Back to Journals » Drug Design, Development and Therapy » Volume 13
Postconditioning Protection Against Myocardiocyte Anoxia/Reoxygenation Injury From Penehyclidine Hydrochloride
Authors Ren JY, Lin DM ![]() , Wang CB, Yang YL, Wang ZQ, Cui BQ, Ma J
, Wang CB, Yang YL, Wang ZQ, Cui BQ, Ma J
Received 23 July 2019
Accepted for publication 19 October 2019
Published 26 November 2019 Volume 2019:13 Pages 3977—3988
DOI https://doi.org/10.2147/DDDT.S224282
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Tin Wui Wong
JY Ren,* DM Lin,* CB Wang, YL Yang, ZQ Wang, BQ Cui, J Ma
Department of Anesthesiology, Beijing Anzhen Hospital, Capital Medical University-Beijing Institute of Heart Lung and Blood Vessel Diseases, Beijing, People’s Republic of China
*These authors contributed equally to this work
Correspondence: J Ma
Department of Anesthesiology, Beijing Anzhen Hospital, Capital Medical University-Beijing Institute of Heart Lung and Blood Vessel Diseases, No.2 Anzhen Road, Chaoyang District, Beijing 100029, People’s Republic of China
Tel +8613370103571
Email [email protected]
Background/Aims: To investigate the postconditioning protective effect of penehyclidine hydrochloride (PHC) against anoxia/reoxygenation (A/R) injury in H9c2 cells along with the involved mechanism and timing effect.
Methods: We divided H9c2 cells into 7 groups: control group, A/R group and PHC+A/R groups at 0 min, 5 mins, 10 mins, 20 mins, 30 mins, respectively (treated with 0.1 μm/L PHC at 0 min, 5 mins, 10 mins, 20 mins, 30 mins after the reoxygenation procedure began). Cell apoptosis, oxidative stress, intracellular Ca2+ concentration, mitochondrial membrane potential and mitochondrial permeability transition pore (MPTP) opening were explored. Bcl-2, Bax, Cyt C, caspase-3 and caspase-9 levels were measured.
Results: A/R significantly increased both cell injury and cell apoptosis. PHC showed postconditioning protective effect by attenuating superoxide production, decreasing Ca2+ overload, restraining MPTP activities, restoring mitochondrial membrane potential, regulating cell apoptosis proteins and modulation of mitochondrial pathway. Earlier administration of PHC offered greater postconditioning protective effect.
Conclusion: H9c2 cells were protected by PHC from A/R injury regardless of timing of PHC administration (0 min, 5 mins, 10 mins, 20 mins, 30 mins). However, earlier administration of PHC resulted in better PHC postconditioning protection.
Keywords: penehyclidine hydrochloride, PHC, anoxia/reoxygenation injury, A/R injury, cell apoptosis, oxidative stress, mitochondrial pathway
Introduction
Ischemic heart disease is one of the most severe cardiovascular diseases with high morbidity and mortality. The most effective means of minimizing myocardial ischemic injury is early reperfusion of ischemic tissues. However, myocardial reperfusion has its own inherent risks; alternations in myocardial metabolic, electrophysiologic and cellular functions are not uncommon. This is referred to as ischemia-reperfusion injury (IRI).1–3 For the past decade, researchers have explored ways of reducing IRI, including preconditioning and postconditioning. In order to understand the chain of events that ultimately result in IRI, as well as the mechanism from which IRI could be protected by postconditioning treatment, we designed an in vitro experiment using H9c2 cell models through A/R procedures.
Penehyclidine Hydrochloride (PHC) is a selective anticholinergic agent launched by the Institute of Pharmacology and Toxicology, a division within the Chinese Academy of Military Medical Sciences. It was found to have a protective effect against myocardial ischemia-reperfusion injury while having minimal cardiovascular side effects.4 Our previous studies demonstrated that PHC offered reperfusion protection by decreasing calcium overload, minimizing the formation of reactive oxygen species, impeding mitochondrial MPTP opening and reducing inflammatory response in vitro and in vivo. We previously determined that the most effective concentration was 0.1 μm/L.5,6 This current investigation focused on the effect of timing that PHC had on postconditioning. We also designed this in vitro experiment to further elucidate the mechanism of A/R injury in the early reoxygenation period. We specifically looked at whether earlier administration of PHC provided significantly better protection against cellular ischemia-reperfusion injury. We hope our results will lead to clinical applications in the treatment of patients with ischemic heart disease and offer protection against reperfusion injury in surgical patients.
Materials And Methods
H9c2 cell line was purchased from Shanghai Cell Library of China, which was originally obtained from ATCC, Manassas, VA, USA. The cells were cultured at 37°C in a chamber consisting of 5% CO2/95% air. When cells' density reached 90%, the cells were harvested for our experiment.
H9c2 cells were divided into seven groups, as described in Figure 1A:
- Control group: H9c2 cells were first incubated in Tyrode solution followed by cultivation in a chamber with 5% CO2/95% air mixture for 3 hrs. The culture medium of H9c2 cells was then quickly replaced by low-sugar DEME medium outside the chamber. The H9c2 cells were quickly returned to the cell chamber consisting of 5% CO2/95% air for 2 hrs.
