")
Back to Journals » Clinical Ophthalmology » Volume 12
Post-marketing surveillance study of the safety of dexamethasone intravitreal implant in patients with retinal vein occlusion or noninfectious posterior segment uveitis
Authors Tufail A, Lightman S, Kamal A , Pleyer U , Gajate Paniagua NM , Dot C , Li XY, Jiao J , Lou J , Hashad Y
Received 24 July 2018
Accepted for publication 2 November 2018
Published 6 December 2018 Volume 2018:12 Pages 2519—2534
DOI https://doi.org/10.2147/OPTH.S181256
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Scott Fraser
Adnan Tufail,1 Sue Lightman,1 Ahmed Kamal,2 Uwe Pleyer,3 Nuria María Gajate Paniagua,4 Corrine Dot,5,6 Xiao-Yan Li,7 Jenny Jiao,7 Jean Lou,7 Yehia Hashad7
For the CONSTANCE Study Group
1Moorfields Eye Hospital, London, UK; 2Aintree University Hospital, Liverpool, UK; 3University Eye Hospital, Uveitis Center, Charité – Universitätsmedizin Berlin, Corporate Member of Freie Universität Berlin, Humboldt University Berlin, and Berlin Institute of Health, Berlin, Germany; 4University Hospital of Burgos, Burgos, Spain; 5Department of Ophthalmology, Desgenettes Military Hospital, Lyon, France; 6French Military Health Service Academy, Val de Grâce, Paris, France; 7Allergan Plc, Irvine, CA, USA
Purpose: To evaluate the long-term safety of dexamethasone intravitreal implant (DEX) in patients treated for macular edema associated with retinal vein occlusion (RVO) or noninfectious posterior segment uveitis (NIPSU) in clinical practice.
Patients and methods: Multicenter (102 sites in France, Germany, Spain, UK), prospective, observational, post-authorization safety study in adult patients treated with DEX. Data collected up to 2 years after enrollment included serious adverse events (SAEs) and adverse events of special interest (AESIs; adverse drug reactions that are considered important risks associated with DEX and listed in the European Union Ozurdex Risk Management Plan).
Results: Overall, 803 patients (652 RVO, 151 NIPSU) received on-study DEX treatment, and 73.1% completed 24 months of follow-up; 72.6% were DEX-naïve. Median number of on-study injections per treated eye was 2 (range, 1–7); median reinjection interval was 27.1 weeks. Nonocular SAEs affected 9.5% of patients; none were considered DEX-related. Ocular SAEs (most common: cataract progression) occurred in 3.2% of treated eyes. SAEs were similar in eyes stratified by previous DEX use and number of on-study DEX injections (≤2 or >2), in both RVO and NIPSU. The most common AESIs were cataract formation and progression (20.0% and 19.2% of treated phakic eyes, n=551), increased intraocular pressure (19.0% of treated eyes), and vitreous hemorrhage (3.3% of treated eyes). Cataract progression was more frequent in baseline phakic eyes that were previously treated with DEX or received >2 on-study DEX injections.
Conclusion: The long-term safety profile of DEX was acceptable. No new safety concerns were identified.
Keywords: corticosteroid, dexamethasone, drug delivery device, retinal vein occlusion, safety profile, uveitis
Introduction
Macular edema associated with retinal vein occlusion (RVO) and noninfectious posterior segment uveitis (NIPSU) are prevalent vision-threatening conditions.1,2 Because inflammation is involved in the pathogenesis of macular edema after RVO3 and NIPSU is an intraocular inflammatory disease per se, corticosteroids such as dexamethasone are a rational approach to treatment. Dexamethasone intravitreal implant (DEX; Ozurdex®, Allergan plc, Dublin, Ireland) is a biodegradable implant containing 0.7 mg dexamethasone; the implant is administered by intravitreal injection using a single-use applicator.4 DEX provides sustained release of dexamethasone into the vitreous over several months.5 Studies have shown that aqueous humor levels of pro-permeability factors6 and proinflammatory cytokines7 are reduced after DEX treatment in patients with RVO.
DEX received marketing authorization from the European Medicines Agency for treatment of macular edema following branch or central RVO in 2010 and for treatment of inflammation of the posterior segment of the eye presenting as noninfectious uveitis in 2011. DEX demonstrated an acceptable risk–benefit profile in its registration studies for these indications. In the GENEVA study in patients with RVO-associated macular edema, significant improvements in central retinal thickness and best-corrected visual acuity were observed for 90 days and 6 months, respectively, after a single implant treatment.8 In the HURON study in patients with NIPSU, a single DEX treatment provided improvement in intraocular inflammation and best-corrected visual acuity that persisted for 6 months.9 The most common adverse events associated with DEX vs sham procedure in the pooled 6-month results from these studies were increases in intraocular pressure (IOP) (25% vs 2% of patients), conjunctival hemorrhage (22% vs 16% of patients), and eye pain (8% vs 5% of patients).10 Additionally, in the GENEVA study, the incidence of cataract in baseline phakic patients was higher after 1 year in patients who received repeated DEX treatment (29.8%) than in patients initially treated with sham who received DEX at month 6 (10.5%).11
Drug exposure and duration of follow-up in these registration studies were limited. Patients in the GENEVA study received one or two implants at 6-month intervals and were followed for 1 year,11 whereas in the HURON study, patients received one implant and were followed for 6 months.9 In real-world practice, patients may be treated with multiple implants over an extended period. The mean number of implants received was 2.6 and the mean reinjection interval was 6.6 months in a prospective, 2-year study (LOUVRE) of real-world use of DEX in 375 patients with RVO-associated macular edema in France.12 In a retrospective study of the IOP-related safety of DEX use in clinical practice in France (SAFODEX), the mean number of implants administered was 2.4 in 142 eyes with RVO and 3.1 in 58 eyes with NIPSU over a mean follow-up of 17 and 21.5 months, respectively.13 A retrospective study of real-world use of DEX in 289 patients with RVO-associated macular edema in the United States who received at least two implants in the study eye (SHASTA) reported a mean of 3.2 implants administered and a mean reinjection interval of 5.6 months.14 In another retrospective study of use of DEX in clinical practice conducted in Canada (CHROME), for 23 patients with uveitis, the mean number of implants received in affected eyes was 1.7, with a mean interval of 4.7 months between the first and second injections and 3.4 months between the second and third injections.15
There is a need to better define the long-term safety profile and patterns of use of DEX in patients with RVO-related macular edema and NIPSU, including patients who receive multiple repeat treatments, in routine clinical practice. The present post-authorization safety study was undertaken to fulfill a European Union (EU) regulatory commitment. The primary objective of this study was to evaluate the long-term safety of DEX, including identified and potential risks as listed in its Risk Management Plan, in patients with macular edema after RVO or NIPSU who are treated with DEX in routine clinical practice. The secondary objective of this study was to describe treatment patterns for patients receiving DEX for treatment of RVO-associated macular edema or NIPSU in clinical practice.
Methods
Study design
This was a multicenter, 2-year, prospective, observational, post-authorization safety study conducted from March 2012 to March 2016 at 102 sites in France, Germany, Spain, and the UK. The study was carried out in accordance with the tenets of the Declaration of Helsinki. A central institutional review board or ethics committee approved the study protocol for each site. The study protocol was approved by the National Research Ethics Service Committee Yorkshire & the Humber – Bradford (a central ethics committee) in the UK, the Ethik-Kommission der Universitaet Ulm (a central ethics committee) in Germany, the Comitè Etic Investigacio Clinica (a central ethics committee) in Spain, and the Comité Consultatif sur le Traitement de l’information Recherche/Santé, the Commission Nationale de l’Informatique et des Libertés, and the Conseil National de l’Ordre des Médecins in France. All participating patients provided written informed consent before the initiation of data collection. The study is registered at ClinicalTrials.gov with the identifier NCT01539577.
