cell cultures containing either paclitaxel alone or the SAMTA7-paclitaxel complex (final paclitaxel concentration 5 μg/mL, molar ratio 1:6) were added to the scratched plates and then incubated at 37°C in a 5% CO2 atmosphere for 24 hours. A final concentration of 2% dimethyl sulfoxide was titrated into the cell lines treated with paclitaxel alone. The distances between the two edges of the scratched cells were observed using an ICC50 HD microscope (Leica Microsystems Ltd., Nunningen, Switzerland). The rate of inhibition of metastasis was calculated using the formula: inhibition rate = (distance of experimental group after inhibition – distance of control group after inhibition – distance prior to inhibition)/distance prior to healing ×100.
Treatment of mice bearing human tumor xenografts
Female DBA/2 mice aged 6 weeks were obtained from the Shanghai Laboratory Animal Company (China Academy of Sciences, Shanghai, People’s Republic of China). All animal studies were conducted in accordance with the protocol approved by the AAALAC (Association for Assessment and Accreditation of Laboratory Animal Care) guidelines in Tianjin Good Laboratory Practice Laboratory (Tianjin, People’s Republic of China). Female DBA/2 mice (weight 16–20 mg) were housed in barrier facilities on a 12-hour light/dark cycle. Food and water were supplied ad libitum. On day 0, five groups of mice were inoculated by subcutaneous implantation of HT1080, HEp2, or U-87MG tumor cells (2×105) in 0.5 mL of Roswell Park Memorial Institute 1640 medium. Tumors were then allowed to grow to about 100 mm3 on the backs of the mice. This experimental protocol in mice with human tumor xenografts is designed to mimic the clinical situation where treatment begins after the tumor has already been established. The animals were treated with free paclitaxel or the SAMTA7-paclitaxel complex by intravenous administration of a specified dose (3 mg/kg body weight) every 5 days. Controls were treated with SAMTA7 alone. Survival time was recorded in days after tumor injection, and weight and tumor size were monitored three times a week during the 30-day study period.
Tumor size was estimated as an ellipsoid volume according to the formula: size = width2 × length × π/6. Mean and median survival time and the statistical significance of the results were determined using a two-tailed Wilcoxon rank test. All data obtained for repeat experiments were pooled and used in the statistical analysis.
Simulation of molecular dynamics
The initial system containing SAMTA7 peptides was constructed using the Molecular Operating Environment system. Arg(R) and Ala(A) have a positive charge while Asp(D) has a negative charge. Therefore, the terminal residuals were formed the construction-like “hairpin” through the Molecular Dynamics simulation. The paclitaxel and six SAMTA7 peptides were then immersed in an anhydrous simulated box for MS simulation simultaneously for 10 msec for the anhydrous system. The time step was 1 fsec, and the trajectories were saved every 20 psec for further analysis. The GROMOS96 53a6 force field was used. The reference temperature was set to 310 K.
Statistical analysis
Statistical significance was evaluated using the Kruskal–Wallis H test. P<0.05 was considered to be statistically significant.
Results
Seven modified peptides were synthesized and validated for the PVGLIG sequences, and are listed in Table 1. Initially, three strategies were considered for characterizing the self-assembling characteristics of the PVGLIG peptide, including additional EAK fragments, RADA fragment(s), and amphipathic modification.
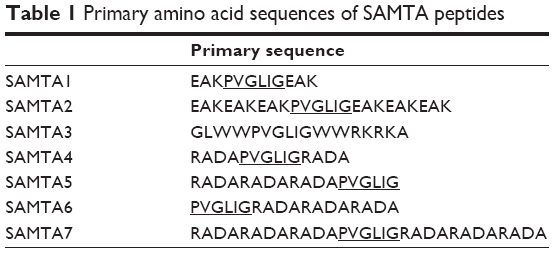 | Table 1 Primary amino acid sequences of SAMTA peptides |
The designed peptides were incubated with free paclitaxel, followed by an HPLC study in order to identify which peptide is capable of forming a supramolecule with paclitaxel. The chromatographic results suggest that the self-assembling properties of SAMTA7 were significant. Figure 1A and 1B show the HPLC spectra for SAMTA7 and free paclitaxel, respectively. SAMTA7 appeared not to interact with free paclitaxel at a molar ratio at 1:1, similarly in Figure 1C. The HPLC spectra for the complex of free paclitaxel and SAMTA7 at a molar ratio at 1:5 is shown in Figure 1D, where a peak retention time of 13.5 minutes is identified, as well as the peak indicating free paclitaxel is diminished (at 30 minutes) suggesting that a complex of SAMTA7 and paclitaxel was formed. In order to confirm the components of the peak, the fraction collected was treated with 100 mM ammonium acetate. The sample was then loaded onto the HPLC-MS system for analysis and the spectrum showed two distinct peaks; the MS result confirmed these to be for paclitaxel and SAMTA7. In addition, it was revealed that the components of collected peak at 13.5 minutes were consisted of SAMTA7 and paclitaxel upon the molar ratio at 6:1. The complex was prepared accordingly and then analyzed by HPLC. The spectra showed a distinct peak at 13.5 minutes and the peaks of SAMTA7 and paclitaxel disappeared (see Figure 1E).