- A/R group: H9c2 cells were incubated in Tyrode solution with a gas mixture of 5% CO2/95% N2 for 3 hrs. The cell culture medium was then replaced with a low-sugar DEME solution and returned back to a gas chamber with a mixture of 5% CO2/95% air at 37°C for 2 hrs of reperfusion treatment.
- PHC postconditioning group: Previous steps were the same as in the A/R groups, except with low-sugar DEME containing 0.1 μm/L PHC replacing low-sugar DEME without PHC at 0 min, 5 mins, 10 mins, 20 mins and 30 mins, respectively, after commencement of reoxygenation. (0 min indicated the time during which Tyrode solution was directly replaced by PHC solution when the anoxia period stopped.) Next, low-sugar DEME solution containing no PHC replaced the PHC solution after a 90 mins treatment period. The total anoxia period was 3 hrs with a reoxygenation period of 2 hrs in both the A/R group and PHC postconditioning group. The total exposure time to PHC in the PHC postconditioning group was 90 mins. Reoxygenation was carried out in a mixture of 5% CO2/95% air at 37°C. H9c2 cells and the upper fluid in each group were collected immediately after the reoxygenation period.
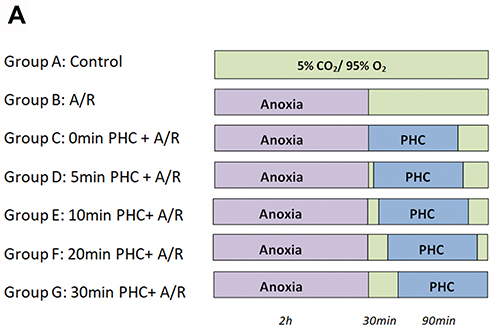 |
Figure 1 Illustration of experimental procedure. H9c2 cells were divided into seven groups: Control group, A/R group and PHC postconditioning group (0 min, 5 mins, 10 mins, 20 mins and 30 mins) (A). |
LDH And CK Activity Assay
LDH and CK are released from cytoplasm upon cellular injury and death. As such, the presence of LDH and CK in medium supernatant can be used as biomarkers of cellular injury or death.7,8 At the end of the experiment, the culture supernatant in each group was collected and subjected to spectrophotometric analysis (Yoke, Shanghai, China) to detect the presence of LDH absorbance at 450 nm and CK absorbance at 660 nm, respectively.9 Each measurement was performed six times.
Cell Viability Assays
At the end of this experiment, 0.2 μL MTT was added to each well. H9c2 cells were treated with MTT at 37°C for 4 hrs. 150 μL dimethyl sulfoxide (DMSO) would then replace the MTT medium. DMSO was used to dissolve the purple formazan crystals produced by viable cells into an aqueous solution. The optical density at 570 nm wavelength of this purple solution was analyzed by a microplate reader (BioTek, Winooski, Vermont, USA). Each assay was performed six times. The mean result was expressed as a ratio of the OD values of the experimental group to the control group.
Annexin-V/PI Assay
Fluorescein isothiocyanate (FITC)-Annexin V and propidium iodide (PI) were used to detect the apoptotic rate of H9c2 cells. At the end of our experiment, H9c2 cells (310 g) were centrifuged for 5 mins, followed by washing and resuspension in PBS twice. After a third centrifugation, H9c2 cells were gently resuspended in 500 μL of binding buffer solution. AnnexinV-FITC (5 μL) and PI (10 μL) were added to double-stain the H9c2 cells. After a treatment period of 15 mins in darkness, cellular apoptotic rate was measured by flow cytometry (BD Bioscience, Franklin Lakes, New Jersey, USA). The Annexin-V(+)/PI(-) cells (which showed up in the Q2-4 region) represented the early apoptotic cells. Annexin-V(+)/PI(+) cells (which showed up in the Q2-2 region) represented the late apoptotic cells. Each assay was performed six times. The mean result of the apoptotic rate was shown as a ratio of the number of apoptotic cells to the total number of cells.
Measurement Of Intracellular Ca2+ Concentrations
At the end of the experiment, the final solution was replaced with HBSS containing 4 μM Fluo-4AM. The H9c2 cells were then cultured at 37°C for 20 mins. Five times the volume of the HBSS containing 1% FBS was added, and the H9c2 cells were incubated for another 40 mins. The cells were then washed three times with HEPES buffer saline followed by resuspension in HEPES-buffered saline. The intracellular calcium level was analyzed with FACSCalibur flow cytometry at an excitation wavelength of 494 nm and emission wavelength of 510 nm. The fluorescence intensity of fluo-3 represented the concentration of intracellular Ca2+. Each assay was performed six times.
Measurement Of Intracellular Reactive Oxygen Species (ROS)
At the end of our experiment, the medium was pipetted out and replaced with 1 mL of 2’,7’-dichlorofluorescin diacetate (DCFHDA) solution. The cells were incubated at 37°C for 20 mins in darkness and were then washed three times with PBS to sufficiently remove the intracellular DCFH-DA. The H9c2 cells were then resuspended in 500 μL of PBS and subjected to flow cytometry. The fluorescent intensity of dichlorofluorescin (DCF) was measured by flow cytometry at an excitation wavelength of 480 nm and an emission length of 530 nm. Each assay was performed six times. The mean result of intracellular ROS was expressed as the mean fluorescent intensity of DCF in channel FL-1.
