Back to Journals » Journal of Hepatocellular Carcinoma » Volume 6
Post-translational modifiers of liver kinase B1/serine/threonine kinase 11 in hepatocellular carcinoma
Authors Delgado TC, Lopitz-Otsoa F, Martínez-Chantar ML
Received 4 January 2019
Accepted for publication 24 April 2019
Published 6 June 2019 Volume 2019:6 Pages 85—91
DOI https://doi.org/10.2147/JHC.S169585
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Ahmed Kaseb
Video abstract of "Post-translational modifiers of LKB1 in HCC" [ID 169585].
Views: 357
Teresa Cardoso Delgado, Fernando Lopitz-Otsoa, María Luz Martínez-Chantar
Liver Disease and Liver Metabolism Laboratories, CIC bioGUNE, Centro de Investigación Biomédica en Red de Enfermedades Hepáticas y Digestivas (CIBERehd), Derio 48160, Bizkaia, Spain
Abstract: Liver kinase B1 (LKB1) also referred to as serine/threonine kinase 11 (STK11) encodes a 50 kDa evolutionary conserved serine/threonine kinase that is ubiquitously expressed in adult and fetal tissues. LKB1 is a master kinase known to phosphorylate and activate several kinases including AMP-activated protein kinase, a crucial cellular energy sensor. LKB1 shows pleiotropic activity playing diverse roles in multiple processes, including cell polarity and other processes relevant in cancer pathology, such as energy metabolism, proliferation and apoptosis. In spite of the fact that LKB1 is often considered a tumor suppressor in a wide variety of organs, in the last years, several studies have shown that LKB1 is unexpectedly high in hepatocellular carcinoma (HCC), the most common type of primary liver cancer. Post-translational modifications of LKB1 are potentially relevant in HCC. Herein, we provide a comprehensive revision of post-translational modifications of LKB1 in HCC and how they modulate LKB1 function by different mechanisms such as regulation of its activity, localization or stability. Overall, the signature post-translational modifications of LKB1 in HCC appear to play an important role in the rather unique role of LKB1 as an oncogenic driver in liver cancer and may provide an alternative valuable therapeutic approach to regulate LKB1 expression and/or activity in HCC.
Keywords: post-translational modifications, SUMO, NEDD8, STRADα, AMPK
Introduction
Liver kinase B1 (LKB1) also referred to as serine/threonine kinase11 (STK11) (located on chromosome 19p13.3) encodes a 50 kDa evolutionary conserved serine/threonine kinase that is ubiquitously expressed in adult and fetal tissues, particularly in pancreas, liver, testis and skeletal muscle (Figure 1).1,2 LKB1 possesses a nuclear localization sequence at its N-terminal noncatalytic region that can promote the shuttling of LKB1 between the cytoplasm and the nucleus.3 Lacking a nuclear export domain of its own, LKB1 cellular localization is mediated by cofactors, such as the pseudokinase Ste20-related adaptor (STRADα) and the scaffolding protein MO25.4–6 STRADα adopts a closed conformation typical of active protein kinases and binds LKB1 as a pseudosubstrate. STRADα promotes the active conformation of LKB1 which is stabilized by MO25 interacting with the LKB1 activation loop.7 Binding of LKB1 to STRADα–MO25 complex induces relocalization of LKB1 from the nucleus to the cytoplasm stimulating its catalytic activity.4,5 Moreover, by facilitating the binding of exportins to LKB1 and acting as a competitor for importin-α/β, STRADα prevents the nuclear relocalization of LKB1 (Figure 2).8 The cellular localization of LKB1 plays an essential role on its activity.
