")
Back to Journals » International Journal of Nephrology and Renovascular Disease » Volume 17
Post-Transplant Glomerulonephritis: Challenges and Solutions
Authors de Sousa MV
Received 10 December 2023
Accepted for publication 12 March 2024
Published 15 March 2024 Volume 2024:17 Pages 81—90
DOI https://doi.org/10.2147/IJNRD.S391779
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Pravin Singhal
Marcos Vinicius de Sousa
University of Campinas, School of Medical Sciences, Department of Internal Medicine, Division of Nephrology, Renal Transplant Unit, Transplant Research Laboratory, Campinas, SP, Brazil
Correspondence: Marcos Vinicius de Sousa, Renal Transplant Unit, Division of Nephrology, School of Medical Sciences, University of Campinas, Rua Tessália Vieira de Camargo 126, Cidade Universitária Zeferino Vaz, Campinas, São Paulo, 13083-970, Brazil, Email [email protected]
Abstract: Glomeruli can be damaged in several conditions after kidney transplantation, with a potential impact on the graft function and survival. Primary glomerulonephritis, a group of glomerular immunological damage that results in variable histological patterns and clinical phenotypes, can occur in kidney transplant recipients as a recurrent or de novo condition. Specific immunologic conditions associated with kidney transplantation, such as acute rejection episodes, can act as an additional trigger after transplantation, impacting the incidence of these glomerulopathies. The post-transplant GN recurrence ranges from 3% to 15%, varying according to the GN subtype and post-transplant time, mainly occurring after 3– 5 years of kidney transplantation. Advances in the knowledge of glomerulonephritis pathophysiology have provided new approaches to pre-transplant risk evaluation and post-transplant monitoring. Glomeruli can be affected by several systemic viral infections, such as human immunodeficiency virus (HIV), hepatitis C virus (HCV), hepatitis B virus (HBV), severe acute respiratory syndrome coronavirus 2 (SARS-COV-2), cytomegalovirus (CMV), and BK virus. The diagnosis of these infections, as well as the identification of possible complications associated with them, are important to minimize the negative impacts of these conditions on kidney transplant recipients’ outcomes.
Keywords: glomerulonephritis, viral infection, kidney transplantation, chronic kidney disease
Introduction
Glomeruli can be damaged in several conditions after kidney transplantation, such as recurrent or de novo glomerulopathies and viral infections. Primary glomerulonephritis (GN) is a group of glomerular immunological damage that results in variable histological patterns and clinical phenotypes.1 GN comprises one of the leading etiologies of chronic kidney disease (CKD) worldwide, varying according to geographic and demographic characteristics and kidney biopsy indications. GN frequently causes end-stage kidney disease (ESKD), and its occurrence after transplantation is a significant cause of graft failure. The post-transplant GN recurrence ranges from 3% to 15%, varying according to the GN subtype and post-transplant time, mainly occurring after 3–5 years of kidney transplantation.1 The clinical manifestations and the histological characteristics of GN post-transplant are influenced by histologic changes secondary to the donor’s comorbidities, immunosuppressive and nephrotoxic drugs, and previous episodes of acute or chronic graft rejections. Therefore, all this information is essential for establishing the correct diagnosis and prognostic evaluation of the graft in case of post-transplant GN suspicion.
Individuals who have undergone a kidney transplant and are receiving immunosuppressive therapy can be more vulnerable to viral infections and their effects on various organs. The glomerular cells can be affected by several systemic viral infections.2 Histological evidence of glomerular disease has been described in several viral infections, such as human immunodeficiency virus (HIV), hepatitis C virus (HCV), hepatitis B virus (HBV), severe acute respiratory syndrome coronavirus 2 (SARS-COV-2), cytomegalovirus (CMV), and BK virus.2–4 This paper reviews the challenges and solutions regarding screening, monitoring, and treating post-transplant glomerulonephritis. These points are summarized in Table 1.
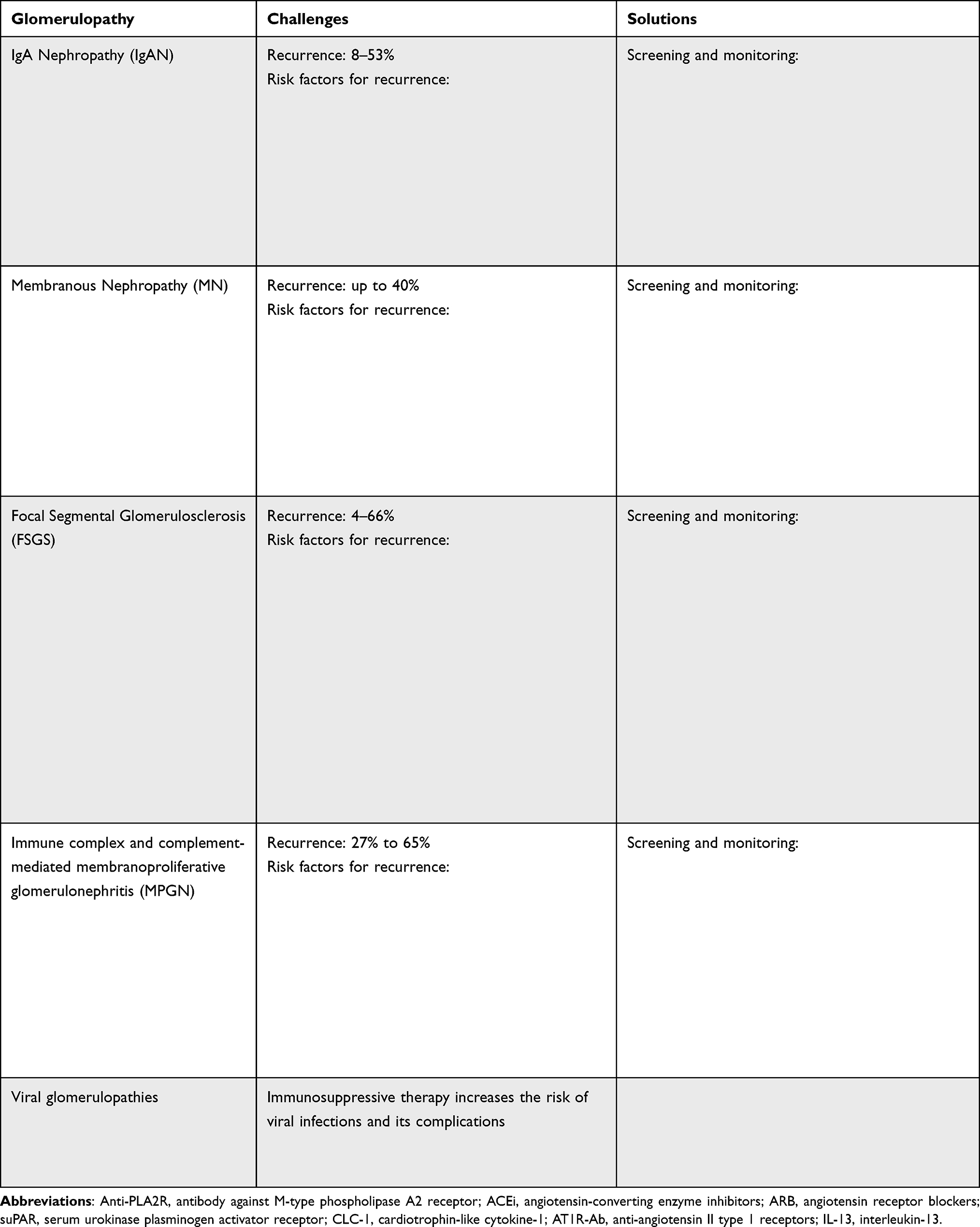 |
Table 1 General Characteristics, Challenges, and Solutions of Post-Transplant Glomerulonephritis |
IgA Nephropathy
IgA nephropathy (IgAN) is the most prevalent GN globally. It can progress to ESKD at around 10% in 10 years and 20% in 20 years.5,6 Genetically predisposed individuals produce galactose deficient IgA (GD-IgA) by mucosa after some stimulus, such as bacterial infection. This GD-IgA elicits auto-antibodies IgG or IgA production, leading to circulating immune complexes and deposition in the glomerular mesangium. This, in turn, activates an inflammatory response and causes tissue damage.6 The immune complexes increase the complement activation, mainly the alternative and lectin pathways.7 Although some studies have shown an association between mesangial C3 deposits and a worse prognosis, the data available in the literature are conflicting. The mesangial C1q deposition has been associated with treatment failure in native IgAN and IgAN recurrence, suggesting a possible role of classical pathway activation in this process.7 The clinical presentations of IgAN range from hematuria during upper respiratory or gastrointestinal infections to proteinuria, hypertension, and acute kidney dysfunction.5 IgAN is confirmed by kidney biopsy, showing the presence of mesangial expansion and hypercellularity in light microscopy and a predominant IgA deposit pattern in the mesangium, associated or not with IgG or IgM on the immunofluorescence assay. The current modified Oxford MEST-C classification is recommended to estimate IgAN prognosis in native kidneys according to the evaluation of mesangial hypercellularity (M), endocapillary hypercellularity (E), segmental glomerulosclerosis (S), tubulointerstitial fibrosis (T), and crescentic lesions (C).8,9 The MEST-C histological score and clinical variables measured at the time of kidney biopsy were incorporated into an International IgAN Prediction Tool, available as an online calculator to quantify the risk of progression and discuss outcomes with patients.10