Measurement Of Intracellular MDA And SOD
MDA, one of the reactive products oxygen radical and unsaturated fatty acid, can be used as a maker to estimate lipid oxidative stress. When reacted with thiobarbituric acid, a chromogen which absorbs at 532 nm is formed.10 Superoxide can react with this chromogen to form a soluble formazan. SOD, one of the anti-oxidative enzyme, can inhibit the production of this water-soluble formazan.11 The intracellular ROS and MDA levels were quantified using BCA standard curve method.12 MDA was measured at 532 nm, and SOD was measured at 450 nm per protocol.13,14 Each measurement was performed six times.
Measurement Of Mitochondrial Membrane Potential (Δψm)
JC-1 is an ideal fluorescent probe which can measure mitochondrial membrane potential. When mitochondrial membrane potential is high, JC-1 forms J-aggregates in the mitochondria and emits red fluorescence. When the mitochondrial membrane potential is low, Jc-1 remains a monomer and emits green fluorescence. The drop of ΔΨm is correlated with the reduction in the red/green fluorescence ratio.15 The H9c2 cells were then collected, washed and incubated in Jc-1 solution at 37° in darkness for 20 mins. Next, the H9c2 cells in the incubation buffer were measured with flow cytometry at 488 nm excitation. The result was shown as the red/green fluorescence ratio. Each assay was performed six times.
MPTP Activity Assay
MPTP (mitochondrial permeability transition pore) is a nonspecific channel located between the mitochondria inner and outer membrane. It plays an important role during cell apoptosis and necrosis.14 BBcellprobe TM CA1 can easily penetrate the cell membrane into the cytoplasm and mitochondria and emits a green fluorescence. Upon adding a quenching agent to the fluorescence probe, the cytoplasmic green fluorescence would vanish, leaving only mitochondrial fluorescence intact. When MPTP opens, the green fluorescence would leak out of the mitochondria into the cytoplasm. Therefore, MPTP activity is indicated by mitochondrial green fluorescence changes. The lesser the amount of mitochondrial green fluorescence, the more active MPTP is. Each measurement was performed six times.
Western Blot Analysis
40 μg protein samples mixed with PBS and 5× loading buffer were dissolved in a SDS-polyacrylamide gel. The gel-electropheresedprotein bands were then transferred onto PVDF (polyvinylidene fluoride) membranes. These membranes were incubated with specific primary antibodies (anti-VDAC-1, anti-Bcl-2, anti-Bax, anti-caspase-3, anti-caspase-9). Anti-rabbit IgG-HRP was then added to the membrane. The blot was then visualized with ECL-Plus reagent. For protein loading controls, their membranes were stripped and re-probed with anti-β-actin and anti-COX IV antibodies. Densitometric analysis was used to quantify the protein bands, and the relative ratio of the optical density between study and control groups was calculated.
Statistical Analysis
Data were presented as mean ± standard deviation (S.D.). Statistical Package for the Social Sciences Software (SPSS.23.0 for windows) was used for statistical analysis. Tukey’s test with one-way ANOVA was used for multiple comparisons. A P value of <0.05 was considered significant.
Results
PHC Reduced LDH And CK After A/R Injury
LDH and CK indicate H9c2 cells' necrosis, as depicted in Figure 2A and B. When compared with the control group, A/R significantly increased LDH and CK levels (P<0.01, respectively). PHC reduced LDH and CK levels in all postconditioning groups compared to A/R group (P<0.01). Even though there were no significant differences in CK levels amongst the PHC treated groups, the PHC postcondition groups revealed significant differences in LDH levels (0 min vs 20 mins, P<0.01; 0 min vs 30 mins, P<0.05 and 5 mins vs 20 mins, P<0.05). The results may indicate that cell necrosis had happened not only during the early period of the reoxygenation (within 30 mins), but during the late (over 30 mins) period of the reoxygenation stage as well.
 |
Figure 2 Effect of PHC postconditioning on LDH and CK. A/R significantly increase LDH and CK level compared with the control group (P<0.01). PHC postconditioning significantly decrease LDH and CK level compared with A/R group (P<0.01) (A and B). However, no significant difference of CK level has been found in those different time of PHC postconditioning groups (P>0.05) (A). Yet, there is significant difference of LDH level among those (0 min vs 20 mins, P<0.01; 0 min vs 30 mins, P<0.05 and 5 mins vs 20 mins, P<0.05) PHC postcondition groups (B). (n=6; ##P < 0.01 versus control group; $$P < 0.01 versus A/R group. Data are shown as mean±S.D.). |
PHC Protected H9c2 Against Cell Viability And Apoptosis
After A/R injury, H9c2 cell viability changed significantly compared with the control groups (P<0.01). Postconditioning administration of PHC at 0 min, 5 mins, 10 mins, 20 mins after the reoxygenation procedure all attenuated cell viability changes significantly compared to A/R group (P<0.01). However, postconditioning at 30 mins failed to demonstrate significant protective effect compared to A/R group (P = 0.80). There were no significant differences among 0 min, 10 mins and 20 mins postcondition groups (P>0.05, respectively) (Figure 3A).