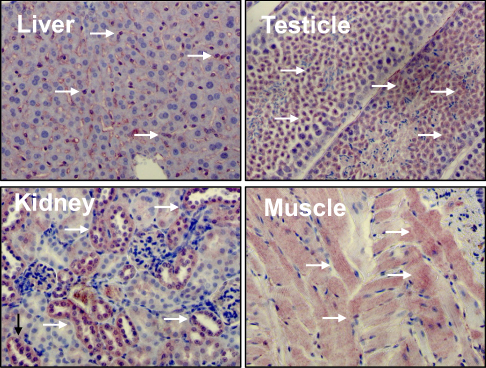 | Figure 1 Liver kinase B1 (LKB1) is ubiquitously expressed in adult tissues. Distribution of LKB1 by immunohistochemistry staining in mouse tissues. Arrows indicate LKB1 staining. |
 | Figure 2 The nucleocytoplasmic shuttling of liver kinase B1 (LKB1) is a tightly regulated process important in the modulation of its activity. The cellular localization of LKB1 plays an essential role in its activity and ability to activate AMP-activated protein kinase (AMPK). LKB1 cellular localization is mediated by cofactors, such as the Ste20-related adaptor (STRADα) and MO25. Binding of LKB1 to STRADα–MO25 complex induces relocalization of LKB1 from the nucleus to the cytoplasm stimulating its catalytic activity, for example promoting the phosphorylation of AMPK. Although LKB1 is imported into the nucleus by importin-alpha/beta, STRADα and MO25 passively diffuse between the nucleus and the cytoplasm. STRADα facilitates nuclear export of LKB1 by serving as an adaptor between LKB1 and exportins CRM1 and exportin7.4,5,8 |
Liver kinase B1 in cancer
LKB1 shows pleiotropic activity being involved in the regulation of cell polarity and the regulation of energy metabolism, proliferation and apoptosis. LKB1 is a master kinase known to phosphorylate and activate 14 kinases, including AMP-activated protein kinase (AMPK) and microtubule-associated protein/microtubule affinity-regulating kinases (MARK).9 AMPK is a crucial nutrient and cellular energy sensor that maintains energy homeostasis mainly by regulating metabolism and favoring the expression of proteins involved in catabolism and switching off biosynthetic pathways under falling of energy status.10 In cells bearing intact LKB1/AMPK signaling, energy depletion promotes growth arrest, whereas cells lacking LKB1, often showing chronic AMPK activation, dysregulated cellular proliferation occurs. Therefore, a role for LKB1 in cancer has been suggested in the last years. Indeed, LKB1 was first identified as a tumor-suppressor gene as germline mutations or deletions in the LKB1 gene were found to be responsible for the Peutz–Jeghers syndrome (PJS), an inherited cancer-prone disorder.1 Moreover, somatic mutations of LKB1 are involved in the development of non-small-cell lung cancer,11 cervical cancer,12 and prostate cancer,13 among others. Although LKB1 is classified as a tumor suppressor, some recent evidence highlight that LKB1 may exhibit cell type-specific functions and an ambivalent role in tumorigenesis.
Liver cancer is the sixth most prevailing cancer worldwide.14 Common risk factors for hepatocellular carcinoma (HCC), the most common types of primary liver cancer include chronic hepatitis B or C infection, metabolic syndrome, Type 2 diabetes and alcohol consumption.15 Of special concern, HCC patients have a poor and gloomy prognosis, with a 5-year survival rate of less than 10%, making liver cancer the second leading cause of global cancer-related deaths.16 In the last years, we and others have shown that LKB1 is unexpectedly high both in animal models of HCC and in liver biopsies of HCC patients.17–19 To date, the levels of LKB1 in HCC have been addressed without taking into consideration the etiology of HCC, being that in most studies presented samples from hepatitis C, alcoholic steatohepatitis (ASH) and nonalcoholic steatohepatitis (NASH) have been used.17,20 Further studies should be undertaken to address the expression of LKB1 in HCC samples from different etiologies. Moreover, LKB1 knockdown in hepatoma cells induces tumor cell death,17,21 whereas in vivo silencing of LKB1 in a xenograft mouse model ameliorated hepatoma tumor growth.17 The mechanisms underlying the overexpression of LKB1 in HCC will be further explored.
Liver kinase B1 regulation in HCC
Protein expression may be modulated in several ways, from the DNA–RNA transcription step to post-translational modification of a protein. Although previous studies have shown that the biallelic inactivation of the Lkb1 gene in mice leads to multiple hepatic nodular foci and HCC,22 genetic alterations of the LKB1 gene together with one LKB1 missense mutation and allelic loss were only sporadically found in clinical HCC.23 Likewise, the frequencies of DNA methylation, a hallmark of many cancer cells, were similar between HCC and the corresponding noncancerous tissues.24 Therefore, other mechanisms, such as post-translational modifications of LKB1, are potentially relevant in HCC.