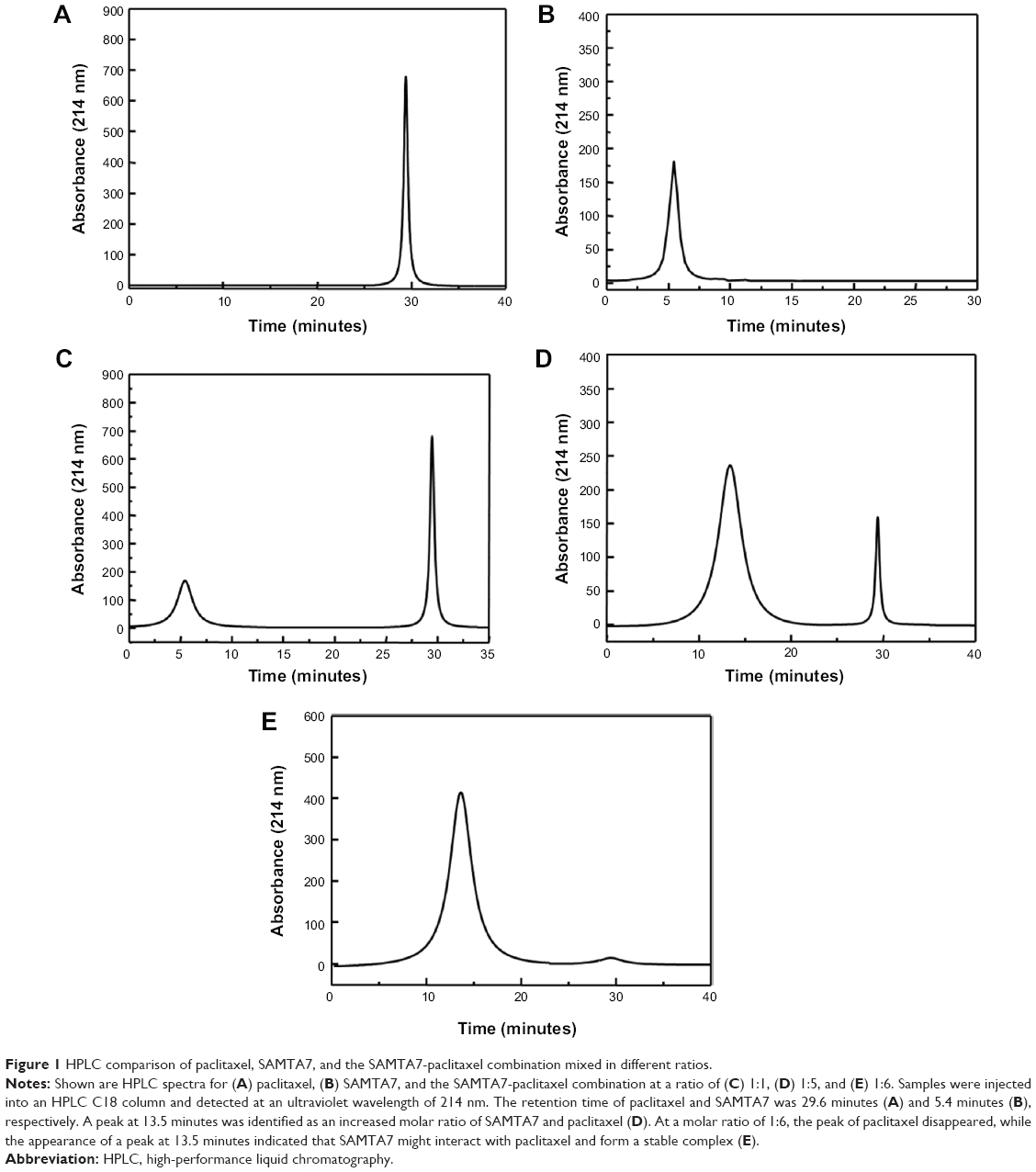 | Figure 1 HPLC comparison of paclitaxel, SAMTA7, and the SAMTA7-paclitaxel combination mixed in different ratios. |
Upon formation of the SAMTA7 and paclitaxel supramolecule, it was necessary to investigate whether this supramolecule is able to release paclitaxel in the presence of the MMP-2 enzyme. A proteolysis assay and an MS study were performed to detect release of paclitaxel from the supramolecule over 24 hours. As seen in Figure 2, the supramolecule formed by SAMTA7 started releasing the drug after incubation with MMP-2 for 2 hours. Paclitaxel seemed to be fully released from the supramolecules by 24 hours; result indicated that SAMTA7 provided complete wrapping for paclitaxel, compared with other peptides.