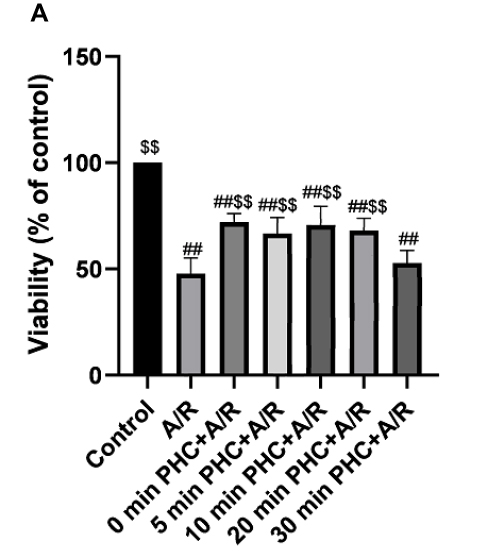 |
Figure 3 Effect of PHC postconditioning on cell viability. A/R significantly increase cell viability compared with the control group (P<0.01). PHC postconditioning (0 min, 5 mins, 10 mins, 20 mins and 30 mins) significantly decrease cell viability compared with the A/R group (P<0.01). Postconditioning at 30 mins has a less protecting effect compared with 0 min, 10 mins and 20 mins postcondition groups (P<0.01, respectively). There is no significant difference among 0 min, 10 mins and 20 mins postcondition group (P>0.05, respectively) (A). (n=6, ##P < 0.01 versus control group; $$P < 0.01 versus A/R group. Data are shown as mean±S.D.). |
Cell apoptosis was detected by analyzing PHC protective ability. Annexin V-FITC and PI double staining were used to measure the percentage of the apoptotic H9c2 cells. Q2-4 and Q2-2 represented early and late apoptotic H9c2 cells, respectively. (Figure 4A) A/R injury significantly increased cellular apoptosis compared to control group (P<0.01). PHC treatment decreased the apoptosis rate (P<0.01), with PHC administration at 0 min showing the most protective effect compared to other groups (P<0.01). Administering PHC 30 mins post reoxygenation showed the least protective effect compared to the 0 min, 5 mins, 10 mins, 20 mins groups (P<0.01), indicating that cell apoptosis happened during the earlier stage of the reoxygenation procedure (Figure 4B).
 |
Figure 4 Effect of PHC postconditioning on cell apoptosis. Q2-4 and Q2-2 represented early and later apoptotic H9c2 cells, respectively (A). A/R significantly increase cell apoptosis compared with the control group (P<0.01). PHC postconditioning significantly decrease cell apoptosis rate compared with the A/R group (P<0.01). PHC postconditioning at 0 min shows the best protective effect compared with other postcondition groups (P<0.01). PHC postconditioning at 30 mins shows the least protective effect compared with other postcondition groups (P<0.01) (B). (n=6; ##P < 0.01 versus control group; $$P < 0.01 versus A/R group. Data are shown as mean±S.D.). Cell apoptosis played an important role in A/R procedure, and cell apoptosis occupied a large percentage of the cell death event in the early period of A/Rprocedure (C). |
Cell apoptosis and cell necrosis are the two main types of cell death. As the A/R procedure progressed, H9c2 cell death increased as indicated by the changes in cell viability. Figure 2A, B and 4B indicated that cell apoptosis was affected more by timing effect of PHC postconditioning in the early stage of reoxygenation in comparison to cell necrosis (which indicated by CK and LDH level). Therefore, we believe H9c2 cell apoptosis occurred primarily in the very early period of postconditioning stage (especially within 30 mins), while H9c2 cells necrosis occurred not only during the early period of the postconditioning stage but throughout the reoxygenation phase as well. Hence, early PHC administration would inhibit both cell apoptosis and necrosis. Even though administration of PHC at 30 mins after reoxygenation did not significantly affect cell apoptosis rate, PHC may still offer some protective effect toward cell injury and cell necrosis. Cell apoptosis occupied a large percentage of cell death procedure in the early A/R period (as is shown in Figure 4C) and cell necrosis and other types of cell death also play an important part in A/R procedure. Therefore, PHC postconditioning therapy attenuated cell death not only through cell apoptosis pathway but via cell necrosis pathway as well. (Figure 4C was drawn by using the mean level of each group in Figures 3A and 4B. Cell variability changes indicated cell death. The result was shown as difference value in A/R group and each postconditioning group compared with the control group.)
PHC And H9c2 Cellular Oxidative Protection
Oxidation plays a significant role in ischemia-reperfusion injury.16 ROS, SOD and MDA were measured to analyze the protective effect of PHC. MDA, the end product of lipid oxidation, increased significantly in the A/R group compared to the control group (P<0.01). PHC decreased MDA levels in all the groups (P<0.01). But 0 min, 5 mins and 10 mins groups exhibited better protection compared to 20 mins and 30 mins groups (P<0.05).