The recurrence of IgAN after transplantation can range from 8–53% depending on the indications of graft biopsy and the length of follow-up.5,7 Several risk factors for IgAN recurrence have been identified, such as kidney recipients from a living donor, younger recipients, steroid-avoidance immunosuppressive therapy, and non-use of induction therapy.1,5 Preemptive transplants and the presence of DSA at the time of transplantation were also associated with the recurrence of IgA deposits in previous studies.11 Hematuria is absent in 64% of patients with IgAN recurrence, and most patients with IgAN detected in protocol biopsies do not show clinical signs of recurrence.5 A diagnosis of IgAN recurrence is based on histological evidence of IgA deposition in a recipient with biopsy-proven IgAN in their native kidneys.5 Although protocol biopsies have shown up to 60% IgAN recurrence in previous studies, routine graft biopsy in patients without signs of recurrence is not helpful in the treatment or prognosis evaluation.5 A previous retrospective study by our group showed an association between younger age at native IgAN diagnosis and absence of prior induction of immunosuppression with IgAN recurrence, with a high prevalence of mesangial hypercellularity at recurrence.12 Other previous studies showed that a higher MEST-C score and a lower estimated glomerular filtration rate at the time of biopsy were associated with poor prognosis in patients with IgAN recurrence.13 Some biomarkers for the prediction of post-transplant IgAN recurrence have been studied, such as IgA and IgA-complexes (IgA-IgG and IgA-soluble CD89), galactose-deficient IgA1 (Gd-IgA1), and anti-Gd-IgA1 autoantibodies, although its clinical application is not entirely known.14 Graft dysfunction related to recurrence is rare before three years of follow-up.15 After that, the recurrence becomes clinically relevant, reaching 10–15% of recurrence-related graft dysfunction at five years.15 A study by Moroni et al showed a death-censored graft survival at 15 years 10% lower in IgAN recipients compared with non-diabetic controls.16
There is no effective therapy for preventing recurrent IgAN, and current treatment recommendations are based on native kidney disease.14,17 Previous studies showed a reduction in recurrence risk in cases of induction of immunosuppression with antithymocyte globulin compared to interleukin-2 receptor antagonist.17 There is no preferable maintenance immunosuppressive therapy to avoid IgAN recurrence. The optimal strategy to treat recurrence remains under study.11 The current recommendation in cases of recurrence includes nonpharmacologic supportive care, anti-hypertensive drugs, antiproteinuric therapy, and immunosuppressive agents.6 Blood pressure may be controlled to achieve a systolic blood pressure of 120–130 mmHg, with a blood pressure threshold of 130/80 in patients with proteinuria < 1g/d and 125/75 in patients with proteinuria >1g/d.5,10 Sustained proteinuria is the main predictor of long-term kidney outcomes, and reducing proteinuria was independently associated with improved kidney outcomes in previous studies.10 The renin-angiotensin system (RAS) blockade improves kidney outcomes in native IgAN. However, the combined use of angiotensin-converting enzyme inhibitors (ACEi) and angiotensin receptor blockers (ARB) is not usually indicated. There is limited data on the benefit of routine use of ACEi or ARB in the IgAN recurrence, but these drugs can be considered in the case of proteinuria.17 Sodium-glucose cotransporter-2 (SGLT-2) inhibitors are indicated for native IgAN with proteinuria or impaired renal function, and their use can be considered in cases of post-transplant recurrence.13 Several glucocorticoid regimens were proposed in clinical for patients presenting proteinuria higher than 0.75–1 g/day despite optimized supportive care.10 However, the clinical benefit of glucocorticoids in IgAN has not been established.10 Patients with biopsy-proven recurrence and rapidly rising serum creatinine or nephrotic-range proteinuria despite the use of ACEIs or ARBs may be treated with high-dose glucocorticoids (prednisone 1 mg/kg/day) for eight weeks, followed by a taper to low doses commonly used to prevent rejection. Treatment with cyclophosphamide is not recommended in IgAN unless in the setting of rapidly progressive disease.10 An oral target-release formulation of budesonide has been approved for treating IgAN in native kidneys, reducing proteinuria with lower systemic side effects. However, a similar benefit was not observed in cases of IgAN recurrence.18
Membranous Nephropathy