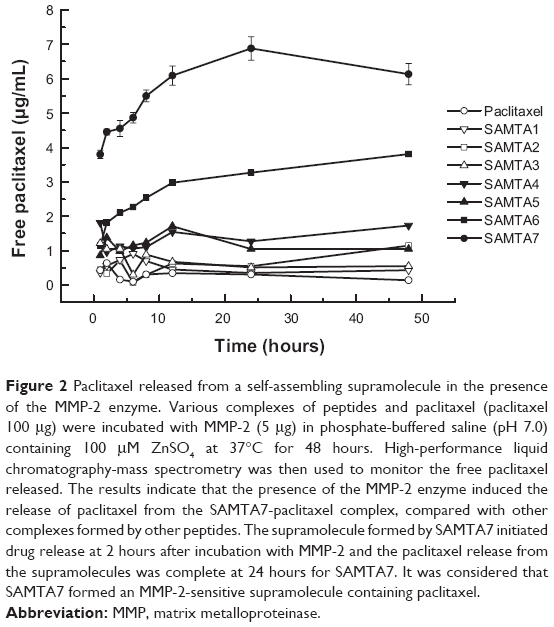 | Figure 2 Paclitaxel released from a self-assembling supramolecule in the presence of the MMP-2 enzyme. Various complexes of peptides and paclitaxel (paclitaxel 100 μg) were incubated with MMP-2 (5 μg) in phosphate-buffered saline (pH 7.0) containing 100 μM ZnSO4 at 37°C for 48 hours. High-performance liquid chromatography-mass spectrometry was then used to monitor the free paclitaxel released. The results indicate that the presence of the MMP-2 enzyme induced the release of paclitaxel from the SAMTA7-paclitaxel complex, compared with other complexes formed by other peptides. The supramolecule formed by SAMTA7 initiated drug release at 2 hours after incubation with MMP-2 and the paclitaxel release from the supramolecules was complete at 24 hours for SAMTA7. It was considered that SAMTA7 formed an MMP-2-sensitive supramolecule containing paclitaxel. |
Thereafter, cellular MTT and scratch assays were used to assess the antitumor activity of the supramolecule formed by SAMTA7 and paclitaxel. Using the MTT assay, the antiproliferative activity of the SAMTA7-paclitaxel complex was MMP-2-specific. In the HT1080 cell line, where MMP-2 is highly expressed, treatment with the SAMTA7-paclitaxel supramolecule resulted in significant loss of cell viability when compared with HEp2 cells, which do not express MMP-2 (see Table 2). From the MTT result, it is clear that the SAMTA7-paclitaxel supramolecule has the ability to inhibit activity in cell lines overexpressing MMP-2, such as HT1080. In addition, in the MTT assay, it was found that there were crystals of paclitaxel surrounding the HT1080 cells only (Figure S1). It was presumed that the complex released the paclitaxel specific in the presence of MMP-2 paclitaxel was oversaturated and then crystallized. This finding provided another explanation that the SAMTA7-paclitaxel complex can release paclitaxel in MMP-2 specific degradation.
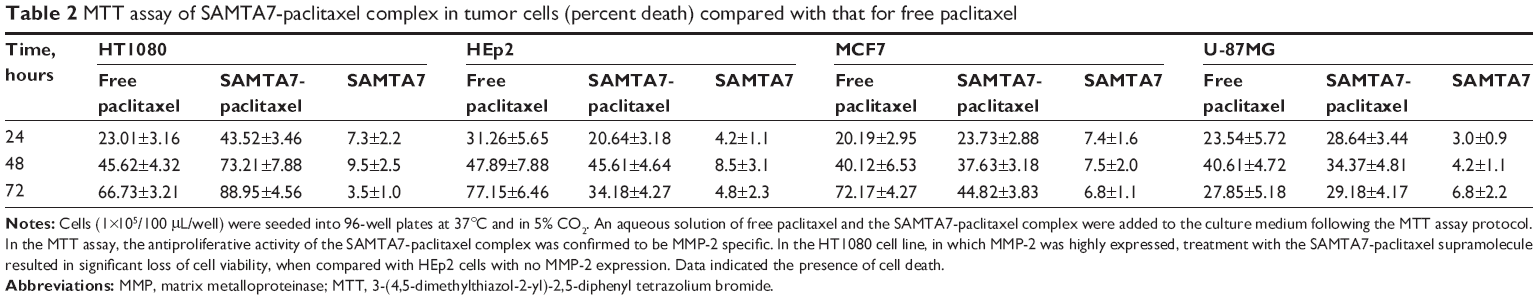 | Table 2 MTT assay of SAMTA7-paclitaxel complex in tumor cells (percent death) compared with that for free paclitaxel |
Since MMP-2 plays a critical role in tumor progression, tumor angiogenesis, and metastasis, the ability of the SAMTA7-paclitaxel supramolecule to inhibit tumor cell metastasis was evaluated using the cellular scratch assay. The results of this assay showed that the SAMTA7-paclitaxel supramolecule significantly inhibited migration of HT1080 cells but not HEp2 cells (Figure 3). Compared with the migration inhibiting effect of free paclitaxel in HT1080 cells, the supramolecule showed a 5.3-fold increase in antimigratory activity, along with the specific targeting properties as it was presumed that MMP-2 specific self assembling peptide could provide a specific drug release upon the presence of MMP-2 protein. It was assumed that the inhibitory effect of the supramolecule on the cellular migrating phenotype is due to the specific release of paclitaxel in the presence of the MMP-2 enzyme.