A qualification survey was used to select physicians and sites for the study. Ophthalmologists routinely involved in the care and treatment of patients with macular edema and NIPSU, and sites with projected availability of eligible patients and availability of site personnel to complete the case report forms, were invited to participate. To the extent possible, the sites selected were representative of sites in the participating countries with respect to size, type, and training.
Patient selection
Patients eligible for the study were adults (age ≥18 years) with a diagnosis of NIPSU or macular edema following branch or central RVO who received DEX treatment. The diagnosis was based on the clinical judgment of the investigator. Patients who had received DEX treatment in a completed clinical trial were eligible, but patients currently participating in any clinical study were excluded. All patients presenting during the enrollment period were assessed for eligibility, and eligible patients were consecutively proposed for enrollment. Eligible patients were enrolled into the study at the time of presentation for a routine clinic visit.
Treatment
All treatment decisions and follow-up were at the physician’s and patient’s discretion. The study protocol did not recommend or mandate any specific treatments or injection frequency, and the study sponsor did not provide study medication.
Outcome measures
The primary outcome measures were serious adverse events (SAEs) and adverse events of special interest (AESIs, determined based on important identified or potential risks of DEX listed in the Ozurdex EU Risk Management Plan). An SAE was defined as an untoward medical occurrence that was life-threatening; or that resulted in death, persistent or significant disability/incapacity, or permanent impairment or damage to a nontrivial body function or structure; or that resulted in a condition necessitating medical or surgical intervention to prevent damage to a nontrivial body structure; or that required hospitalization or prolongation of existing hospitalization; or that was a diagnosis of cancer or a congenital anomaly/birth defect; or that was an important medical event that may not fall into one of the above categories but may jeopardize the patient and may require intervention to prevent one of the outcomes above; or that was associated with the suspected transmission of an infectious agent by the product. The AESIs were increased IOP, glaucoma, and ocular hypertension; cataract formation and progression; retinal tear or detachment; vitreous hemorrhage or detachment; endophthalmitis (infectious or noninfectious); retinitis secondary to reactivation of latent viral or other ophthalmic infections; significant vitreous leak or hypotony; systemic corticosteroid effects; mechanical failure of device; and implant misplacement and dislocation. Implant misplacement refers to injection of the implant into a tissue other than the vitreous, and implant dislocation refers to implant migration from where it was injected (primarily to the anterior chamber) after injection. AESIs were reported regardless of their seriousness or severity. As is common in clinical trials conducted for regulatory agency review of drug safety, there were no predetermined criteria for adverse events. Adverse events were determined based on that clinical judgment of the investigator, because individual patient circumstances influence whether a finding constitutes an untoward medical occurrence (ie, adverse event).
Secondary outcome measures included the number of DEX injections per patient and per treated eye, the number of DEX injections per person-year on study, and the average interval (weeks) between DEX injections.
Study visits and data collection
No clinic visits were mandated by the study protocol. All assessments were intended to be performed at the time of a routine clinic visit or by referencing the patient’s medical record. Structured questionnaires were administered by the physician to elicit data of interest.
SAEs and AESIs, including those directly observed as well as those reported by patients to the study physician or site staff, were monitored throughout the study period. At each RVO- or NIPSU-related follow-up visit up to 2 years after enrollment, the SAEs and AESIs occurring since the previous clinic visit, and their duration, severity, and suspected relationship to treatment, were recorded on the case report form. Patients not seen within 6 months of their previous visit were contacted by telephone for these data. If a patient had been seen by a nonstudy physician, the physician was contacted to obtain the necessary information concerning the adverse event.
Demographic information and medical and ophthalmic histories were collected at baseline (the visit with the first DEX injection after study enrollment). Additional data collected at baseline and at 6-month (±1 month) intervals during follow-up included RVO-related macular edema and NIPSU characteristics on ophthalmic examination, visual acuity or best-corrected visual acuity measured by the method used in usual patient care, prior and concomitant treatments for RVO or NIPSU (including DEX and any concomitant treatments), and concurrent medications/procedures and diseases. For each of these parameters, cumulative data were collected for the period since the previous study visit with data collection.
Analysis
The analysis set included all enrolled patients who provided informed consent. All patient-level analyses of safety were performed using the ATP (according to protocol) population, defined as all patients in the analysis set who received at least one DEX injection during the study. Treated eyes were defined as eyes that received at least one DEX injection during the study. Patients could contribute one or two eyes to the analyses of treated eyes.
Data were compiled and summarized using descriptive statistics. Event rates per person-year were calculated based on the sum of time from the first DEX injection to the event onset for patients with the event and the follow-up time for patients without the event. SAEs and AESIs were coded using the Medical Dictionary for Regulatory Activities nomenclature and are presented by primary system organ class (SOC) and preferred term. Analyses of SAEs and AESIs were preplanned to include stratification by indication (RVO vs NIPSU), history of DEX use (DEX-naïve vs previously treated with DEX), and number of DEX injections during study (≤2 vs >2). This threshold was chosen to fulfill a need for data on DEX safety in patients who receive more than two injections, as no patients received more than two injections in the global registration studies of DEX for treatment of RVO-associated macular edema and NIPSU. Observed data were used in the analyses. There was no imputation for missing values, except in estimation of dates for treatments and start/stop dates of adverse events, when partial data were available.
The planned enrollment was at least 650 patients (500 with RVO and 150 with NIPSU) across participating countries, with at least 40% of patients receiving more than 2 DEX injections on study (during the study) in the same eye. The sample size was selected to permit sufficient precision to detect AESIs that had the lowest observed incidence (ie, ~0.1%–0.2%) in the registration studies and claims analyses.
Results
A total of 853 eligible patients provided informed consent and were enrolled in the study. Among these patients, 803 (652 with RVO and 151 with NIPSU) received on-study DEX treatment and were included in the ATP population for analysis. Figure 1 shows patient disposition stratified by indication (RVO vs NIPSU), history of DEX use (DEX-naïve vs previous DEX treatment), and number of on-study DEX injections (≤2 vs >2). The overall 2-year study completion rate for patients in the ATP population was 73.1% and was similar for patients with RVO and those with NIPSU (Figure 1). The most common reason for early study exit was lost to follow-up (Figure 1).
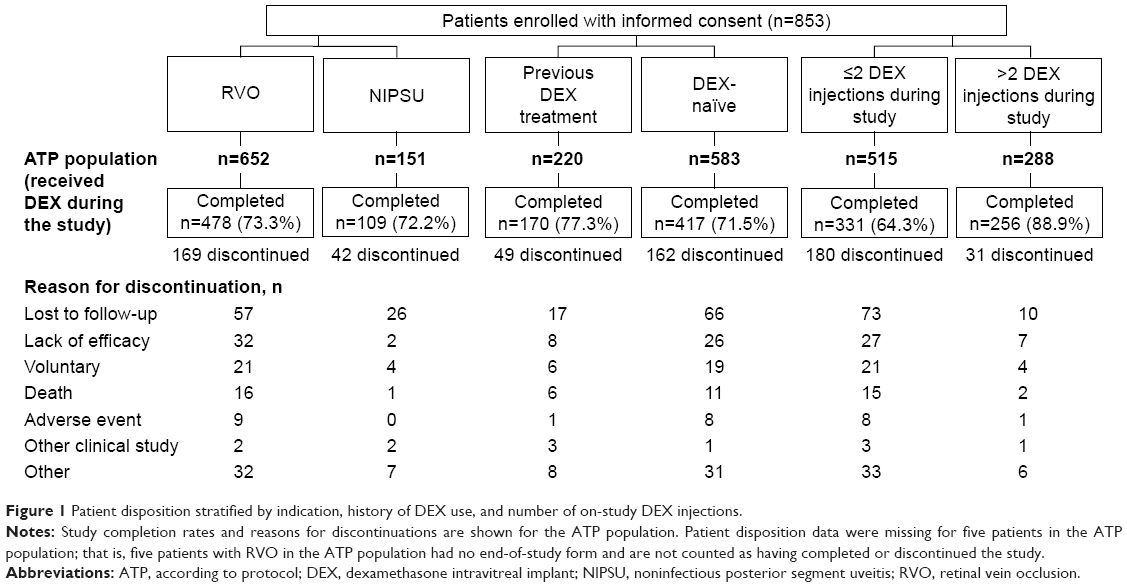 | Figure 1 Patient disposition stratified by indication, history of DEX use, and number of on-study DEX injections. |
Baseline characteristics of patients and treated eyes
Baseline patient characteristics in the ATP population are listed in Table 1. The patient population was predominantly white with similar numbers of men and women. The mean age of patients was 68 years. The majority (62.1%) were enrolled in the UK, 81.2% were diagnosed with RVO, and 72.6% had not used DEX prior to the study. Baseline characteristics were generally similar in patient stratifications by indication, history of DEX use, and number of DEX injections during the study. However, patients with NIPSU were younger (mean age 54.9 years vs 71.0 years) and a larger proportion were female (62.9% vs 45.4%) compared with RVO patients (Table 1).