Membranous nephropathy (MN) is characterized by the thickening of the glomerular capillary walls and projections of the glomerular basement membrane between deposits, with complement components C3 and C4 often present.19 It affects patients of all ages and ethnicities and is present in about 20% of nephrotic syndrome cases in adults.19 MN is divided into primary or idiopathic MN and secondary MN. Primary MN is an autoimmune disease, with the production of autoantibodies against antigens located in the kidney. The M-type phospholipase A2 receptor (PLA2R) is the major target antigen identified in primary MN, eliciting autoantibody production, mainly IgG4.20 Soluble phospholipase A2 is considered a potent pro-inflammatory enzyme.21 PLA2R exerts an anti-inflammatory activity and regulates cellular senescence throughout the production of reactive oxygen species and activation of DNA-damage pathways.21 The second most frequent autoantigen involved in the MN pathogenesis is the thrombospondin type-1 domain-containing 7A (THSD7A), responsible for 5–10% of idiopathic MN cases.19 PLA2R and THSD7A are present in podocytes and colocalized within subepithelial deposits with IgG4.19 Secondary MN can occur in association with other clinical disorders, such as hepatitis virus B infection, systemic lupus erythematosus, cancer, and drugs. In the secondary MN, non-podocyte antigens are mainly responsible, with the participation of non-IgG4 autoantibodies subclasses and C1q deposits.19 In native kidneys, the clinical course of MN can vary, with 30% to 40% of cases experiencing spontaneous remission and 30% to 40% progressing to ESKD within ten years of diagnosis.22 A high level of anti-PLA2R antibodies is associated with a lower rate of spontaneous remission and a higher incidence of nephrotic syndrome and ESKD.23
Post-transplant MN can be a recurrent or de novo disease. MN is the glomerulopathy that recurs most frequently post-transplantation, affecting up to 40% of recipients.19 Patients with high levels of proteinuria and detected anti-PLA2R antibodies before transplantation are at a higher risk of recurrence.24,25 Circulating autoantibodies can be absent at the time of transplant and reemerge after transplantation because of the reduction of immunosuppression or second hits such as infections.26 The post-transplant rise of anti-PLA2R may also help identify the possibility of MN recurrence and may be associated with disease progression and resistance to treatment.14,27 The diagnostic and prognostic application of measuring other autoantibodies, such as THSD7A or glomerular staining of PLA2R, is still undetermined.14 The post-transplant surveillance measurement of proteinuria or albuminuria is recommended to identify the recurrence early, especially among patients with a high risk of recurrence.24 Proteinuria should be evaluated monthly for at least the first year post-transplant in cases of MN not associated with anti-PLA2R antibodies, and graft biopsy is indicated in cases of proteinuria higher than 1 g/d.10 For patients with PLA2R-associated MN, regular measurement of anti-PLA2R antibodies is recommended in the first 6–12 months.10 When recurrent MN is associated with PLA2R antibodies, it usually has a lower likelihood of spontaneous remission and requires adjuvant immunosuppressive therapy earlier.27 Previous studies have shown that higher tacrolimus exposure before MN recurrence was associated with spontaneous remission, suggesting that maintenance immunosuppressive therapy may impact the clinical course of the disease.27 About 20% of post-transplant MN cases are de novo disease associated with infection, malignancy, renal infarction, ureteral obstruction, toxicity, or antivascular endothelial growth factor therapy.26 Previous studies also suggested a possible triggering role of antibody-mediated rejection episodes on de novo MN development.28–31 The impact of MN recurrence on graft survival is variable among previous studies, with progression to graft loss ranging from 45% to 65% within six years of follow-up after diagnosis.27
The damage caused by the immune system can disrupt the glomerular filtration barrier, leading to proteinuria in varying degrees.27 The treatment goals for recurrent idiopathic MN are to control symptoms and prevent the progression of podocyte injury.14 The nonspecific treatment of MN includes dietary sodium restriction, dietary protein intake reduction, blood pressure control, and antiproteinuric therapy with ACEi and ARB.22 Although SGLT-2 inhibitors have been shown to decrease the risk of CKD progression, their use on patients with nephrotic syndrome and/or immunosuppressed is lacking.31 Anti-CD20 antibodies have been used to treat MN in native kidneys and its recurrence after transplantation, with remission in up to 80% of patients with recurrent disease.14 However, the criteria for starting treatment with anti-B cell therapy and the frequency and dose recommended remain under discussion.14 For native MN, patients with mild to moderate disease usually undergo spontaneous remission, and immunosuppressive therapy should be reserved for those who are at the highest risk for developing progressive kidney failure.32 In patients with native MN presenting proteinuria less than 3 g/d, serum albumin higher than 30 g/l, and eGFR higher than 60 mL/min/1.73m², immunosuppressive therapy is not required.10 However, post-transplant MN recurrence usually progresses even in patients with a small amount of proteinuria, and early treatment may be considered.14 There is insufficient data to support a preemptive treatment with rituximab.10 In cases of proteinuria over 1g/day, treatment of recurrent MN with rituximab may be helpful, but its effectiveness in cases of lower proteinuria remains unknown.10,27
Focal Segmental Glomerulosclerosis
Focal segmental glomerulosclerosis (FSGS) is a type of kidney injury primarily affecting the podocyte. It is characterized by the deposition of extracellular matrix, which leads to the obliteration of a portion of the glomerular tuft in some glomeruli.33,34 FSGS can occur at any age, and it is present in approximately 20% to 30% of adults with nephrotic syndrome, more frequent in male and black patients.34 FSGS can be classified into primary, genetic, or secondary forms, including maladaptive, virus-associated, and drug-induced FSGS.33,34 Primary FSGS is an immunological disease caused by serum molecules known as circulating factors, which selectively target and damage the glomerular barrier, especially podocytes.33,34 Several factors have been considered as biomarkers of podocyte injury because of their ability to induce podocyte cytoskeleton reorganization in vitro and induce albuminuria in animal model studies. Some of these biomarkers include serum urokinase plasminogen activator receptor (suPAR), urine apolipoprotein A-1b, cardiotrophin-like cytokine-1 (CLC-1), anti-angiotensin II type 1 receptors (AT1R-Ab), anti-CD40, and interleukin-13.35 Genetic FSGS is associated with gene mutations in podocyte development and structural protein-encoding.33 In all FSGS forms, injured podocytes are detached from the basement membrane, leaving it uncovered and allowing the interaction of capillary loops and parietal epithelial cells. The remaining podocytes suffer hypertrophy, and intracapillary hypertension results in changes in podocyte, endothelial, and mesangial cells, with progressive focal and segmental sclerosis.34 Spontaneous remission is rare in primary FGSG, occurring in about 5% of the cases.33 About 40–70% of patients with FSGS course with ESKD despite treatment within 10 to 20 years after diagnosis.36