Post-translational modifications of liver kinase B1 in HCC
Post-translational modifications are considered key mechanisms regulating protein homeostasis and function in eukaryotic cells. These modifications extend the diversity of the proteome by inducing structural and functional changes in proteins through different mechanisms like covalent binding of functional groups, cleavage of regulatory subunits and degradation of other proteins. The most common post-translational modifications include phosphorylation, methylation, acetylation, glycosylation, ubiquitination and ubiquitin-like protein (UBLs)-mediated post-translational modifications.
Phosphorylation of liver kinase B1 in HCC
Reversible protein phosphorylation, mainly on serine, threonine or tyrosine residues, is one of the most well-studied post-translational modifications. In the context of liver cancer, phosphorylation of LKB1 at Ser428 was previously observed in liver tumors of mice that spontaneously develop HCC, the mice deficient in methionine adenosyl transferase 1 (Mat1A-/-). Likewise, SAMe-D (SAMe-Deficient) cells, a cell line derived from Mat1A-/- mice, OKER cells, hepatic tumor cells derived from the HCC mouse model deficient in glycine N-methyltransferase (Gnmt) (Gnmt−/- mice), together with several human hepatoma cells lines, express high levels of phosphorylated LKB1 at Ser428.17,21 In hepatoma SAMe-D cells, LKB1 phosphorylation regulates Akt-mediated survival in a process regulated by p53, HAUSP and HuR.21 Moreover, Ras-mediated hyperphosphorylation of LKB1, concomitant with expression of Ras guanyl-releasing protein-3 (RASGRP3), promoted proliferation of OKER hepatoma cells and required mitogen-activated protein kinase-2 (ERK) and ribosomal protein S6 kinase polypeptide-2 (p90RSK).17 Importantly, HCC tumors with the poorer prognosis have the highest levels of phosphorylated LKB1 (Ser428).21 Overall, these results suggest that LKB1 phosphorylation at Ser428 is involved in a pro-survival mechanism of hepatoma cells accounting for aberrant tumor growth.
Ubiquitination of liver kinase B1 in HCC
The ubiquitination of proteins is a post-translational modification that is involved in many different cellular processes in addition to its well-known function during protein degradation. LKB1 ubiquitination has been implicated in HCC. The polyubiquitination of LKB1 takes place on five lysine residues (K41, K44, K48, K62 and K63) at the N-terminus of LKB1. Indeed, Lee et al have described that LKB1 is polyubiquitinated by the Skp2-SCF ubiquitin ligase being that overexpression of Skp2 and LKB1 is observed in late-stage HCC, and their overexpression predicts poor survival outcomes.19 Mechanistically, the polyubiquitination of LKB1 is crucial by maintaining the integrity of the LKB1-STRADα–Mo25 complex, which plays an important role in the regulation of LKB1 nucleocytoplasmic export and concomitant kinase activity. Furthermore, oncogenic Ras acts upstream of Skp2 to promote LKB1 polyubiquitination by activating Skp2-SCF ubiquitin ligase.19 In summary, ubiquitination of LKB1 is a hallmark of late stages HCC.
Neddylation of liver kinase B1 in HCC
The NEDD8 conjugation pathway, NEDDylation, is similar to that described for ubiquitination, resulting in the reversible covalent conjugation of a molecule of NEDD8 to a lysine residue of the substrate protein. NEDDylation conjugation was shown to be aberrant in liver biopsies of HCC patients in comparison with healthy controls,18,25 where a strong positive correlation was observed between the levels of LKB1 and NEDD8.18 Indeed, Barbier-Torres et al have performed some in vitro experiments in pro-tumoral mouse hepatocytes, suggesting that LKB1 is directly NEDDylated and that NEDD8 directly stabilizes LKB1 in HCC.18 Further studies are necessary to find the enzymes involved in the NEDDylation and deNEDDylation pathway of LKB1 together with the residues that are relevant for NEDDylation. Importantly, when pre-tumoral hepatocytes were treated either with Pevonedistat,26 a small pharmacological inhibitor of NEDDylation, or by silencing NEDD8 using molecular approaches, LKB1 levels were reduced and tumor cell death was induced.18 Likewise, when liver tumor-bearing mice were treated with Pevonedistat, the levels of LKB1 fell.18 Liver tumor cell apoptosis induced by Pevonedistat was reduced by overexpressing LKB1, seconding the important role of LKB1 as an oncogenic driver in HCC. Overall, NEDD8-mediated post-translational modifications of LKB1 are relevant in HCC and offer a novel druggable therapeutic approach for the regulation of LKB1 levels.