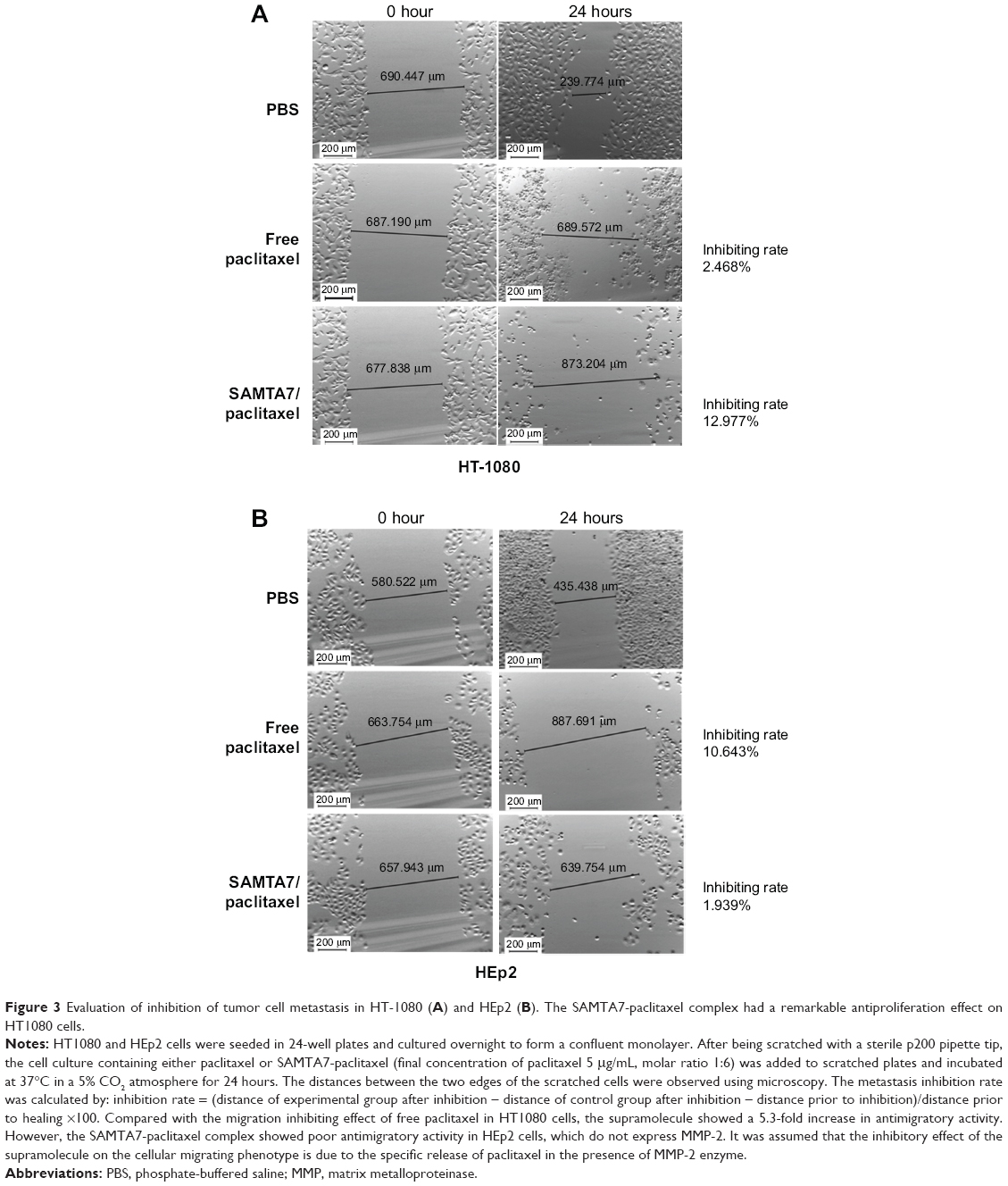 | Figure 3 Evaluation of inhibition of tumor cell metastasis in HT-1080 (A) and HEp2 (B). The SAMTA7-paclitaxel complex had a remarkable antiproliferation effect on HT1080 cells. |
In order to study the effect of the SAMTA7-paclitaxel supramolecule, mice bearing HT1080, HEp2, or U-87MG xenografts were treated with the supramolecule and free paclitaxel, respectively. The antitumor activity of the SAMTA7-paclitaxel supramolecule was determined by monitoring tumor volume after treatment in HT1080 tumor-bearing mice (see Figure 4). The supramolecule containing paclitaxel was administered at a paclitaxel dose of 3 mg/kg body weight for 35 days. The free paclitaxel was given at an identical dose and frequency as the control. Figure 4A shows the reduction in tumor size in the mice bearing HT1080 cells and the results revealed that the tumor size was remarkably reduced compared to that of free paclitaxel. However, the supramolecule had poor antitumor activity in mice bearing HEp2 and U-87MG tumors, as shown in Figure 4B and C.