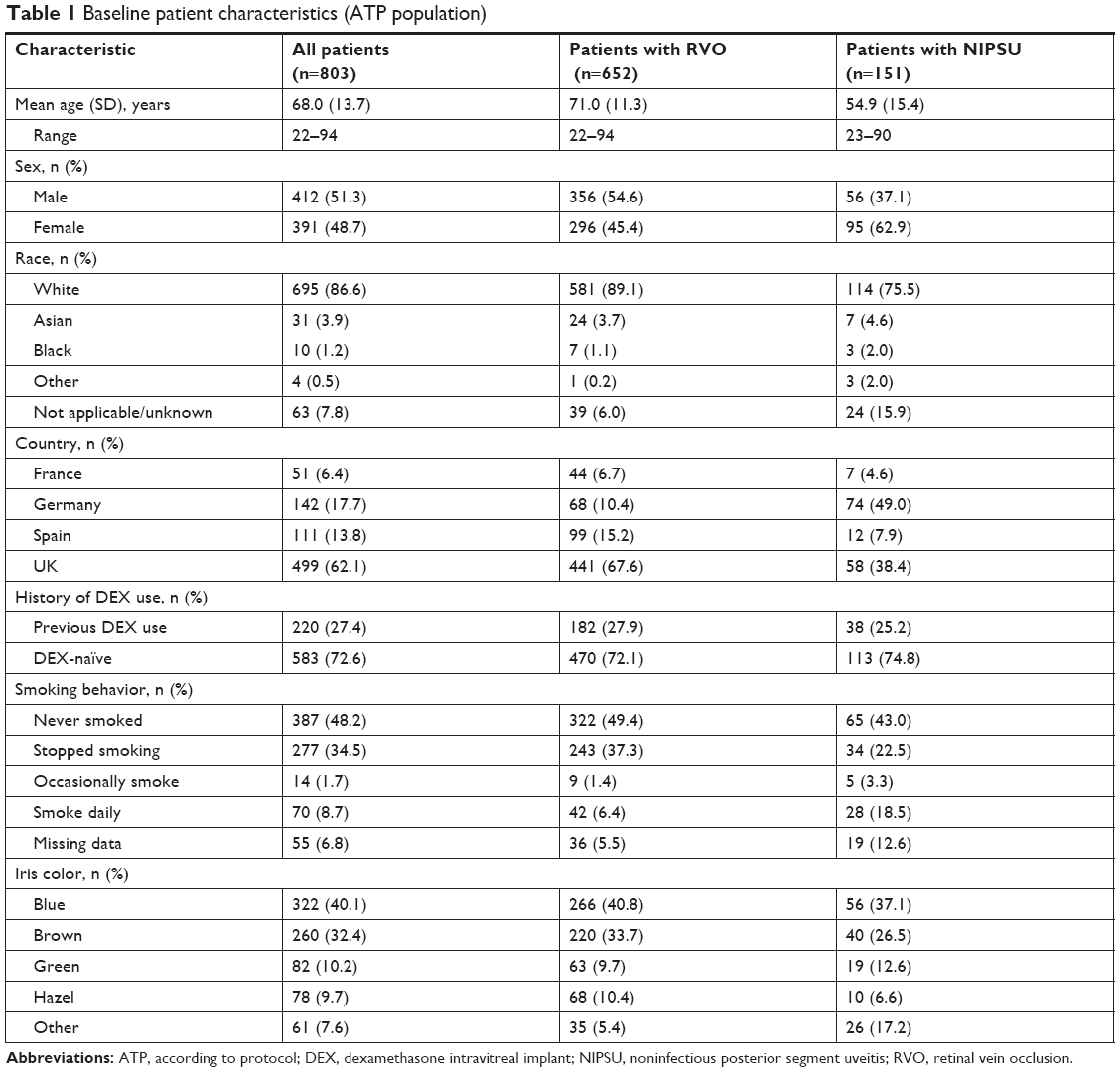 | Table 1 Baseline patient characteristics (ATP population) |
Table 2 shows baseline characteristics of treated eyes. Most of these eyes were treatment-naïve at baseline. In previously treated eyes, the most common previous treatments were DEX injections (24.5% of eyes), anti-vascular endothelial growth factor injections (6.5% of eyes), and laser photocoagulation (5.5% of eyes). Baseline lens status was phakic in 63.2% and pseudophakic in 27.6% of treated eyes. Among treated eyes with RVO, 57.0% were diagnosed with branch RVO, 38.1% were diagnosed with central RVO, and 4.9% had missing data on the type of RVO. Baseline characteristics of treated eyes were generally similar across stratifications of treated eyes by indication, history of DEX use, and number of DEX injections during the study, except that treated eyes diagnosed with NIPSU that were previously treated with DEX, or that received >2 DEX injections during the study, were more likely to be pseudophakic.