FSGS can recur or occur de novo after kidney transplantation, and it may be primary or secondary to hyperfiltration, drugs, or infection.34 The FSGS form’s accurate distinction is essential in pre-transplant evaluation patients.37 Genetic forms of FSGS, with podocin or structural podocyte protein mutations, have shown a significantly lower recurrence rate after transplantation.1,33 Therefore, genetic testing may provide valuable information for patients with FSGS who plan to undergo a kidney transplant.10 The reported FSGS recurrence rate after transplantation ranges from 4 to 66%, varying according to the population and study design.34,36 Some risk factors for recurrence have been described, such as younger age at transplantation, higher level of proteinuria before kidney transplantation, related donor, nephrectomy of native kidneys, a history of recurrence in a previous allograft, severe hypoalbuminemia at presentation, and the absence of a positive family history of FSGS.34,36 Idiopathic FSGS can recur any time after transplantation, but it is more frequent early after transplantation.17 The recurrence of FGSG usually occurs in the first days after transplantation, suggesting the presence of a circulating factor against podocytes. Some initiatives have been developed to predict the risk for FSGS recurrence, such as a panel of circulating autoantibodies related to the disease, but in most cases, it remains unpredictable.35 The circulating biomarkers of podocyte injury or permeability factors can be presented high before or after transplantation and may predict those at risk of recurrence.14 FSGS recurrence is most common in the first two years post-transplant, and patients with recurrent FSGS progress to graft failure more quickly.34,36 The recurrence usually courses with proteinuria, commonly heavy.17 The time to the disappearance of native proteinuria post-transplant is variable, and interpreting the post-transplant proteinuria requires knowledge of the pretransplant values.17 Proteinuria screening may be done early post-transplant to detect potential recurrent cases.17 The FSGS recurrence negatively impacts graft survival, with a graft loss rate approximately three times higher among patients with recurrence than those who do not recur.34 A previous multicenter international study showed a graft loss rate of 39% among recipients with FSGS recurrence post-transplant.37
The treatment strategies in cases of FSGS recurrence focus on removing pathogenic circulating factors and anti-B cell therapy.14 Although the benefit of prophylactic plasmapheresis remains controversial, therapeutic plasmapheresis is considered the first line of treatment in recurrent FSGS.34,37 A multicenter study showed a complete remission of recurrent FSGS in 57% of cases treated with plasmapheresis with or without rituximab.37 The number of plasma-exchange treatments required to reduce proteinuria in cases of FSGS recurrence remains unclear.17 In cases of non-response to the plasma-exchange treatment or non-nephrotic proteinuria, treatment with ACEi or ARB may be beneficial.17 The efficacy of adjunctive therapy with rituximab and calcineurin inhibitor (CNI) remains unclear.1 Cyclosporine is considered helpful in patients with FSGS since it is thought to act directly on podocyte cytoskeleton stabilization and is a potent vasoconstrictor, reducing albuminuria.35 The efficacy of tacrolimus in case of FSGS recurrence, in turn, is unclear.35 For patients already receiving a calcineurin inhibitor drug for maintenance immunosuppression who develop FSGS recurrence, it is not recommended to change to an alternative CNI.1 However, patients receiving a mammalian target of rapamycin (mTOR) inhibitor may benefit from the change for a CNI drug.1
Immune Complex and Complement-Mediated Membranoproliferative Glomerulonephritis
Membranoproliferative glomerulonephritis (MPGN) is characterized by glomerular injury, mesangial hypercellularity, endocapillary proliferation, and capillary wall remodeling with double-contours.38 There are two forms of idiopathic MPGN based on pathogenesis: immune complex-mediated MPGN and MPGN associated with dysregulation of the alternative complement pathway.39 Immune complex-mediated MPGN is caused by the deposition of immune complexes in the glomeruli, triggering an activation of the classical pathway of complement and the deposition of factors of the classical and terminal complement pathways along the capillary wall.38 Several factors, such as chronic virus B or C infections, systemic lupus erythematosus, Sjogren syndrome, rheumatoid arthritis, mixed connective-tissue disorders, and monoclonal gammopathy, can trigger this.38 MPGN associated with dysregulation of the alternative pathway, on the other hand, can result from mutations or autoantibodies directed against complement-regulating proteins.38 The dysregulation of the alternative pathway results in activated complement products, which deposit in the mesangium and subendothelial regions, causing inflammation.38 Cases of MPGN due to dysregulation of the alternative