SOD, a dismutase which can eliminate oxygen free radical in vivo, has protective effect against ischemia-reperfusion injury.17 MDA can be used as a maker to estimate lipid oxidative stress. Our results showed that A/R decreased SOD and increased MDA levels compared to the control group (P<0.01). PHC increased SOD and decreased MDA levels (P<0.01) versus the A/R group. There were significant differences in SOD and MDA levels among 0 min vs 20 mins and 0 min vs 30 mins in PHC postconditioning groups (Figure 5A and B).
 |
Figure 5 Effect of PHC postconditioning on SOD and MDA level. A/R decreased SOD and increased MDA level compared with control group (P<0.01), and PHC increased SOD and decreased MDA level (P<0.01) compared with A/R group (A and B). There is significant difference of SOD level among 0 min vs 20 mins and 0 min vs 30 mins in PHC postcondition groups (P<0.05, respectively)(A), and there is significant difference of MDA level among 10 mins vs 20 mins and 20 mins vs 30 mins in PHC postcondition groups (B). (n=6; ##P < 0.01 versus control group; $$P < 0.01 versus A/R group. Data are shown as mean±S.D.). |
ROS, analyzed with flow cytometry, was found to be elevated in A/R group compared to the control group. PHC was able to decrease ROS levels, with administration within 20 mins having a more significant effect compared to 30 mins group (P<0.01). However, there were no significant differences among the other PHC postconditioning groups (0 min, 5 mins, 10 mins, 20 mins, P>0.05, respectively) (Figure 6A and B).
 |
Figure 6 Effect of PHC postconditioning on ROS level. ROS level was detected by flow cytometry with DCF staining technique (A). ROS was found increasing in A/R group compared with the control group, and PHC can drop ROS level compared with the A/R group, yet administering PHC within 20 mins has more significant effect compared with 30 mins group (P<0.01). However, there are no significant differences among the other PHC postconditioning groups (0 min, 5 mins, 10 mins, 20 mins, P>0.05, respectively) (B). (n=6; ##P < 0.01 versus control group; $$P < 0.01 versus A/R group. Data are shown as mean±S.D.). |
PHC Protects H9c2 Cells From Ca2+ Overload
Ca2+ overload plays an important part in A/R injury.18 A/R significantly increased Ca2+ overload (P<0.01). PHC postconditioning attenuated Ca2+ overload in all of the PHC postconditioning groups. However, administering PHC at 30 mins after reoxygenation had the least effect compared to other groups (P<0.01). There were no significant differences among the other PHC postconditioning groups (0 min, 5 mins, 10 mins, 20 mins) (P>0.05, respectively) (Figure 7A and B).
 |
Figure 7 Effect of PHC postconditioning on Ca2+ overload. A/R increased Ca2+ overload compared with the control group. PHC postconditioning significantly decrease overload no matter when PHC was administered compared with the A/R group. However, administering PHC within 20 mins has a more significant effect compared with 30 mins group (P<0.01). There are no significant differences among the other PHC Postconditioning groups (0 min, 5 mins, 10 mins, 20 mins)(P>0.05, respectively) (A and B). (n=6; ##P < 0.01 versus control group; $$P < 0.01 versus A/R group. Data are shown as mean±S.D.). |
PHC Restored A/R-Induced Mitochondrial Membrane Potential
Mitochondrial membrane (potential) drops in the apoptotic pathway can further accelerate cell death and cell apoptosis.19 JC-1 assay was used to detect ΔΨm (Figure 8A). Whereas A/R decreased mitochondrial membrane potential, PHC restored cell membrane potential (P<0.01). However, administering PHC within 10 mins had more significant effect versus the 20 mins and 30 mins groups (P<0.01). Administering PHC at 30 mins after the reoxygenation exhibited the least effect compared to other postcondition groups (P<0.01). The result was shown in a red/green fluorescence ratio (Figure 8B).
 |
Figure 8 Effect of PHC postconditioning on mitochondrial membrane potential. A/R significantly decrease mitochondrial membrane potential compared with the control group (p<0.01). PHC postconditioning significantly restore mitochondrial membrane potential compared with the A/R group. However, administering PHC within 10 mins has more significant effect compared with 20 mins and 30 mins group (P<0.01). Administering PHC at 30 mins after the reoxygenation showed the least effect compared with other postcondition groups (P<0.01). The result was shown in a red/green fluorescence ratio (A and B). (n=6; ##P < 0.01 versus control group; $$P < 0.01 versus A/R group. Data are shown as mean±S.D.). |
PHC Affect Mitochondrial Pathway During Cell Apoptosis In A/R Injury
Mitochondria have an important part in apoptosis. Death signals were integrated through Bcl-2 family20 Cyt-C21 MPTP opening22 and the caspase family23 Cyt-C was released, MPTP activity was enhanced, and caspase family was activated in ischemia-reperfusion injury. Therefore, we measured Bcl-2, Bas, caspase-3 and caspase-9 level to determine whether or not PHC could regulate mitochondrial pathways, as depicted in Figure 9A. The results revealed a decrease in Bcl-2, and an increase in Bax, caspase-3 and caspase-9 in A/R group versus the control group (P<0.01). As depicted by Western blot, PHC significantly attenuated the apoptosis pathway, and administering PHC at 30 mins after the reoxygenation was shown to have the least effect compared to other postcondition groups (P<0.01, respectively), as shown in Figure 9B–F.