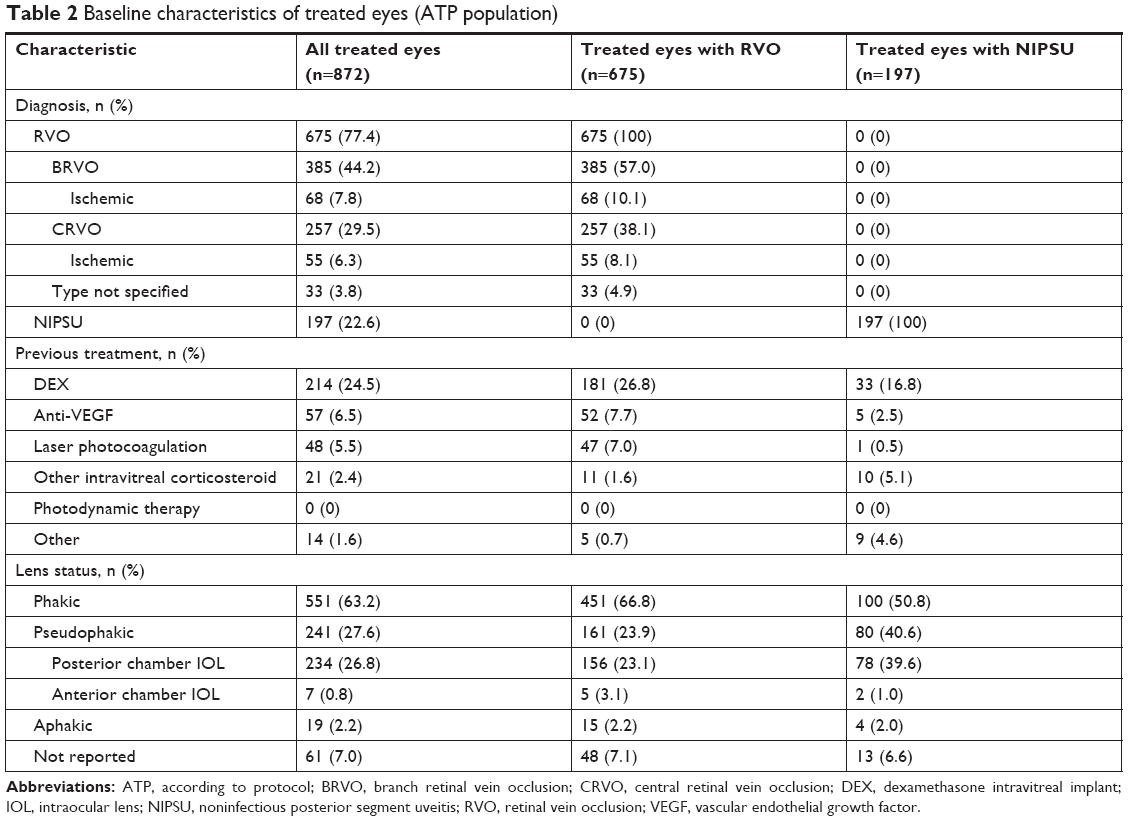 | Table 2 Baseline characteristics of treated eyes (ATP population) |
The most common conditions in patients’ medical histories at baseline were hypertension, hyperlipidemia, and diabetes (Table 3). The distribution of medical conditions was generally similar in patient stratifications by indication, history of DEX use, and number of DEX injections during the study, with the exception that patients with RVO were more likely than patients with NIPSU to have a history of hypertension (61.7% vs 27.2%) and hyperlipidemia (39.9% vs 17.2%) because they are common comorbidities of RVO.16 The distribution of ocular conditions other than RVO and NIPSU in patient histories was generally similar in patient stratifications by indication, history of DEX use, and number of DEX injections during the study. Cataract, glaucoma, and ocular hypertension were the most common conditions in patients’ ocular histories (Table 3).
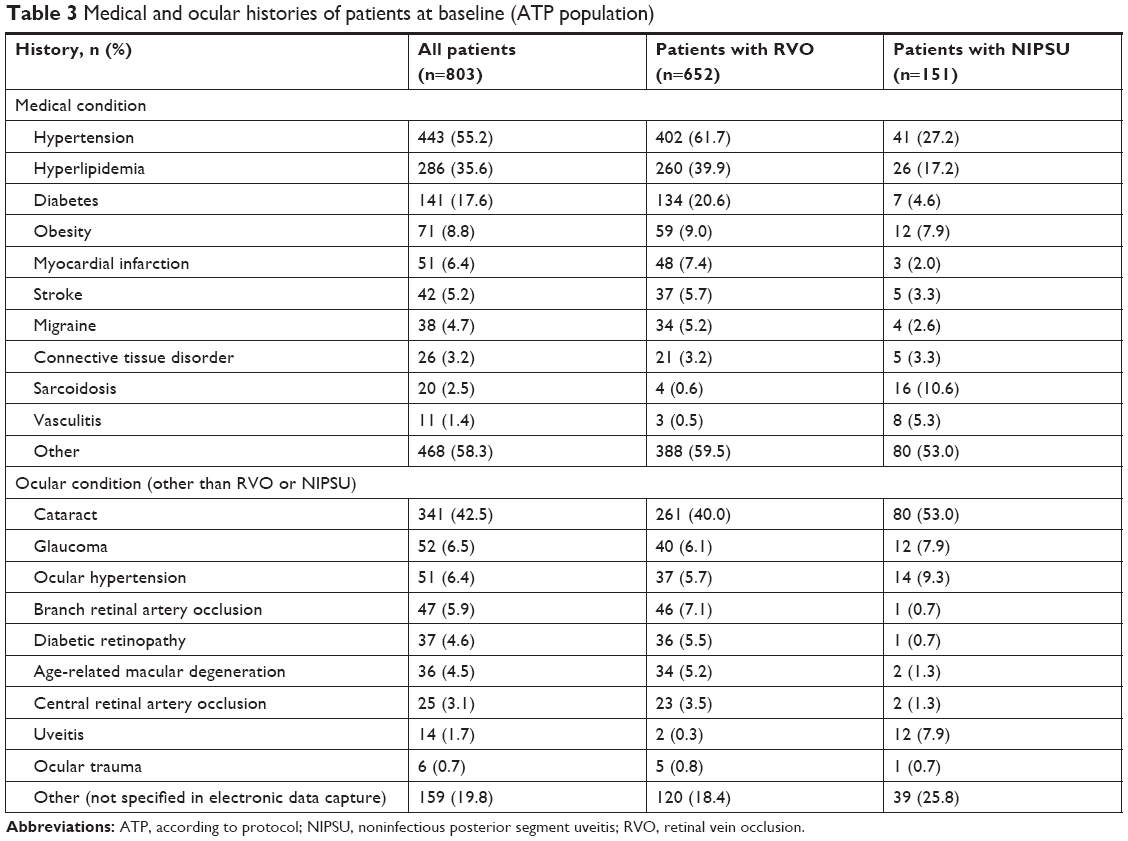 | Table 3 Medical and ocular histories of patients at baseline (ATP population) |
Serious adverse events
The overall incidence of any nonocular SAE in the ATP population was 9.5% (76/803), and the overall incidence rate of nonocular SAEs was 0.066 per person-year. Nonocular SAEs were most frequently classified in the primary SOC of infections and infestations (pneumonia, lower respiratory tract infection, and urinary tract infection were the most common nonocular SAEs in this class). All nonocular SAEs were considered to be unrelated to DEX. The incidence of nonocular SAEs classified by primary SOC in patient stratifications by indication, history of DEX use, and number of DEX injections during the study is shown in Table 4. The distribution of nonocular SAEs was similar in patients with or without previous DEX use and in those who received ≤2 or >2 DEX injections during the study, regardless of indication (RVO or NIPSU).