pathway are characterized by a proliferative histologic lesion with C3 deposition at least two orders of magnitude greater than any other immune reactant on immunofluorescence.10 This condition may be subdivided into dense-deposit disease (DDD) and C3 glomerulonephritis (C3GN).38 The DDD is characterized by electron-dense deposits on the glomerular basement and mesangium. In contrast, on the C3GN, its deposits are in mesangial, subendothelial, subepithelial, and intramembranous spaces.38 Patients with MPGN usually present proteinuria, dysmorphic hematuria, and hypertension, with variable progressive decline in renal function. Persistently decreased serum levels of complement factors are commonly seen in patients with MPGN. In the immune complex-mediated MPGN, low C3 and C4 are usually present, while a low C3 with normal C4 pattern is common in the alternative-pathway dysfunction cases.38 Patients with chronic infections, autoimmune diseases, or monoclonal gammopathies should be treated for underlying conditions.38 The best treatment of complement-mediated MPGN remains under investigation. Immunosuppressive therapy with glucocorticoids or rituximab or treatment with inhibitors of the membrane-attack complex formation, such as eculizumab, have been used in complement-mediated MPGN cases. Patients with MPGN due to a deficiency of complement-regulating proteins might benefit from plasma or factor H infusions.38
The suspicion of MPGN recurrence occurs in recipients with MPGN as the primary cause of ESKD who present a new onset of proteinuria, hematuria, and impaired graft function during the follow-up. The rate of MPGN recurrence ranges from 27% to 65%, varying according to the underlying cause.38,39 A retrospective study by our group, including recipients of kidney transplantation with CKD secondary to MPGN, showed a recurrence rate of 25%, most recipients of kidneys from living donors, like that observed in other studies.40 The MPGN recurrence rate is higher among patients with complement-mediated MPGN than immune complex-mediated MPGN.41 The recurrence rate of DDD is high, reaching 100% in some studies, with a 5-year rate of graft failure of 50%.39 Data on the C3GN recurrence rate are scarce.38 The MPGN recurrence is associated with poor allograft outcome, mainly in cases of complement-mediated disease.39 A previous multicentric retrospective cohort study including 34 complement-mediated and 186 cases of immune complex-mediated MPGN found a recurrence rate of 25%.41 Of the recurrent cases, 61% experienced graft loss. Early recurrence, low estimated glomerular filtration rate, and hypoalbuminemia were considered the main determinants of no remission.
Pre-transplant evaluation of the MPGN cases is essential since treatment of underlying causes can prevent post-transplant recurrence in some cases.39 There is no preferable immunosuppressive therapy for preventing MPGN recurrence.39 Patients with MPGN recurrence presenting stable renal function and mild proteinuria may receive treatment with ACEi or ARB. In cases of proteinuria 1.5–3.5 g/day, therapy with high-dose steroids (prednisone 1 mg/kg) for 16 weeks is indicated, followed by a taper over several weeks. Several MPGN recurrent cases with rapidly declining graft function, proteinuria > 3.5g/day, or presence of crescentic disease, treatment with plasmapheresis, pulse intravenous methylprednisolone, and cyclophosphamide can be considered. Treatment of C3 glomerulopathy in kidney transplant recipients with a combination of steroids and mycophenolate mofetil seems to be less effective than the observed among native kidney disease.41 A case series including patients with C3 glomerulopathy showed that the currently available therapies with rituximab and eculizumab had limited success in treating disease recurrence.42 Some new therapeutic strategies for patients presenting C3 glomerulopathy recurrence are under study. For cases of immune complex-mediated MPGN with the identification of the underlying trigger, the most effective therapy is to treat the primary disease.10 For patients with idiopathic immune complex-mediated MPGN presenting proteinuria less than 3.5 g/d, absence of nephrotic syndrome, and normal estimated glomerular filtration, the treatment involves supportive therapy with renin-angiotensin-aldosterone system inhibition.10 For patients with idiopathic immune complex-mediated MPGN presenting abnormal kidney function and active urine sediment, treatment with high-dose steroids can be considered.10
Viral Glomerulopathies
The glomeruli can suffer damage during an HIV infection through several processes, including glomerular and tubular epithelial cell infection, immune complex-mediated injury, thrombotic microangiopathy, and nephrotoxicity secondary to antiretroviral therapy.2 Previous studies showed that HIV can infect kidney allograft cells even in patients with undetectable viral loads and receiving antiretroviral therapy.43 The immune complex-mediated glomerulopathy is the lead form in patients receiving antiretroviral therapy.2 It results from the association of immunoglobulins and viral antigens with their deposition on subendothelial, subepithelial, intramembranous, or mesangial spaces. Patients with this form of HIV-associated glomerulopathy can course with proteinuria, hematuria, or acute kidney injury. The treatment of immune-complex-associated glomerulopathy includes renin-angiotensin-aldosterone system (RAAS) blockage, and antiretroviral therapy is usually ineffective in slowing progression to ESKD.2 HIV-associated nephropathy (HIVAN), in turn, is caused by infection of epithelial cells and is characterized by collapsing focal segmental glomerulosclerosis and proliferation of glomerular epithelial cells, typically occurring in patients with low CD4 cell counts. Patients with this form of glomerulopathy usually present nephrotic proteinuria, and the RAAS blockade is indicated.