 |
Figure 9 Western blot analysis of VDAC1, Bcl-2, Bax, cleaved caspase-3 and cleaved caspase-9 (A). As depicted in the above pictures, there is a decrease of Bcl-2, an increase of Bax, caspase-3 and caspase-9 in A/R group compared with the control group. As depicted by Western blot, PHC significantly attenuated the apoptosis pathway, and administering PHC at 30 mins after the reoxygenation showed the least effect compared with other postcondition groups (P<0.01, respectively). (B–F) (n=6; ##P < 0.01 versus control group, $$P < 0.01 versus A/R group. Data are shown as mean±S.D.). |
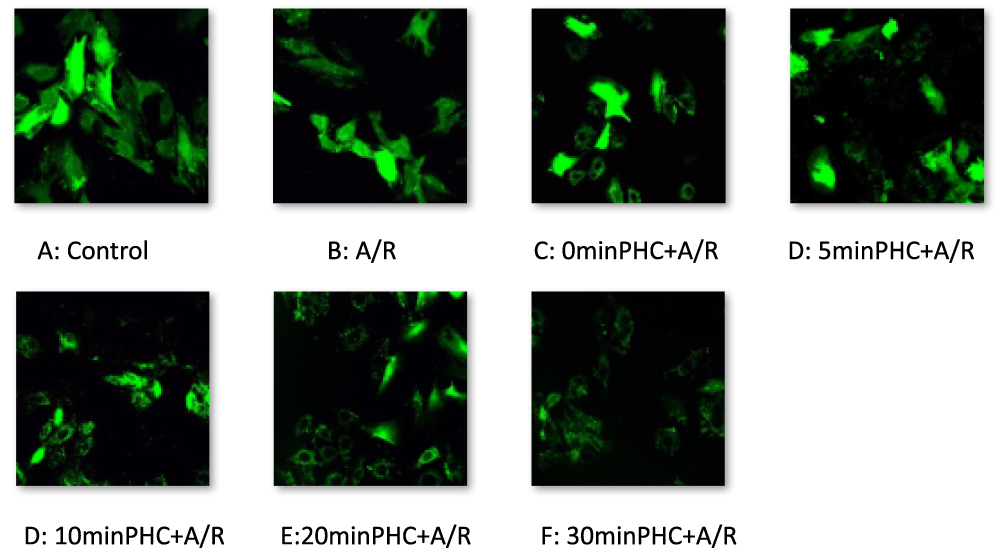 |
Figure 10 MPTP activity observed by confocal microscopy and with fluorescent staining technique. A/R promotes MPTP opening, and PHC attenuated MPTP activity. |
PHC Attenuates MPTP Activity And VDAC Level
Ischemia-reperfusion (injury) accelerates MPTP opening and plays an important part in accelerated cell apoptosis and cell death.24 We therefore aimed to find out if PHC would inhibit MPTP activity and protect H9c2 cell from A/R injury. Confocal microscopy and fluorescent staining technique were used to observe MPTP activity. When MPTP opened, the green fluorescence would leak out of the mitochondria. Therefore, MPTP activity was indicated by mitochondrial green fluorescence change. There was inverse relationship between the amount of mitochondrial green fluorescence and MPTP activity, as shown in Figure 10A. VDAC, component of MPTP, also influenced cell death and survival.25,26 Western blot technique was used to access VDAC level (Figure 9A). As a result, A/R increased VDAC level, and promoted MPTP opening. PHC decreased VDAC level and attenuated MPTP activity. Earlier administration of PHC offered greater postconditioning protective effect (Figures 9B and 10A).
Discussion
Our experiment showed that PHC postconditioning had a positive effect in protecting H9c2 cells from A/R injury. Earlier PHC administration resulted in better protection. Our previous investigation showed that Penehyclidine Hydrochloride offered this protective effect via limiting calcium overload, decreasing formation of reactive oxygen species, restraining mitochondrial MPTP opening and reducing inflammatory response in vitro and in vivo.5,6 The most effective dose of Penehyclidine Hydrochloride on H9c2 cell against anoxia/reoxygenation injury was determined to be 0.1 μm/L in our research group.9
In 2012, Barrere-Lemaire et al found out the effective time window for delayed postconditioning treatment in rats model was 30 mins,27 and our previous animal study determined the protective time window of PHC postcondition was within 10 mins after the reperfusion stage began.6 We hypothesized that IRI could be attenuated in the early period of the reperfusion stage. PHC was administered directly in the cell culture, which effected H9c2 cells directly, would offer more direct protective effect compared with those in vivo studies. In this cell culture experiment, we focused on the variation tendency of each mechanism in the very early reperfusion stage (from 0 min to 30 mins) to better understand the time effect of each mechanism in A/R procedure in the treatment of PHC. There were not many differences in CK and LDH level from 0 min to 30 mins PHC postconditioning treatment, while there were huge differences in cell apoptosis, mitochondrial pathway and Ca2+ concentration. Regardless of when PHC was administered within the first 30 mins after reoxygenation began, H9c2 cells were protected via almost all of the mechanisms that contributed to A/R injury. Our results indicate that postcondition treatment should be applied as early as possible, preferable within 20 mins after reoxygenation began. Oxidative stress, calcium overload, MPTP opening and cell apoptosis all started in the early reoxygenation stage and persisted, resulting in cell injury and necrosis. Administering PHC during any period in the reoxygenation stage could attenuate oxidative stress, calcium overload, mitochondrial pathway and cell death, which would offer some degree of cellular protection. Earlier treatment of PHC postconditioning offer greater protective effect.