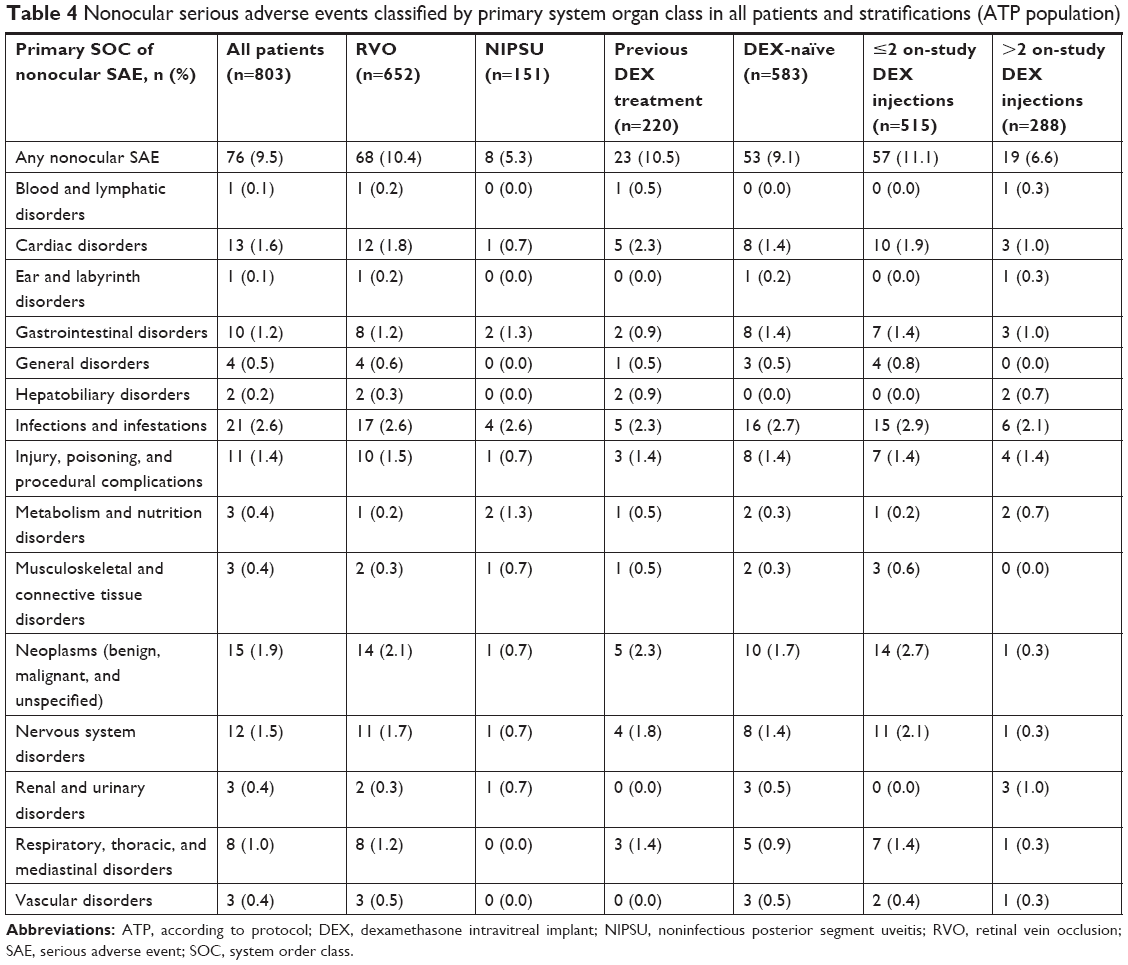 | Table 4 Nonocular serious adverse events classified by primary system organ class in all patients and stratifications (ATP population) |
Ocular SAEs were reported in 3.2% (28/872) of treated eyes, and the overall incidence rate of ocular SAEs was 0.023 per person-year. The most common ocular SAE in treated eyes was cataract progression (Table 5). There were two SAE reports of increased IOP and one SAE of glaucoma in treated eyes. All three SAEs were considered potentially related to DEX treatment and resolved after treatment without sequelae. In one patient, IOP had increased to 40 mm Hg at 2.5 months after DEX treatment and 3 weeks after grid laser treatment for branch RVO-associated macular edema. The patient began topical treatment with timolol and latanoprost in the affected eye. Three months later, the topical IOP-lowering medications were discontinued and IOP was in the normal range. In a second patient, an IOP of 50 mm Hg was measured 85 days after administration of DEX for treatment of uveitis. The patient initiated treatment with oral and topical IOP-lowering medication, and IOP in the affected eye was reduced to 20 mm Hg. In a third patient, steroid-induced glaucoma was reported at 39 days after DEX treatment. A peak IOP of 57 mm Hg was measured 18 days later. The patient had a history of primary open-angle glaucoma, and there was no evidence for or against optic nerve or visual field damage. The patient was hospitalized, and the increase in IOP resolved after cyclophotocoagulation.
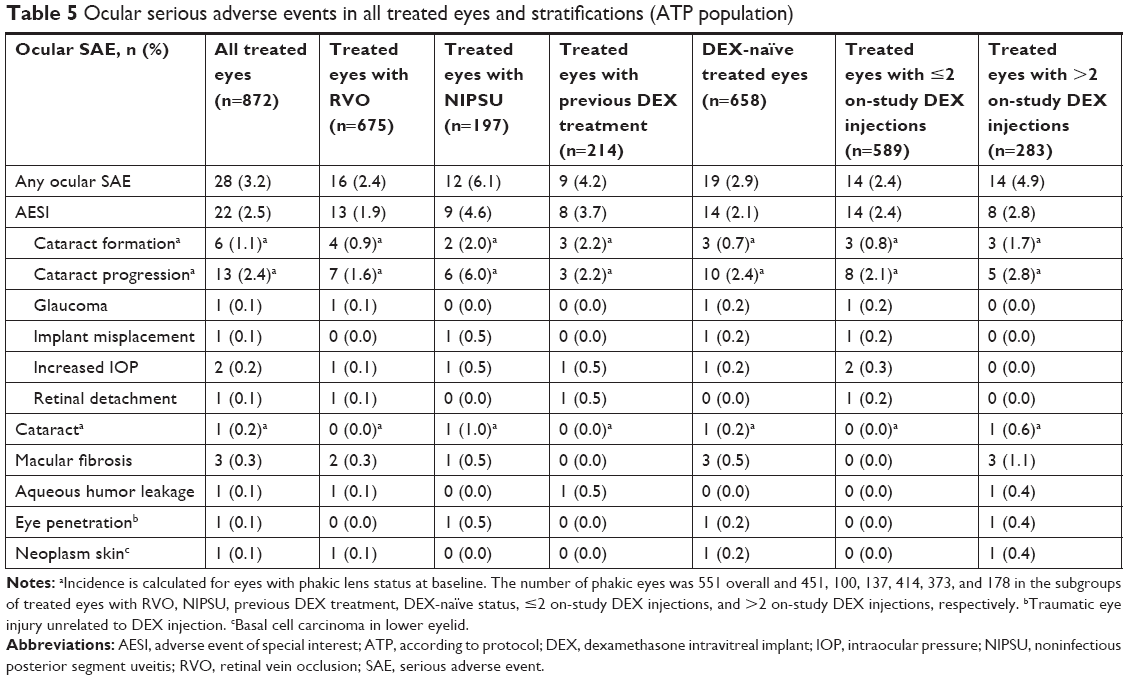 | Table 5 Ocular serious adverse events in all treated eyes and stratifications (ATP population) |
Table 5 shows the incidence of ocular SAEs in all treated eyes and in treated eye stratifications. The distribution of ocular SAEs was similar in eyes with RVO and those with NIPSU, in DEX-naïve eyes and those previously treated with DEX, and in eyes treated with ≤2 and >2 on-study DEX injections (Table 5). Consistent results were obtained in an analysis of the incidence rates of ocular SAEs per patient-year in study (Table S1).
Adverse events of special interest
The most common AESIs reported were cataract formation (20.0% of treated baseline phakic eyes), cataract progression (19.2% of treated baseline phakic eyes), increased IOP (19.0% of treated eyes), vitreous hemorrhage (3.3% of treated eyes), ocular hypertension (3.2% of treated eyes), and glaucoma (1.3% of treated eyes). All other AESIs occurred in ≤0.5% of treated eyes. There were no reports of mechanical failure of device, retinitis secondary to reactivation of latent viral or other ophthalmic infections, retinal tear, significant vitreous leak, or systemic corticosteroid effects.
Table 6 shows the incidence of AESIs overall and in treated eyes stratified by indication, history of DEX use, and number of on-study DEX injections. The distribution of AESIs was similar in treated eyes with RVO and those with NIPSU. Among treated eyes that were phakic at baseline, those previously treated with DEX had a higher incidence of cataract progression (32.8%) compared with those that were DEX-naïve (14.7%). The distribution of all other AESIs was similar in treated eyes stratified by history of DEX use. Eyes that received >2 DEX injections during the study had a higher incidence of cataract progression compared with eyes that received one or two on-study DEX injections (32.0% vs 13.1% in baseline phakic eyes). Vitreous hemorrhage was reported in 2.0% of eyes that received one or two on-study DEX injections and 6.0% of eyes that received >2 on-study DEX injections. Other AESIs were similar in eyes stratified by number of on-study DEX injections. Consistent results were obtained in an analysis of the incidence rates of AESIs per patient-year in study (Table S2).