Recipients of a kidney transplant can present clinical complications associated with HCV or HBV infection due to de novo infections or chronic disease. Chronic infection with HCV and HBV can lead to glomerular disease through the production and deposition of immune complexes in the glomerulus and podocyte dysregulation.44 Chronic HCV infection usually causes mixed cryoglobulinemia, a pattern of polyclonal IgG and monoclonal IgM, which can cause a cryoglobulinemic glomerulonephritis.44 HCV infection can course with other glomerular diseases, like non-cryoglobulinemic MPGN, MN, FSGS, IgAN, and polyarteritis nodosa (PAN), and these glomerulopathies can also occur in kidney allografts. The most frequent glomerulopathies associated with chronic HBV infection are MN, MPGN, and PAN.44 Advances in therapy for HCV and HBV have created opportunities for effective treatment of these patients before the kidney transplantation of patients without advanced liver disease.45 For patients with advanced liver disease, a simultaneous kidney-liver disease can be considered.45 Before the direct-acting antiviral (DAA) agents, HCV was associated with lower graft and patient survival after kidney transplantation.44 Nowadays, patients receiving hemodialysis should be treated with DAA before transplantation, and this treatment is safe for kidney transplant recipients.44 Treatment with DAA is recommended for the treatment of patients with HCV-associated glomerulopathy, with additional treatment with immunosuppressive drugs in case of nephrotic syndrome or rapidly progressive kidney failure.2 All kidney transplant candidates with HBsAg positive should be treated before transplantation to maintain undetectable HBV DNA and avoid complications associated with the disease before transplantation.44 It is recommended that patients with HBV-associated glomerulopathy be treated with a nucleoside/nucleotide analog drug.2
The SARS-CoV-2 is the etiologic agent of the coronavirus disease 2019 (COVID-19) pandemic, which negatively impacted the survival of kidney transplant recipients, especially those who had presented acute kidney injury (AKI).46 SARS-CoV-2 can cause AKI through cellular toxicity, complement activation, immune dysregulation, and coagulopathy.47 Previous studies from patients with COVID-19 who had their kidneys biopsied showed that collapsing focal segmental glomerulosclerosis was the most frequent glomerular pathology in native kidneys and allografts. However, a causal relationship between COVID-19 and the glomerular lesion remains unclear.47 However, it has been suggested that cytokine-mediated effects of the disease and immune responses are implicated in this process.2 Besides collapsing glomerulopathy, COVID-19 may elicit immune responses exacerbating immune-mediated glomerular conditions, such as MN, lupus nephritis, and anti-glomerular basement membrane disease.2
CMV is a type of herpesvirus that is widespread across the world. It is mainly transmitted through the urine and saliva of young children but can also be transmitted sexually, by blood transfusion, or by organ transplantation. Primary infection occurs in patients without immunity against the virus, and the CMV remains latent, enabling posterior reactivations. A person with a previous CMV infection can also present a reinfection when in contact with infected individuals. Symptoms are usually absent during CMV infection, but immunocompromised individuals may have high viral loads, leading to organ damage to the lungs, gastrointestinal tract, central nervous system, and retina.48 Although CMV renal infection is rare, some studies have shown that the virus can affect the glomeruli, with CMV viral inclusion in the glomerular endothelial cell nuclei.3
The BK virus is a non-enveloped double-stranded DSA virus belonging to the Polyomaviridae family. It is a common infection in the general population that can be transmitted through the oral, gastrointestinal, and respiratory tracts. After an initial viremia, the virus can reside in the kidney and uroepithelial cells, leading to a latent infection.49 However, the virus can start replicating in patients who have undergone kidney transplantation and are on immunosuppressive therapy, leading to viruria, viremia, and allograft nephropathy (BKVAN). Most cases of BKVAN occur within the first year of transplantation.49 The main histologic features of BKVAN include enlarged and hyperchromatic nuclei with intranuclear inclusions in tubular epithelial cells.49 Although less frequent, glomerular damage caused by BKVAN has also been observed, with evidence of infection of the Bowman’s capsular epithelium, associated with a variable increase in mesangial matrix and aneurysmal dilatation of glomerular capillaries.4 The glomerular visceral epithelial cells seem resistant to BK virus infection.4 The treatment of BKVAN involves a reduction in the intensity of immunosuppression, with a decrease in the dose of antimetabolite by half, followed by a drop of calcineurin-inhibitor though goals if persistence of infection (4–6 ng/mL for tacrolimus and 50–100 ng/mL for cyclosporine).49 Adjunctive therapies for treating BKVAN include quinolones, cidofovir, leflunomide, and intravenous immunoglobulin.49 However, the nephrotoxicity of some of these drugs limits its clinical application.49
Conclusion
Several conditions can affect the glomeruli of kidney transplant recipients, including recurrence or de novo glomerulopathies and viral infections. The impact of these conditions on the graft outcomes varies according to the primary kidney disease and the severity of the recurrence. The knowledge on viral glomerulopathy is based on case series, most referring to the native kidney disease. Early identification of viral infections, correct management of immunosuppressive treatment, and treatment of infections are essential to minimize glomerular damage from these infections.