Sumoylation of liver kinase B1 in HCC
Small ubiquitin-related modifier (SUMO)-mediated modifications appear to be upregulated in many types of cancer, including HCC.27–29 LKB1 contains 3 high scoring sites for possible SUMO-binding sites in its amino acid sequence. Recently, Ritho et al have described for the first time LKB1 SUMO-mediated modifications and its implication under metabolic stress circumstances.30 Moreover, a recent report by Zubiete-Franco et al highlights that in human hepatoma cells and preclinical mouse models of liver cancer as well as in clinical biopsies of HCC patients, LKB1 is modified by the SUMO-2 paralogue in the Lys178. Noteworthy, LKB1 SUMOylation, which is more prevalent during hypoxic stimuli, offers growth advantage to hepatoma cells and is a signature of more aggressive HCC clinical tumors. Under these circumstances, LKB1 SUMOylation appears to hamper the binding of LKB1 to STRADα, thereby diminishing the LKB1 nucleocytoplasmic shuttling and reducing LKB1 kinase activity.20 Even though LKB1 functions in the nucleus are not totally understood to date, they appear to play an important role in the regulation of HCC tumor cell growth and survival during hypoxia. Further studies are necessary to fully understand the mechanisms underlying the oncogenic role of SUMOylated LKB1 in HCC.
Acetylation of liver kinase B1 in HCC
LKB1 has been shown to be acetylated in rat liver. Indeed, LKB1 activity was 33% higher and its total acetylation at Lys48 was 60% lower in the liver of starved rats where LKB1 acetylation at Lys48 prevents LKB1 binding to STRADα hampering LKB1 nucleocytoplasmic export.31 It is well-described that protein acetylation can compete with SUMO-mediated modifications for the same lysine residue, resulting in a SUMO/acetylation switch32 or in opposite, acetylation switch can regulate SUMO-dependent interaction networks.33 In human hepatoma cells, we have recently shown that LKB1 acetylation at Lys48 is essential for its posterior SUMOylation at Lys178 by SUMO-2.20 On this basis, unpublished data from our laboratory indicate that in human hepatoma cells during hypoxia, LKB1 acetylation is not reduced (data not shown). More studies are necessary to unequivocally address the role of LKB1 acetylation in HCC.
Conclusion
Although LKB1 has been previously described in a wide variety of organs as a tumor suppressor, in the last years, several groups have shown that LKB1 expression is augmented in HCC. Under these conditions, LKB1 overexpression does not appear to be regulated at the transcriptional level being that post-translational modifications of LKB1 appear to play a role in the regulation of the levels of LKB1 in HCC. To date, five different post-translational modifications of LKB1 have been implicated in HCC which appear to regulate the life cycle of LKB1 (Figure 3). The time course relative to disease progression and the nature of the relationship (competition/collaboration) between the post-translational modifications of LKB1 in HCC is not completely known. We although we can hypothesize that NEDD8-stabilization of LKB1, a process occurring very early in the progression of chronic liver disease and HCC18,34,35 increasing the protein half-life time, may result that LKB1 is a more easily accessible target to other post-translational modifications in HCC. On this basis, NEDDylated LKB1 could be further acetylated and then SUMOylated, being that LKB1 SUMOylation promotes a more predominant nuclear localization of LKB1 associated with growth and survival advantage to the tumor cells and inducing tumor growth.20 Taking into consideration the nuclear localization of LKB1 and the fact that nucleocytoplasmic shuttling is a mechanism relevant in the mediation of the transcriptional response, the role of LKB1 as a modulator of the transcriptional activity should be further explored. In alternative, NEDDylated LKB1 can also be phosphorylated17,21 or polyubiquitinated19 inducing proliferation and survival. In the near future, more research should focus on the study of the intermediates and players involved in these post-translational modifications of LKB1 in HCC. The recent advances on the development of pharmacological inhibitors of post-translational modifications, such as Pevonedistat, a specific inhibitor of NEDDylation,26 several small molecule inhibitors of the ubiquitination cascade,36 and inhibitors targeting the SUMO pathway37 further support these studies in order to find novel therapeutic targets to tackle HCC.