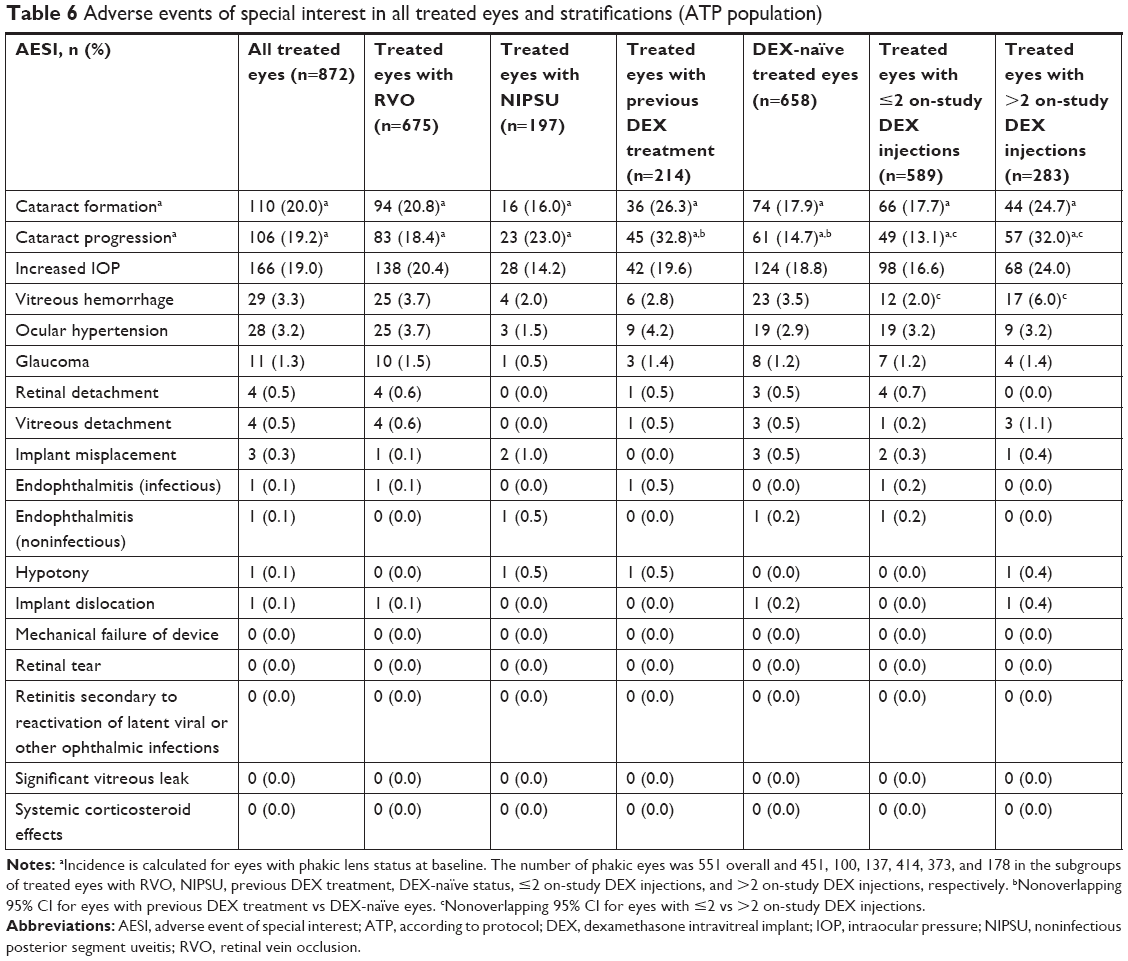 | Table 6 Adverse events of special interest in all treated eyes and stratifications (ATP population) |
The occurrence of AESIs was generally similar in France, Germany, Spain, and the UK, with the exception that the incidence of increased IOP was higher among patients in the UK (23.7%) compared with patients in Spain (6.8%), and the incidence of cataract formation was higher among patients in the UK (17.0%) compared with patients in Spain (5.9%).
DEX exposure
The mean (±SD) number of on-study DEX injections per patient was 2.3±1.4 (median, 2.0; range, 1–10); the mean number of on-study DEX injections per treated eye was 2.2±1.3 (median, 2.0; range, 1–7). The number of injections per person-year on study was 1.5. The median time between sequential injections in retreated eyes was 27.1 (range, 7.1–111.6) weeks, with a median interval between injections of 26.1 weeks for eyes with RVO and 31.1 weeks for eyes with NIPSU.
The number of on-study DEX injections per patient and per treated eye was comparable for patients with NIPSU and patients with RVO, as well as for patients previously treated with DEX and DEX-naïve patients. The number of injections per person-year on study was 1.4 for patients with RVO and 1.7 for patients with NIPSU. Stratification by country showed that the number of injections per person-year on study was 1.9 in France, 1.7 in Germany, 1.5 in Spain, and 1.4 in the UK. The mean (±SD) number of on-study DEX injections per treated eye was 2.8±1.4 in France, 2.1±1.4 in Germany, 2.4±1.5 in Spain, and 2.0±1.1 in the UK.
Ocular procedures
During the 2-year study, 50.6% of eyes treated with DEX underwent ocular procedures other than DEX injections, most commonly retinal laser photocoagulation in eyes with RVO, and procedures related to cataract surgery and intraocular lens implantation (Table 7). With respect to procedures for IOP control in treated eyes, three (0.3%) eyes underwent iridotomy, two (0.2%) eyes underwent filtering surgery, and one (0.1%) eye underwent cryotherapy. One of the filtering surgeries was performed in a patient with a history of glaucoma who had increased IOP, which could not be controlled with topical and oral medication, in both eyes 10 days after initiating oral prednisolone therapy (30 mg twice daily) for asthma. The investigator reported the increased IOP as an SAE in the untreated eye. The SAE occurred 4 months after the last DEX treatment and was determined by the investigator to be unrelated to DEX treatment. The patient underwent trabeculectomy, considered to be related to the oral corticosteroid, in both eyes. No laser trabeculoplasty procedures were performed during the study in treated eyes.
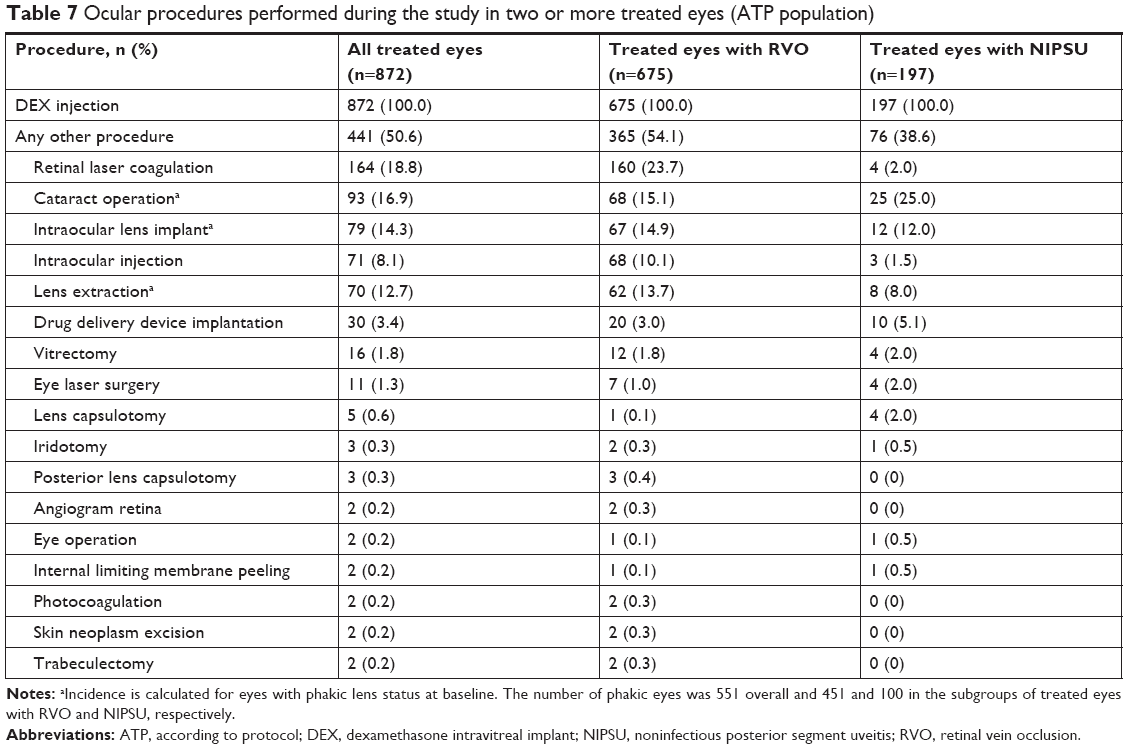 | Table 7 Ocular procedures performed during the study in two or more treated eyes (ATP population) |
Discussion
This study was designed to elucidate the long-term safety profile of DEX when used in the treatment of patients with RVO-associated macular edema and NIPSU in clinical practice in France, Germany, Spain, and the UK. The long-term safety profile of DEX was acceptable, and no new safety concerns were identified. The safety profile was generally similar in patients and eyes stratified by indication, previous treatment with DEX, and number of DEX injections received during the study, with the exceptions that previous DEX treatment and a larger number of on-study DEX injections (>2) were associated with a higher incidence of cataract progression, and a larger number of on-study DEX injections was also associated with a higher incidence of vitreous hemorrhage.