Funding
There is no funding to report.
Disclosure
The author reports no conflicts of interest in this work.
References
1. Lim WH, Shingde M, Wong G. Recurrent and de novo glomerulonephritis after kidney transplantation. Front Immunol. 2019;10:1944. doi:10.3389/fimmu.2019.01944
2. Deoliveira M, Sikri H, Yu SMW, He JC. Viral glomerulopathy. Glomerular Dis. 2023;148–154. doi:10.1159/000531434
3. Posadas Salas MA, Thompson J, Kadian M, Ngo T, Bruner E, Self S. Cytomegalovirus renal infection: rare manifestation of a common post‐transplant viral infection—A case series. Transplant Infect Dis. 2019;21(6):e13169. doi:10.1111/tid.13169
4. Celik B, Randhawa PS. Glomerular changes in BK virus nephropathy. Hum Pathol. 2004;35(3):367–370. doi:10.1016/j.humpath.2003.09.009
5. Wyld ML, Chadban SJ. Recurrent iga nephropathy after kidney transplantation. Transplantation. 2016;100(9):1827–1832. doi:10.1097/TP.0000000000001093
6. Caster DJ, Lafayette RA. The treatment of primary igA nephropathy: change, change, change. Am J Kidney Dis. 2024;83(2):229–240. doi:10.1053/j.ajkd.2023.08.007
7. Hayashi A, Kawabe M, Yamamoto I, et al. Clinical and pathological significance of mesangial c1q deposition in kidney transplant recipients with recurrent iga nephropathy and patients with native iga nephropathy. Nephron. 2023;147(Suppl. 1):80–88. doi:10.1159/000530916
8. Cattran DC, Coppo R, Cook HT, et al. The Oxford classification of IgA nephropathy: rationale, clinicopathological correlations, and classification. Kidney Int. 2009;76(5):534–545. doi:10.1038/ki.2009.243
9. Trimarchi H, Barratt J, Cattran DC, et al. Oxford classification of iga nephropathy 2016: an update from the iga nephropathy classification working group. Kidney Int. 2017;91(5):1014–1021. doi:10.1016/j.kint.2017.02.003
10. Kidney Disease: Improving Global Outcomes (KDIGO) Glomerular Diseases Work Group. KDIGO 2021 clinical practice guideline for the management of glomerular diseases. Kidney Int. 2021;100(4S):S1–276.
11. Uffing A, Pérez-Saéz MJ, Jouve T, et al. Recurrence of iga nephropathy after kidney transplantation in adults. Clin J Am Soc Nephrol. 2021;16(8):1247–1255. doi:10.2215/CJN.00910121
12. Vasconcelos A de S, Mazzali M, de Sousa MV. IgA nephropathy and kidney transplantation according to the Oxford classification. Braz J Nephro. 2023;45(3):350–356.
13. Alachkar N, Delsante M, Greenberg RS, et al. Evaluation of the modified oxford score in recurrent iga nephropathy in north American kidney transplant recipients: the Banff recurrent glomerulonephritis working group report. Transplantation. 2023;107(9):2055–2063. doi:10.1097/TP.0000000000004640
14. De Souza L, Prunster J, Chan D, Chakera A, Lim WH. Recurrent glomerulonephritis after kidney transplantation. Curr Opin Organ Transplant. 2021;26(4):360–380. doi:10.1097/MOT.0000000000000887
15. Floege J, Grone HJ. Recurrent IgA nephropathy in the renal allograft: not a benign condition. Nephrol Dial Transplant. 2013;28(5):1070–1073. doi:10.1093/ndt/gft077
16. Moroni G, Longhi S, Quaglini S, et al. The long-term outcome of renal transplantation of IgA nephropathy and the impact of recurrence on graft survival. Nephrol Dial Transplant. 2013;28(5):1305–1314. doi:10.1093/ndt/gfs472
17. Kidney Disease: Improving Global Outcomes (KDIGO) Transplant Work Group. KDIGO clinical practice guideline for the care of kidney transplant recipients. Am J Transplant. 2009;9(Suppl 3):S1–157.