During cardiomyocyte anoxic period, oxidative phosphorylation halts as a result of anoxia in the mitochondrial respiratory chain. Under anaerobic conditions, the glycolysis becomes the dominant pathway of ATP generation. However, glycolysis would lead to lactic acid accumulation. As blood flow stops and CO2 elimination fails, continued cellular acidosis would lead to a drop in cellular pH. The increasing H+ in the cell will open the Na+/H+ channels. The increase in cytoplasmic Na+ will stimulate Na+/Ca2+ exchange, resulting in calcium overload.28 Even with restoration of adequate oxygen supply during reoxygenation, mitochondrial respiratory chain complexes still could not play its role. As a result, a large amount of reactive oxygen species (ROS) was generated, inflammatory response was activated, calcium overload ensued and mitochondrial permeability transition pore (MPTP) was activated.29 Mitochondria played the central role of cell death and apoptosis. With MPTP opening, mitochondria starts to swell and inner membrane begins to depolarize. This leads to the release of cytochrome C and activation of caspase. Ultimately, cell death and apoptosis occur.30
Acute myocardial ischemia is a common occurrence all around the world, and it is difficult to predict when it will happen. This in vitro experiment may offer some insight into treating ischemia-reperfusion injury with postconditioning method. Our result showed that PHC postcondition in 30 mins still offered its protective effect, which may provide improved prognosis for patients with myocardial infarction or with other organ shock. Further in vitro and clinical research utilizing PHC are needed. We hope this current in vitro experiment would increase confidence for researchers, and physicians in future exploration of postconditioning ischemia treatment.
Conclusion
PHC postconditioning protected H9c2 cells by attenuating superoxide production, decreasing Ca2+ overload, restraining MPTP activities, restoring mitochondrial membrane potential, regulating cell apoptosis proteins and modulating the mitochondrial pathway. The most effective dose of PHC on H9c2 cell against IRI was determined to be 0.1 μm/L. H9c2 cells were protected by PHC from A/R injury regardless of when PHC was administered (0 min, 5 mins, 10 mins, 20 mins, 30 mins). However, earlier administration of PHC resulted in better PHC postconditioning protective effect.
Acknowledgment
This research was supported by grants from Beijing Municipal Administration of Hospitals Clinical Medicine Development of Special Funding Support (ZYLX 201810), the National Natural Science Foundation of China (No.81471902, No.81871592), and Beijing Hospitals Authority Youth Programme (QML20180602).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Houang EM, Bartos J, Hackel BJ, et al. Cardiac muscle membrane stabilization in myocardial reperfusion injury. JACC Basic Transl Sci. 2019;4(2):275–287. doi:10.1016/j.jacbts.2019.01.009
2. Kalogeris T, Baines CP, Krenz M, Korthuis RJ. Cell biology of ischemia/reperfusion injury. Int Rev Cell Mol Biol. 2012;298:229–317.
3. Kalogeris T, Baines CP, Krenz M, Korthuis RJ. Ischemia/reperfusion. Compr Physiol. 2016;7(1):113–170.
4. Wang Y, Gao Y, Ma J. Pleiotropic effects and pharmacological properties of penehyclidine hydrochloride. Drug Des Devel Ther. 2018;12:3289–3299. doi:10.2147/DDDT
5. Lin D, Ma J, Xue Y, Wang Z. Penehyclidine hydrochloride preconditioning provides cardioprotection in a rat model of myocardial ischemia/reperfusion injury. PLoS One. 2015;10(12):e0138051. doi:10.1371/journal.pone.0138051
6. Tan H, Chen L, Ma J. Penehyclidine hydrochloride post-conditioning reduces ischemia/reperfusion-induced cardiomyocyte apoptosis in rats. Exp Ther Med. 2017;14(5):4272–4278. doi:10.3892/etm.2017.5089
7. Kim S, Simon E, Myers L, Hamm LL, Jazwinski SM. Programmed cell death genes are linked to elevated creatine kinase levels in unhealthy male nonagenarians. Gerontology. 2016;62(5):519–529. doi:10.1159/000443793
8. Kaja S, Payne AJ, Naumchuk Y, Koulen P. Quantification of lactate dehydrogenase for cell viability testing using cell lines and primary cultured astrocytes. Curr Protoc Toxicol. 2017;72:
9. Wang Z, Lin D, Zhang L, Liu W, Tan H, Ma J. Penehyclidine hydrochloride prevents anoxia/reoxygenation injury and induces H9c2 cardiomyocyte apoptosis via a mitochondrial pathway. Eur J Pharmacol. 2017;797:115–123. doi:10.1016/j.ejphar.2017.01.012
10. Lapenna D, Cuccurullo F. TBA test and “free” MDA assay in evaluation of lipid peroxidation and oxidative stress in tissue systems. Am J Physiol. 1993;265(3 Pt 2):H1030–H1032. doi:10.1152/ajpheart.1993.265.3.H1030
11. Lei XG, Zhu JH, Cheng WH, et al. Paradoxical roles of antioxidant enzymes: basic mechanisms and health implications. Physiol Rev. 2016;96(1):307–364. doi:10.1152/physrev.00010.2014
12. Smith PK, Krohn RI, Hermanson GT, et al. Measurement of protein using bicinchoninic acid. Anal Biochem. 1985;150(1):76–85. doi:10.1016/0003-2697(85)90442-7