The incidence of ocular SAEs in treated eyes was low (3.2%). The majority of ocular SAEs in treated eyes were cataract formation or progression. No nonocular SAEs were reported that were considered to be related to DEX, and no differences were evident between patients with RVO and patients with uveitis in the occurrence of ocular or nonocular SAEs.
DEX was initially approved for treatment of RVO-associated macular edema and NIPSU. More recently, DEX was approved in the United States for treatment of diabetic macular edema (DME) and in the EU for the treatment of adult patients with visual impairment due to DME who are pseudophakic or who are considered insufficiently responsive to, or unsuitable for, non-corticosteroid therapy. In the DEX registration study for DME (the MEAD study), patients were treated for up to 3 years with an interval between treatments of at least 6 months. The long-term safety of DEX in patients with DME demonstrated in the MEAD study17 and in the real-life RELDEX study18 was consistent with the long-term safety of DEX demonstrated here in clinical use in patients with RVO-associated macular edema and NIPSU.
Cataract formation and progression and increased IOP were the most commonly reported AESIs in all strata evaluated. These findings are consistent with results from previous studies of long-term DEX use for treatment of RVO-associated macular edema12–14 and cystoid macular edema associated with quiescent NIPSU19 in clinical practice. The increased incidence of cataract progression in patients who received more than two implants was expected. Cataract progression is associated with corticosteroid use but it takes time to develop; a significant association between DEX treatment and cataract was seen in the GENEVA study only in RVO patients treated with two implants, at 12 months after the initial implant.11 Similarly, there was no significant association between cataract AEs and DEX treatment in the 6-month HURON study in patients with NIPSU,9 but cataract progression after multiple implants has been reported during long-term follow-up in patients treated with DEX for NIPSU.20
In this study, increased IOP was reported as an AESI in 20.4% (95% CI, 17.5%–23.7%) of patients with RVO and 14.2% (95% CI, 9.7%–19.9%) of patients with NIPSU. A similar percentage of patients (25.2%) had AE reports of increased IOP in the pooled 6-month registration studies of DEX in patients with RVO and NIPSU.10 Previous real-life studies of DEX use have reported findings consistent with these AE data. In a prospective study in France, increases in IOP of >5 mm Hg were reported in 25.9% (89/343) of eyes with RVO that were treated with DEX and followed for up to 2 years.12 A retrospective chart review study of eyes with RVO that received at least two injections of DEX in the United States reported increases in IOP to ≥25 mm Hg in 33.7% (97/288) of eyes and ≥10 mm Hg increases in IOP from baseline in 32.6% (91/279) of eyes.14 More recently, in a retrospective chart review study (SAFODEX) of patients who were treated with DEX in France, increases in IOP to ≥25 mm Hg were reported in 20.2% (85/421) of eyes and ≥10 mm Hg increases in IOP from baseline were reported in 27.1% (114/421) of eyes followed for an average of 16.8 months after receiving DEX treatment for RVO-associated macular edema, NIPSU, or other indications.13 In the SAFODEX study, ocular hypertension defined as IOP ≥25 mm Hg or a ≥10 mm Hg increase from baseline was reported in 36% of the 142 eyes treated for RVO and 38% of the 58 eyes treated for NIPSU.13 The previous real-life studies, as well as the DEX registration studies, reported that increases in IOP after DEX treatment are usually managed with topical IOP-lowering medications. The IOP increases are usually transient; after stopping DEX treatment, most patients have controlled IOP without a need for topical IOP-lowering medications.21,22 In accordance with previous studies, in the present study, laser or surgical procedures for IOP control were used in <1% of treated eyes.
The frequency of vitreous hemorrhage AESIs was higher in patients who received a larger number of on-study DEX injections (6.0% [95% CI, 3.5%–9.4%] in patients with >2 DEX injections vs 2.0% [95% CI, 1.1%–3.5%] in patients with ≤2 DEX injections). This result was not unexpected, because vitreous hemorrhage can happen with any intravitreal injection. There was also a trend for more increased IOP AESIs in patients who received a larger number of on-study DEX injections. Increased IOP AESIs were reported in 24.0% (95% CI, 19.2%–29.4%) of patients who received >2 DEX injections vs 16.6% (95% CI, 13.7%–19.9%) of patients who received ≤2 injections. However, the overlap in the 95% CIs of these incidence rates indicates a lack of statistical significance. These results are consistent with results of previous studies (MEAD, SAFODEX), which suggested that for patients who experience increased IOP during DEX treatment, the initial IOP increase most frequently occurs after the first or second DEX injection, with reduced risk of an initial IOP increase after subsequent injections.13,23
Rhegmatogenous retinal detachment can occur after any intravitreal injection. A very low incidence of retinal detachment has been reported after intravitreal anti-vascular endothelial growth factor injections (0.013%, 5 occurrences associated with a total of 35,942 injections).24 In the present study, 872 treated eyes received a mean of 2.2 DEX injections per eye, and there were 4 AESI reports of retinal detachment.
In clinical practice, some patients may need DEX retreatment at shorter intervals than the 6-month interval used in the registration trials of DEX for RVO-associated macular edema11 and the 6-month minimum interval used in the registration trials of DEX for DME.17 Accordingly, the interval between sequential DEX injections in retreated eyes in the present study varied widely. The median interval was 27.1 weeks (6.2 months), and the mean interval was 32.4 weeks (7.4 months). In comparison, previous large studies of real-life use of DEX for treatment of RVO-associated macular edema have reported a median reinjection interval of 4.9 months (mean, 5.6 months) (SHASTA study)14 and a mean reinjection interval of 6.6 months (LOUVRE study).12 In the smaller SOLO study, early retreatment before 6 months was indicated in 54% (55/102) of eyes, and the mean reinjection interval in eyes that received early retreatment was ~18 weeks.25
Because inclusion of patients previously treated with DEX could select for patients who have used DEX previously without adverse events, the analysis was stratified to account for the effects of previous DEX treatment. In the ATP population used for analysis, 75.5% of treated eyes had not been treated with DEX prior to the study. With the exception of cataract progression, which was more frequent in eyes with a history of DEX use, no differences were seen in the DEX safety profile between DEX-naïve eyes and eyes previously treated with DEX, suggesting that a patient selection bias did not lead to the favorable safety profile that was observed.