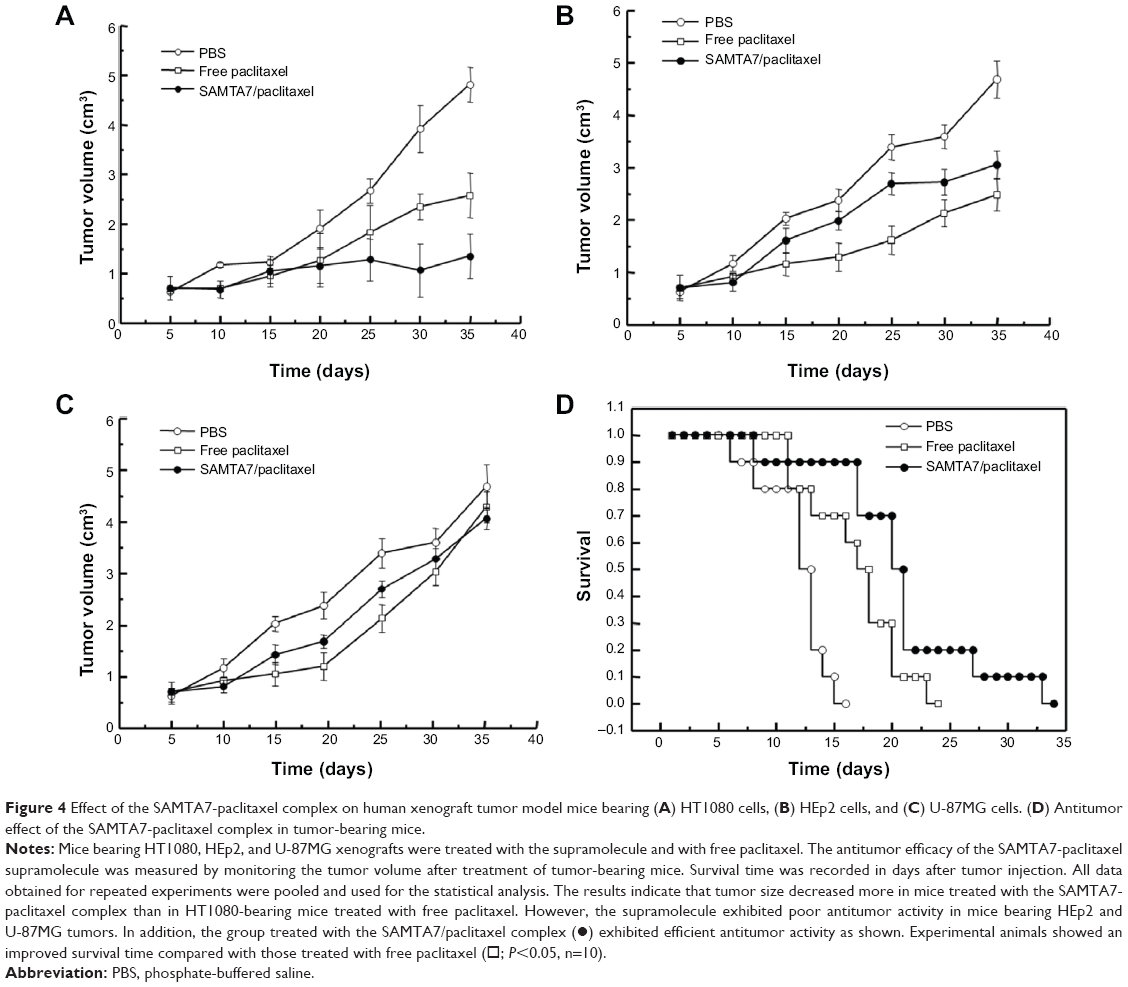 | Figure 4 Effect of the SAMTA7-paclitaxel complex on human xenograft tumor model mice bearing (A) HT1080 cells, (B) HEp2 cells, and (C) U-87MG cells. (D) Antitumor effect of the SAMTA7-paclitaxel complex in tumor-bearing mice. |
Mice bearing HT1080 tumors were administered phosphate-buffered saline, free paclitaxel, and the SAMTA7-paclitaxel complex. Figure 4D shows that the SAMTA7-paclitaxel complex increased the survival rate in these mice.
The SAMTA7 indicated the molecular dynamic simulation in order to clarify its self-assembling properties and its interaction with paclitaxel. Figure 5A shows that the single SAMTA7 was constrained in a “hairpin”-like conformation upon interaction with RADA fragments. This conformation explored that the MMP-2 substrate peptide, PVGLIG fragment, is vulnerable to MMP-2 proteolysis. Six SAMTA7 molecules and one paclitaxel molecule were put into a simulation box for structural analysis of the supramolecule. Figure 5B indicates that SAMTA7 has self-assembling properties and is able to wrap paclitaxel inside. Also, this formation of supramolecule has not altered the phenomenon that PVGLIG remained exposed against MMP-2 protein.