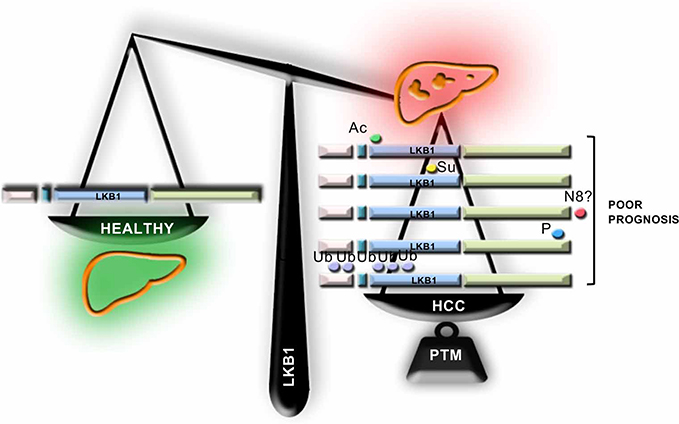 | Figure 3 Liver kinase B1 (LKB1) post-translational modifications in hepatocellular Carcinoma (HCC). In the last years, several evidence highlight that LKB1 expression is induced in HCC, a process that appears to be regulated at the post-translational level. In this regard, the role of post-translational modifications of LKB1, such as acetylation (Ac), SUMOylation (Su), NEDDylation (N8), phosphorylation (P) and ubiquitination (Ub), has been recently described. |
Acknowledgments
This work was supported by grants from ELKARTEK 2016, Departamento de Industria del Gobierno Vasco (to MLMC), MINECO: SAF2017-87301-R integrado en el Plan Estatal de Investigación Cientifica y Técnica y Innovación 2013–2016 cofinanciado con Fondos FEDER (to MLMC), EITB Maratoia BIO15/CA/014; Asociación Española contra el Cáncer (TCD), Daniel Alagille award from EASL (to TCD), Fundación Científica de la Asociación Española Contra el Cancer (AECC Scientific Foundation) Rare Tumor Calls 2017 (to MLMC), and La Caixa Foundation Program (to MLMC). Finally, Ciberehd_ISCIII_MINECO is funded by the Instituto de Salud Carlos III. We thank MINECO for the Severo Ochoa Excellence Accreditation to CIC bioGUNE (SEV-2016-0644).
Disclosure
Dr. Martínez-Chantar advises for Mitotherapeutix LLC. The other authors report no conflicts of interest in this work.