18. Gandolfini I, Alibrandi S, Gentile M, et al. Targeted-release budesonide in recurrent IgA nephropathy after kidney transplantation. Kidney Int. 2023;103(5):995–996. doi:10.1016/j.kint.2023.02.012
19. Ronco P, Debiec H. Pathophysiological advances in membranous nephropathy: time for a shift in patient’s care. Lancet. 2015;385(9981):1983–1992. doi:10.1016/S0140-6736(15)60731-0
20. Beckwith H, Kingsbury M, Horsburgh J. Why do people choose nephrology? Identifying positive motivators to aid recruitment and retention. Clin Kidney J. 2018;11(5):599–604. doi:10.1093/ckj/sfy076
21. Beck LH, Bonegio RGB, Lambeau G, et al. M-type phospholipase A2 receptor as target antigen in idiopathic membranous nephropathy. N Engl J Med. 2009;361(1):11–21. doi:10.1056/NEJMoa0810457
22. Filippone EJ, Farber JL. Membranous nephropathy in the kidney allograft. Clin Transplant. 2016;30(11):1394–1402. doi:10.1111/ctr.12847
23. Cavanaugh C, Okusa MD. The evolving role of novel biomarkers in glomerular disease: a review. Am J Kidney Dis. 2021;77(1):122–131. doi:10.1053/j.ajkd.2020.06.016
24. Grupper A, Cornell LD, Fervenza FC, Beck LH, Lorenz E, Cosio FG. Recurrent membranous nephropathy after kidney transplantation. Transplantation. 2016;100(12):2710–2716. doi:10.1097/TP.0000000000001056
25. Berchtold L, Letouzé E, Alexander MP, et al. HLA-D and PLA2R1 risk alleles associate with recurrent primary membranous nephropathy in kidney transplant recipients. Kidney Int. 2021;99(3):671–685. doi:10.1016/j.kint.2020.08.007
26. Leon J, Pérez-Sáez MJ, Batal I, et al. Membranous nephropathy posttransplantation: an update of the pathophysiology and management. Transplantation. 2019;103(10):1990–2002. doi:10.1097/TP.0000000000002758
27. Buxeda A, Caravaca-Fontán F, Vigara LA, et al. High exposure to tacrolimus is associated with spontaneous remission of recurrent membranous nephropathy after kidney transplantation. Clin Kidney J. 2023;16(10):1644–1655. doi:10.1093/ckj/sfad077
28. de Sousa MV, Fernandes LGR, de Freitas LLL, Zollner R de L, Mazzali M. De novo membranous nephropathy associated with antibody-mediated rejection in kidney transplant recipients. Transplant Proc. 2022;54(5):1270–1277. doi:10.1016/j.transproceed.2021.11.041
29. El Kossi M, Harmer A, Goodwin J, et al. De novo membranous nephropathy associated with donor‐specific alloantibody. Clin Transplant. 2008;22(1):124–127. doi:10.1111/j.1399-0012.2007.00741.x
30. Patel K, Hirsch J, Beck L, Herlitz L, Radhakrishnan J. De novo membranous nephropathy in renal allograft associated with antibody-mediated rejection and review of the literature. Transplant Proc. 2013;45(9):3424–3428. doi:10.1016/j.transproceed.2013.05.011
31. Honda K, Horita S, Toki D, et al. De novo membranous nephropathy and antibody-mediated rejection in transplanted kidney. Clin Transplant. 2011;25(2):191–200. doi:10.1111/j.1399-0012.2010.01213.x
32. Radhakrishnan Y, Zand L, Sethi S, Fervenza FC. Membranous nephropathy treatment standard. Nephrol Dial Transplant. 2023;9:gfad225.
33. Salfi G, Casiraghi F, Remuzzi G. Current understanding of the molecular mechanisms of circulating permeability factor in focal segmental glomerulosclerosis. Front Immunol. 2023;14:1247606. doi:10.3389/fimmu.2023.1247606
34. Shabaka A, Tato Ribera A, Fernández-Juárez G. Focal segmental glomerulosclerosis: state-of-the-art and clinical perspective. Nephron. 2020;144(9):413–427. doi:10.1159/000508099
35. Canaud G, Delville M, Legendre C. Recurrence of focal and segmental glomerulosclerosis after transplantation. Transplantation. 2016;100(2):284–287. doi:10.1097/TP.0000000000000902
36. Bai J, Zhang T, Wang Y, et al. Incidence and risk factors for recurrent focal segmental glomerulosclerosis after kidney transplantation: a meta-analysis. Ren Fail. 2023;45(1). doi:10.1080/0886022X.2023.2201341
37. Uffing A, Pérez-Sáez MJ, Mazzali M, et al. Recurrence of FSGS after kidney transplantation in adults. Clin J Am Soc Nephrol. 2020;15(2):247–256. doi:10.2215/CJN.08970719
38. Sethi S, Fervenza FC. Membranoproliferative glomerulonephritis — a new look at an old entity. N Engl J Med. 2012;366(12):1119–1131. doi:10.1056/NEJMra1108178
39. Sprangers B, Kuypers DR. Recurrence of glomerulonephritis after renal transplantation. Transplant Rev. 2013;27(4):126–134.
40. de Paula LC, Mazzali M, de Sousa MV. Recurrent membranoproliferative glomerulonephritis after kidney transplantation: risk factors and impact on graft survival. Ann Transp. 2023;7(28):e940502.
41. Caravaca-Fontán F, Polanco N, Villacorta B, et al. Recurrence of immune complex and complement-mediated membranoproliferative glomerulonephritis in kidney transplantation. Nephrol Dial Transplant. 2023;38(1):222–235. doi:10.1093/ndt/gfac148
42. Regunathan-Shenk R, Avasare RS, Ahn W, et al. Kidney transplantation in C3 glomerulopathy: a case series. Am J Kidney Dis. 2019;73(3):316–323. doi:10.1053/j.ajkd.2018.09.002
43. Canaud G, Dejucq-Rainsford N, Avettand-Fenoël V, et al. The kidney as a reservoir for HIV-1 after renal transplantation. J Am Soc Nephrol. 2014;25(2):407–419. doi:10.1681/ASN.2013050564
44. Sharma P, Sawtell R, Wang Q, Sise ME. Management of hepatitis c virus and hepatitis b virus infection in the setting of kidney disease. Adv Kidney Dis Health. 2023;30(4):343–355. doi:10.1053/j.akdh.2023.04.003
45. Huskey J, Wiseman AC. Chronic viral hepatitis in kidney transplantation. Nat Rev Nephrol. 2011;7(3):156–165. doi:10.1038/nrneph.2010.192
46. Requião-Moura LR, de S-FTV, Viana LA, et al. High mortality among kidney transplant recipients diagnosed with coronavirus disease 2019: results from the Brazilian multicenter cohort study. PLoS One. 2021;16(7):e0254822. doi:10.1371/journal.pone.0254822
47. Klomjit N, Zand L, Cornell LD, Alexander MP. COVID-19 and glomerular diseases. Kidney Int Rep. 2023;8(6):1137–1150. doi:10.1016/j.ekir.2023.03.016
48. Griffiths P, Reeves M. Pathogenesis of human cytomegalovirus in the immunocompromised host. Nat Rev Microbiol. 2021;19(12):759–773. doi:10.1038/s41579-021-00582-z
49. Kant S, Dasgupta A, Bagnasco S, Brennan DC. BK virus nephropathy in kidney transplantation: a state-of-the-art Review. Viruses. 2022;14(8):1616. doi:10.3390/v14081616
 © 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.