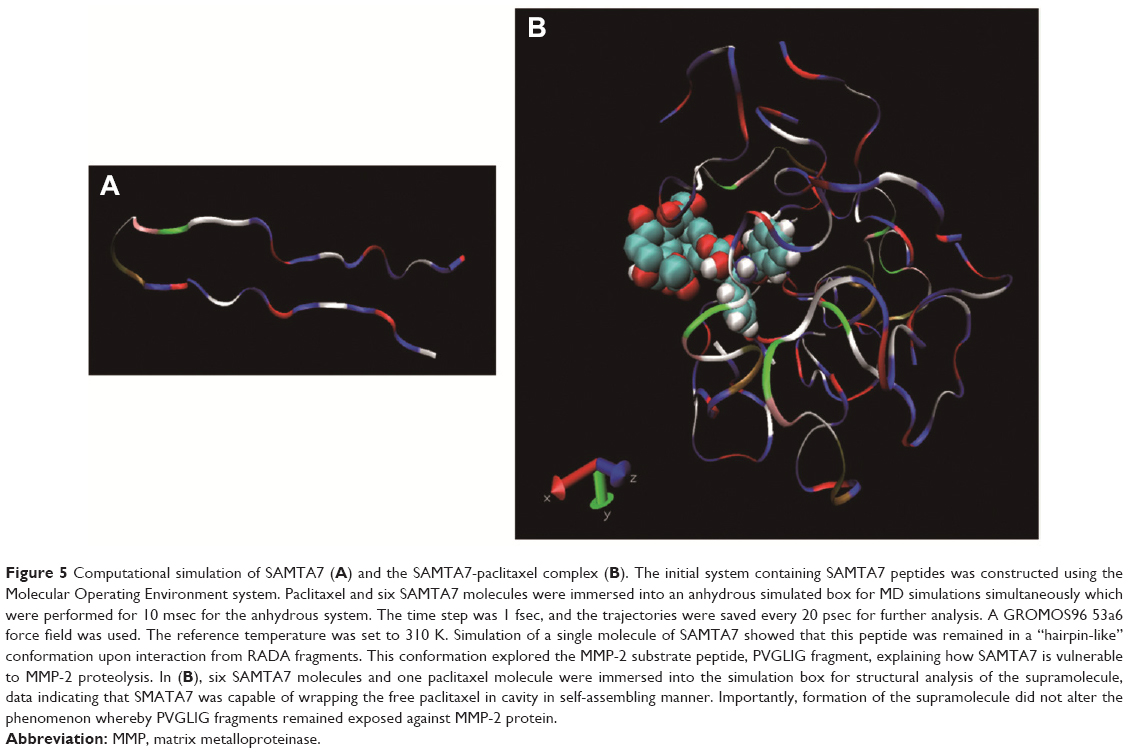 | Figure 5 Computational simulation of SAMTA7 (A) and the SAMTA7-paclitaxel complex (B). The initial system containing SAMTA7 peptides was constructed using the Molecular Operating Environment system. Paclitaxel and six SAMTA7 molecules were immersed into an anhydrous simulated box for MD simulations simultaneously which were performed for 10 msec for the anhydrous system. The time step was 1 fsec, and the trajectories were saved every 20 psec for further analysis. A GROMOS96 53a6 force field was used. The reference temperature was set to 310 K. Simulation of a single molecule of SAMTA7 showed that this peptide was remained in a “hairpin-like” conformation upon interaction from RADA fragments. This conformation explored the MMP-2 substrate peptide, PVGLIG fragment, explaining how SAMTA7 is vulnerable to MMP-2 proteolysis. In (B), six SAMTA7 molecules and one paclitaxel molecule were immersed into the simulation box for structural analysis of the supramolecule, data indicating that SMATA7 was capable of wrapping the free paclitaxel in cavity in self-assembling manner. Importantly, formation of the supramolecule did not alter the phenomenon whereby PVGLIG fragments remained exposed against MMP-2 protein. |
In summary, an MMP-2 targeted drug release system was developed based on the MMP-2-specific substrate peptide of PVGLIG in this study. The hexapeptide, PVGLIG, was wrapped with self-assembling properties via structural modification to obtain SAMTA7, which appears to have both self-assembling properties and MMP-2-specific targeting activity. SAMTA7 is capable of forming a supramolecule in a self-assembling manner and can then wrap a drug (eg, paclitaxel) inside of the cavity of the supramolecule. Formation of this complex was considered to increase the therapeutic index of paclitaxel, due to the enhanced specific targeting activity.
Conclusion
Self-assembling peptides have been investigated extensively over several decades; however, it has only been used as a targeted drug-release carrier recently. In this study, a novel drug carrier was designed for controlled release of paclitaxel. This peptide has self-assembling properties and specific binding activity for MMP-2. By comparison of MMP substrate peptide and drug conjugate, in this study the MMP specific peptide was modified to contain the self-assembling property which could form a complex with free paclitaxel. Our findings indicate that the presence of MMP-2 is able to dispatch the supramolecule containing paclitaxel specifically. The residence time of paclitaxel in blood was significantly increased since it was wrapped inside the supramolecule, leading to lower toxicity. The findings of this study indicate that a tumor-targeting peptide might be a powerful tool in clinical studies after safety evaluation of self-assembling peptide.