References
1. Hemminki A, Markie D, Tomlinson I, et al. A serine/threonine kinase gene defective in Peutz-Jeghers syndrome. Nature. 1998;391(6663):184–187. doi:10.1038/34432
2. Jenne DE, Reimann H, Nezu J, et al. Peutz-Jeghers syndrome is caused by mutations in a novel serine threonine kinase. Nat Genet. 1998;18(1):38–43. doi:10.1038/ng0198-38
3. Nezu J, Oku A, Shimane M. Loss of cytoplasmic retention ability of mutant LKB1 found in Peutz-Jeghers syndrome patients. Biochem Biophys Res Commun. 1999;261(3):750–755. doi:10.1006/bbrc.1999.1047
4. Baas AF, Boudeau J, Sapkota GP, et al. Activation of the tumour suppressor kinase LKB1 by the STE20-like pseudokinase STRAD. Embo J. 2003;22(12):3062–3072. doi:10.1093/emboj/cdg292
5. Boudeau J, Baas AF, Deak M, et al. MO25alpha/beta interact with STRADalpha/beta enhancing their ability to bind, activate and localize LKB1 in the cytoplasm. Embo J. 2003;22(19):5102–5114. doi:10.1093/emboj/cdg490
6. Brajenovic M, Joberty G, Kuster B, Bouwmeester T, Drewes G. Comprehensive proteomic analysis of human Par protein complexes reveals an interconnected protein network. J Biol Chem. 2004;279(13):12804–12811. doi:10.1074/jbc.M312171200
7. Zeqiraj E, Filippi BM, Deak M, Alessi DR, van Aalten DM. Structure of the LKB1-STRAD-MO25 complex reveals an allosteric mechanism of kinase activation. Science. 2009;326(5960):1707–1711. doi:10.1126/science.1178377
8. Dorfman J, Macara IG. STRADalpha regulates LKB1 localization by blocking access to importin-alpha, and by association with Crm1 and exportin-7. Mol Biol Cell. 2008;19(4):1614–1626. doi:10.1091/mbc.e07-05-0454
9. Alessi DR, Sakamoto K, Bayascas JR. LKB1-dependent signaling pathways. Annu Rev Biochem. 2006;75:137–163. doi:10.1146/annurev.biochem.75.103004.142702
10. Hardie DG, Ross FA, Hawley SA. AMPK: a nutrient and energy sensor that maintains energy homeostasis. Nat Rev Mol Cell Biol. 2012;13(4):251–262. doi:10.1038/nrm3311
11. Sanchez-Cespedes M, Parrella P, Esteller M, et al. Inactivation of LKB1/STK11 is a common event in adenocarcinomas of the lung. Cancer Res. 2002;62(13):3659–3662.
12. McCabe MT, Powell DR, Zhou W, Vertino PM. Homozygous deletion of the STK11/LKB1 locus and the generation of novel fusion transcripts in cervical cancer cells. Cancer Genet Cytogenet. 2010;197(2):130–141. doi:10.1016/j.cancergencyto.2009.11.017
13. Pearson HB, McCarthy A, Collins CM, Ashworth A, Clarke AR. Lkb1 deficiency causes prostate neoplasia in the mouse. Cancer Res. 2008;68(7):2223–2232. doi:10.1158/0008-5472.CAN-07-5169
14. Forner A, Llovet JM, Bruix J. Hepatocellular carcinoma. Lancet. 2012;379(9822):1245–1255. doi:10.1016/S0140-6736(11)61347-0
15. Gerbes A, Zoulim F, Tilg H, et al. Gut roundtable meeting paper: selected recent advances in hepatocellular carcinoma. Gut. 2017;67(2)380-388. doi:10.1136/gutjnl-2017-315068
16. Lepage C, Bossard N, Dejardin O, et al. Trends in net survival from rectal cancer in six European Latin countries: results from the SUDCAN population-based study. Eur J Cancer Prev. 2017;26:S48–S55. doi:10.1097/CEJ.0000000000000305
17. Martinez-Lopez N, Garcia-Rodriguez JL, Varela-Rey M, et al. Hepatoma cells from mice deficient in glycine N-methyltransferase have increased RAS signaling and activation of liver kinase B1. Gastroenterology. 2012;143(3):
18. Barbier-Torres L, Delgado TC, Garcia-Rodriguez JL, et al. Stabilization of LKB1 and Akt by neddylation regulates energy metabolism in liver cancer. Oncotarget. 2015;6(4):2509–2523. doi:10.18632/oncotarget.3191
19. Lee SW, Li CF, Jin G, et al. Skp2-dependent ubiquitination and activation of LKB1 is essential for cancer cell survival under energy stress. Mol Cell. 2015;57(6):1022–1033. doi:10.1016/j.molcel.2015.01.015
20. Zubiete-Franco I, Garcia-Rodriguez JL, Lopitz-Otsoa F, et al. SUMOylation regulates LKB1 localization and its oncogenic activity in liver cancer. EBioMedicine. 2018;40:406-421. doi:10.1016/j.ebiom.2018.12.031
21. Martinez-Lopez N, Varela-Rey M, Fernandez-Ramos D, et al. Activation of LKB1-Akt pathway independent of phosphoinositide 3-kinase plays a critical role in the proliferation of hepatocellular carcinoma from nonalcoholic steatohepatitis. Hepatology. 2010;52(5):1621–1631. doi:10.1002/hep.23860
22. Nakau M, Miyoshi H, Seldin MF, Imamura M, Oshima M, Taketo MM. Hepatocellular carcinoma caused by loss of heterozygosity in Lkb1 gene knockout mice. Cancer Res. 2002;62(16):4549–4553.