13. Moore K, Roberts LJ. Measurement of lipid peroxidation. Free Radic Res. 1998;28(6):659–671.
14. Sun Y, Oberley LW, Li Y. A simple method for clinical assay of superoxide dismutase. Clin Chem. 1988;34(3):497–500.
15. Cossarizza A, Baccarani-Contri M, Kalashnikova G, Franceschi C. A new method for the cytofluorimetric analysis of mitochondrial membrane potential using the J-aggregate forming lipophilic cation 5,5’,6,6’-tetrachloro-1,1’,3,3’-tetraethylbenzimidazolcarbocyanine iodide (JC-1). Biochem Biophys Res Commun. 1993;197(1):40–45. doi:10.1006/bbrc.1993.2438
16. Zhou T, Chuang CC, Zuo L. Molecular characterization of reactive oxygen species in myocardial ischemia-reperfusion injury. Biomed Res Int. 2015;2015:864946. doi:10.1155/2015/864946
17. Batinic-Haberle I, Tovmasyan A, Roberts ER, Vujaskovic Z, Leong KW, Spasojevic I. SOD therapeutics: latest insights into their structure-activity relationships and impact on the cellular redox-based signaling pathways. Antioxid Redox Signal. 2014;20(15):2372–2415. doi:10.1089/ars.2012.5147
18. Messner B, Türkcan A, Ploner C, Laufer G, Bernhard D. Cadmium overkill: autophagy, apoptosis and necrosis signalling in endothelial cells exposed to cadmium. Cell Mol Life Sci. 2016;73(8):1699–1713. doi:10.1007/s00018-015-2094-9
19. Zorova LD, Popkov VA, Plotnikov EY, et al. Mitochondrial membrane potential. Anal Biochem. 2018;552:50–59. doi:10.1016/j.ab.2017.07.009
20. Warren C, Wong-Brown MW, Bowden NA. BCL-2 family isoforms in apoptosis and cancer. Cell Death Dis. 2019;10(3):177. doi:10.1038/s41419-019-1407-6
21. Hüttemann M, Helling S, Sanderson TH, et al. Regulation of mitochondrial respiration and apoptosis through cell signaling: cytochrome c oxidase and cytochrome c in ischemia/reperfusion injury and inflammation. Biochim Biophys Acta. 2012;1817(4):598–609. doi:10.1016/j.bbabio.2011.07.001
22. Hausenloy DJ, Duchen MR, Yellon DM. Inhibiting mitochondrial permeability transition pore opening at reperfusion protects against ischaemia-reperfusion injury. Cardiovasc Res. 2003;60(3):617–625. doi:10.1016/j.cardiores.2003.09.025
23. McIlwain DR, Berger T, Mak TW. Caspase functions in cell death and disease. Cold Spring Harb Perspect Biol. 2015;7(4):a008656. doi:10.1101/cshperspect.a026716
24. Ong SB, Dongworth RK, Cabrera-Fuentes HA, Hausenloy DJ. Role of the MPTP in conditioning the heart – translatability and mechanism. Br J Pharmacol. 2015;172(8):2074–2084. doi:10.1111/bph.13013
25. Baines CP, Kaiser RA, Sheiko T, Craigen WJ, Molkentin JD. Voltage-dependent anion channels are dispensable for mitochondrial-dependent cell death. Nat Cell Biol. 2007;9(5):550–555. doi:10.1038/ncb1575
26. Noskov SY, Rostovtseva TK, Chamberlin AC, Teijido O, Jiang W, Bezrukov SM. Current state of theoretical and experimental studies of the voltage-dependent anion channel (VDAC). Biochim Biophys Acta. 2016;1858(7 Pt B):1778–1790. doi:10.1016/j.bbamem.2016.02.026
27. Barrere-Lemaire S, Nargeot J, Piot C. Delayed postconditioning: not too late? Trends Cardiovasc Med. 2012;22:173–179. doi:10.1016/j.tcm.2012.07.016
28. Inserte J, Barba I, Hernando V, Garcia-Dorado D. Delayed recovery of intracellular acidosis during reperfusion prevents calpain activation and determines protection in postconditioned myocardium. Cardiovasc Res. 2009;81(1):116–122. doi:10.1093/cvr/cvn260
29. Widgerow AD. Ischemia-reperfusion injury: influencing the microcirculatory and cellular environment. Ann Plast Surg. 2014;72(2):253–260. doi:10.1097/SAP.0b013e31825c089c
30. Chen Q, Xu H, Xu A, et al. Inhibition of Bcl-2 sensitizes mitochondrial permeability transition pore (MPTP) opening in ischemia-damaged mitochondria. PLoS One. 2015;10(3):e0118834. doi:10.1371/journal.pone.0118834
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.