Acknowledgments
This study was supported by funds from the National Science and Technology Major Project of the Ministry of Science and Technology of China (2014ZX09507005-003) and from the Natural Science Foundation of Tianjin, People’s Republic of China (12JCYBJC31500 and 13RCGFSY19700), and also funded by The National Natural Science foundation (81400932).
Disclosure
The authors report no conflicts of interest in this work.
References
Hagedorn HG, Bachmeier BE, Nerlich AG. Synthesis and degradation of basement membranes and extracellular matrix and their regulation by TGF-beta in invasive carcinomas (Review). Int J Oncol. 2001;18(4):669–681. | ||
Mendes O, Kim HT, Lungu G, Stoica G. MMP-2 role in breast cancer brain metastasis development and its regulation by TIMP2 and ERK1/2. Clin Exp Metastasis. 2007;24(5):341–351. | ||
Kondratiev S, Gnepp DR, Yakirevich E, et al. Expression and prognostic role of MMP-2, MMP-9, MMP13, and MMP14 matrix metalloproteinases in sinonasal and oral malignant melanomas. Hum Pathol. 2008; 39(3):337–343. | ||
Zheng SQ, Huang RQ, Zhang YJ. [Role of matrix metalloproteinase (MMP)-2 and -9 and vascular endothelial growth factor C in lymph node metastasis of breast cancer]. Zhonghua Bing Li Xue Za Zhi. 2010;39(4): 240–244. Chinese. | ||
Negro A, Onisto M, Pellati D, Garbisa S. CNTF up-regulation of TIMP-2 in neuroblastoma cells. Brain Res Mol Brain Res. 1997;48(1):30–36. | ||
Nielsen BS, Sehested M, Kjeldsen L, Borregaard N, Rygaard J, Danø K. Expression of matrix metalloprotease-9 in vascular pericytes in human breast cancer. Lab Invest. 1997;77(4):345–355. | ||
Nguyen M, Arkell J, Jackson CJ. Active and tissue inhibitor of matrix metalloproteinase-free gelatinase B accumulates within human microvascular endothelial vesicles. J Biol Chem. 1998;273(9):5400–5404. | ||
Koivunen E, Arap W, Valtanen H, et al. Tumor targeting with a selective gelatinase inhibitor. Nat Biotechnol. 1999;17(8):768–774. | ||
Droppelmann CA, Gutiérrez J, Vial C, Brandan E. Matrix metalloproteinase-2-deficient fibroblasts exhibit an alteration in the fibrotic response to connective tissue growth factor/CCN2 because of an increase in the levels of endogenous fibronectin. J Biol Chem. 2009;284(20): 13551–13561. | ||
Grant DS, Kinsella JL, Fridman R, et al. Interaction of endothelial cells with a laminin A chain peptide (SIKVAV) in vitro and induction of angiogenic behavior in vivo. J Cell Physiol. 1992;153(3):614–625. | ||
Andey T, Patel A, Jackson T, Safe S, Singh M. 1,1-Bis (3′-indolyl)-1-(p-substitutedphenyl)methane compounds inhibit lung cancer cell and tumor growth in a metastasis model. Eur J Pharm Sci. 2013;50(2): 227–241. | ||
Hu J, Chen C, Su Y, DU J, Qian X, Jin Y. Vascular endothelial growth factor promotes the expression of cyclooxygenase 2 and matrix metalloproteinases in Lewis lung carcinoma cells. Exp Ther Med. 2012;4(6): 1045–1050. | ||
Peng CW, Liu XL, Liu X, Li Y. Co-evolution of cancer microenvironment reveals distinctive patterns of gastric cancer invasion: laboratory evidence and clinical significance. J Transl Med. 2010;15;8:101–107. | ||
Raghuwanshi SK, Smith N, Rivers EJ, et al. G protein-coupled receptor kinase 6 deficiency promotes angiogenesis, tumor progression, and metastasis. J Immunol. 2013;190(10):5329–5336. | ||
Shukla S, MacLennan GT, Fu P, Gupta S. Apigenin attenuates insulin-like growth factor-I signaling in an autochthonous mouse prostate cancer model. Pharm Res. 2012;29(6):1506–1517. | ||