23. Kim CJ, Cho YG, Park JY, et al. Genetic analysis of the LKB1/STK11 gene in hepatocellular carcinomas. Eur J Cancer. 2004;40(1):136–141.
24. Chen H, Zhang T, Sheng Y, et al. Methylation profiling of multiple tumor suppressor genes in hepatocellular carcinoma and the epigenetic mechanism of 3OST2 regulation. J Cancer. 2015;6(8):740–749. doi:10.7150/jca.11691
25. Luo Z, Yu G, Lee HW, et al. The Nedd8-activating enzyme inhibitor MLN4924 induces autophagy and apoptosis to suppress liver cancer cell growth. Cancer Res. 2012;72(13):3360–3371. doi:10.1158/0008-5472.CAN-12-0388
26. Soucy TA, Smith PG, Milhollen MA, et al. An inhibitor of NEDD8-activating enzyme as a new approach to treat cancer. Nature. 2009;458(7239):732–736. doi 10.1038/nature07884
27. Seeler JS, Dejean A. SUMO and the robustness of cancer. Nat Rev Cancer. 2017;17(3):184–197. doi:10.1038/nrc.2016.143
28. Tomasi ML, Tomasi I, Ramani K, et al. S-adenosyl methionine regulates ubiquitin-conjugating enzyme 9 protein expression and sumoylation in murine liver and human cancers. Hepatology. 2012;56(3):982–993. doi:10.1002/hep.25701
29. Li J, Xu Y, Long XD, et al. Cbx4 governs HIF-1alpha to potentiate angiogenesis of hepatocellular carcinoma by its SUMO E3 ligase activity. Cancer Cell. 2014;25(1):118–131. doi:10.1016/j.ccr.2013.12.008
30. Ritho J, Arold ST, Yeh ET. A critical SUMO1 modification of LKB1 regulates AMPK activity during energy stress. Cell Rep. 2015;12(5):734–742. doi:10.1016/j.celrep.2015.07.002
31. Lan F, Cacicedo JM, Ruderman N, Ido Y. SIRT1 modulation of the acetylation status, cytosolic localization, and activity of LKB1. Possible role in AMP-activated protein kinase activation. J Biol Chem. 2008;283(41):27628–27635. doi:10.1074/jbc.M805711200
32. Schimmel J, Eifler K, Sigurethsson JO, et al. Uncovering SUMOylation dynamics during cell-cycle progression reveals FoxM1 as a key mitotic SUMO target protein. Mol Cell. 2014;53(6):1053–1066. doi:10.1016/j.molcel.2014.02.001
33. Ullmann R, Chien CD, Avantaggiati ML, Muller S. An acetylation switch regulates SUMO-dependent protein interaction networks. Mol Cell. 2012;46(6):759–770. doi:10.1016/j.molcel.2012.04.006
34. Zubiete-Franco I, Fernandez-Tussy P, Barbier-Torres L, et al. Deregulated neddylation in liver fibrosis. Hepatology. 2017;65(2):694–709. doi:10.1002/hep.28933
35. Delgado TC, Barbier-Torres L, Zubiete-Franco I, et al. Neddylation, a novel paradigm in liver cancer. Transl Gastroenterol Hepatol. 2018;3:37. doi:10.21037/tgh.2018.06.05
36. Zhang W, Sidhu SS. Development of inhibitors in the ubiquitination cascade. FEBS Lett. 2014;588(2):356–367. doi:10.1016/j.febslet.2013
37. Zhou Y, Ji C, Cao M, et al. Inhibitors targeting the SUMOylation pathway: a patent review 2012-2015 (Review). Int J Mol Med. 2018;41(1):3–12. doi:10.3892/ijmm.2017.3231
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.