Grams F, Reinemer P, Powers JC, et al. X-ray structures of human neutrophil collagenase complexed with peptide hydroxamate and peptide thiol inhibitors. Implications for substrate binding and rational drug design. Eur J Biochem. 1995;228(3):830–841. | ||
Kruse MN, Becker C, Lottaz D, et al. Human meprin alpha and beta homo-oligomers: cleavage of basement membrane proteins and sensitivity to metalloprotease inhibitors. Biochem J. 2004;378 Pt 2:383–389. | ||
Chu QS, Forouzesh B, Syed S, et al. A phase II and pharmacological study of the matrix metalloproteinase inhibitor (MMPI) COL-3 in patients with advanced soft tissue sarcomas. Invest New Drugs. 2007; 25(4):359–367. | ||
Brinker BT, Krown SE, Lee JY, et al. Phase 1/2 trial of BMS-275291 in patients with human immunodeficiency virus-related Kaposi sarcoma: a multicenter trial of the AIDS Malignancy Consortium. Cancer. 2008;112:1083–1088. | ||
Orchard J, Massey A, Brown R, Cardon-Dunbar A, Hofmann J. Successful management of tendinopathy with injections of the MMP-inhibitor aprotinin. Clin Orthop Relat Res. 2008;466(7):1625–1632. | ||
Gapski R, Hasturk H, Van Dyke TE, et al. Systemic MMP inhibition for periodontal wound repair: results of a multi-centre randomized-controlled clinical trial. J Clin Periodontol. 2009;36(2):149–156. | ||
Skulason S, Holbrook WP, Kristmundsdottir T. Clinical assessment of the effect of a matrix metalloproteinase inhibitor on aphthous ulcers. Acta Odontol Scand. 2009;67(1):25–29. | ||
Shi NQ, Gao W, Xiang B, Qi XR. Enhancing cellular uptake of activable cell-penetrating peptide-doxorubicin conjugate by enzymatic cleavage. Int J Nanomedicine. 2012;7:1613–1621. | ||
Bae M, Cho S, Song J, et al. Metalloprotease-specific poly(ethylene glycol) methyl ether-peptide-doxorubicin conjugate for targeting anticancer drug delivery based on angiogenesis. Drugs Exp Clin Res. 2003;29(1):15–23. | ||
Lee GY, Park K, Kim SY, Byun Y. MMPs-specific PEGylated peptide-DOX conjugate micelles that can contain free doxorubicin. Eur J Pharm Biopharm. 2007;67(3):646–654. | ||
Chau Y, Dang NM, Tan FE, Langer R. Investigation of targeting mechanism of new dextran-peptide-methotrexate conjugates using biodistribution study in matrix-metalloproteinase-overexpressing tumor xenograft model. J Pharm Sci. 2006;95(3):542–551. | ||
Chau Y, Padera RF, Dang NM, Langer R. Antitumor efficacy of a novel polymer-peptide-drug conjugate in human tumor xenograft models. Int J Cancer. 2006;118(6):1519–1526. | ||
Chau Y, Tan FE, Langer R. Synthesis and characterization of dextran-peptide-methotrexate conjugates for tumor targeting via mediation by matrix metalloproteinase II and matrix metalloproteinase IX. Bioconjug Chem. 2004;15(4):931–941. | ||
Li Y, Zheng X, Cao Z, Xu W, Zhang J, Gong M. Self-assembled peptide (CADY-1) improved the clinical application of doxorubicin. Int J Pharm. 2012;434(1–2):209–214. |
Supplementary material
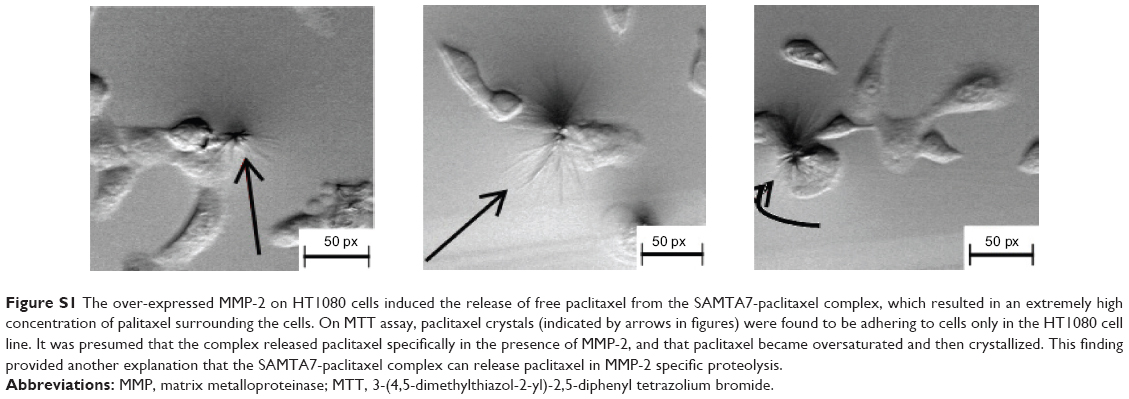 | Figure S1 The over-expressed MMP-2 on HT1080 cells induced the release of free paclitaxel from the SAMTA7-paclitaxel complex, which resulted in an extremely high concentration of palitaxel surrounding the cells. On MTT assay, paclitaxel crystals (indicated by arrows in figures) were found to be adhering to cells only in the HT1080 cell line. It was presumed that the complex released paclitaxel specifically in the presence of MMP-2, and that paclitaxel became oversaturated and then crystallized. This finding provided another explanation that the SAMTA7-paclitaxel complex can release paclitaxel in MMP-2 specific proteolysis. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.