This study had a long duration, with safety data collected through 2 years of follow-up. Other strengths of the study include the inclusion of patients with comorbidities and concomitant medication use that could potentially affect the safety of DEX use in real-world clinical practice, and the stratification of the analyses by characteristics that could potentially have an effect on the safety profile of DEX including history of DEX use, number of injections received, and indication for treatment. A limitation of the study was that AEs that were not among the AESIs were not collected, unless they were serious. Also, data on use of topical glaucoma medications were not reliably collected. In addition, the study completion rate for the ATP population was only 73.1%, with 10.3% of the ATP population lost to follow-up despite repeated attempts to contact patients who repeatedly missed scheduled clinic visits. Low study completion rates are typical in observational studies, but the large number of early discontinuations could have affected the results if the patients who discontinued from the study differed from those who remained in the study in characteristics that affect the safety profile of DEX. Finally, the study sample size, although large, could not be expected to uncover the occurrence of very rare SAEs related to DEX. The study was not designed to evaluate the effectiveness of DEX treatment, and no conclusion can be drawn as to whether the number and frequency of DEX injections administered was sufficient for treatment success.
Conclusion
DEX demonstrated an acceptable safety profile in this 2-year safety study. The long-term safety profile of DEX used in clinical practice in patients with NIPSU or macular edema due to RVO was consistent with the known safety profile of DEX.26 Repeated treatment in the same eye was not associated with any new safety concerns.
Data sharing statement
Allergan will share de-identified patient-level data and study-level data including protocols and clinical study reports for Phase II, III, or IV trials completed after 2008 that are registered to ClinicalTrials.gov or EudraCT after the intervention has received regulatory approval in the United States and/or the European Union in a given indication and the primary manuscript from the trial has been published. To request access to the data, the researcher must sign a data use agreement and any shared data is to be used for noncommercial purposes. More information can be found on http://www.allerganclinicaltrials.com/.
Acknowledgments
This study was sponsored by Allergan plc (Dublin, Ireland) and conducted by Quintiles (Durham, NC, USA), a contract research organization. Writing and editorial assistance was provided to the authors by Kate Ivins, PhD, of Evidence Scientific Solutions (Philadelphia, PA, USA) and funded by Allergan. All authors met the ICMJE authorship criteria. Neither honoraria nor payments were made for authorship. This work was presented in part in a poster at the Annual Meeting of the European Association for Vision and Eye Research (EVER), September 27–30, 2017, Nice, France; the abstract of the poster was published in Acta Ophthalmol. 95: https://doi.org/10.1111/j.1755-3768.2017.0T083. The CONSTANCE Study Group Principal Investigators are Emilio Abecia Martinez, Alfredo Adan Civera, Isabelle Aknin, Winfried Amoaku, Francisco Javier Ascaso, Sanjiv Banerjee, Maria Francisca Bassaganyas, Xavier Benouaich, Christoph Binder, Benjamin Burton, Carlos Cava Valenciano, Shuaib Chaudhary, Jose-Javier Chavarri Garcia, Inigo Corcostegui, Catherine Creuzot-Garcher, Barbara Delas, Christian Delhay, Christoph Deuter, François Devin, Narendra Dhingra, Andrew Dick, Corinne Dot, Louise Downey, Clive Edelsten, Nicole Eter, Catherine Favard, Nuria María Gajate Paniagua, Richard Gale, Maria-Andreea Gamulescu, Carmen Garcia Borque, Faruque Ghanchi, Maged Habib, Arnd Heiligenhaus, Henar Heras Mulero, Edward Hughes, Ahmed Kamal, Niral Karia, Simon Kelly, Javeed Khan, Vineeth Kumar, Gabriele Lang, Katrin Lorenz, Andrew Lotery, Nabeel Malik, Kaveri Mandal, Roslyn Kathryn Manrique Lipa, Simon Morgan, Mohammed Musadiq, Thomas Neuhann, Yinka Osoba, Sergio Pagliarini, Maria Ascension Pardo Muñoz, Nishal Patel, Sudeshna Patra, Dominique Pawlak, Begona Pina Marin, Uwe Pleyer, Elena Rodriguez Neila, Helmut Sachs, Dirk Sandner, Mahmoud Sarhan, Claire Scemama-Timsit, Phillipe Schauer, Harald Schilling, Berthold Seitz, Walter Sekundo, Ramesh Sivaraj, James Talks, Simon Taylor, Ulrich Thelen, Adnan Tufail, Laurent Valasque, Michel Weber, Lars Wagenfeld, Benjamin Wolff.
Disclosure
Financial arrangements of the authors with companies whose products may be related to the present report are listed below, as declared by the authors. Dr Tufail is a consultant for Allergan, Bayer, Novartis, and Roche/Genentech. Dr Lightman serves on an advisory board for AbbVie. Dr Kamal has served as a paid consultant for Alcon, Allergan, Bayer, and Novartis. Dr Pleyer has served as a principal investigator or consultant for AbbVie, Alcon, Allergan, Bausch and Lomb, Bayer, Novartis, Santen, and Thea. Dr Gajate Paniagua has served as a principal investigator for Allergan and Novartis. Dr Dot is a consultant for Bayer, Allergan, and Novartis. Dr Li, Dr Jiao, Dr Lou, and Dr Hashad are full-time employees of Allergan and receive stock/stock options.
References
Whitcup SM, Robinson MR. Development of a dexamethasone intravitreal implant for the treatment of noninfectious posterior segment uveitis. Ann N Y Acad Sci. 2015;1358:1–12. | ||
Ho M, Liu DT, Lam DS, Jonas JB. Retinal vein occlusions, from basics to the latest treatment. Retina. 2016;36(3):432–448. | ||
Deobhakta A, Chang LK. Inflammation in retinal vein occlusion. Int J Inflam. 2013;2013:438412. | ||
Haller JA, Dugel P, Weinberg DV, Chou C, Whitcup SM. Evaluation of the safety and performance of an applicator for a novel intravitreal dexamethasone drug delivery system for the treatment of macular edema. Retina. 2009;29(1):46–51. | ||
Chang-Lin JE, Attar M, Acheampong AA, et al. Pharmacokinetics and pharmacodynamics of a sustained-release dexamethasone intravitreal implant. Invest Ophthalmol Vis Sci. 2011;52(1):80–86. | ||
Campochiaro PA, Hafiz G, Mir TA, et al. Pro-permeability factors after dexamethasone implant in retinal vein occlusion; the Ozurdex for Retinal Vein Occlusion (ORVO) study. Am J Ophthalmol. 2015;160(2):313–321.e19. | ||
Rezar-Dreindl S, Eibenberger K, Pollreisz A, et al. Effect of intravitreal dexamethasone implant on intra-ocular cytokines and chemokines in eyes with retinal vein occlusion. Acta Ophthalmol. 2017;95(2):e119–e127. | ||
Haller JA, Bandello F, Belfort R Jr, et al. Randomized, sham-controlled trial of dexamethasone intravitreal implant in patients with macular edema due to retinal vein occlusion. Ophthalmology. 2010;117(6):1134–1146.e3. | ||
Lowder C, Belfort R, Lightman S, et al. Dexamethasone intravitreal implant for noninfectious intermediate or posterior uveitis. Arch Ophthalmol. 2011;129(5):545–553. | ||
Allergan. Ozurdex Prescribing Information; 2010. Available from: http://www.accessdata.fda.gov/drugsatfda_docs/label/2014/022315s010lbl.pdf. Accessed September 10, 2017. | ||
Haller JA, Bandello F, Belfort R Jr, et al. Dexamethasone intravitreal implant in patients with macular edema related to branch or central retinal vein occlusion twelve-month study results. Ophthalmology. 2011;118(12):2453–2460. | ||
Korobelnik JF, Kodjikian L, Delcourt C, et al. Two-year, prospective, multicenter study of the use of dexamethasone intravitreal implant for treatment of macular edema secondary to retinal vein occlusion in the clinical setting in France. Graefes Arch Clin Exp Ophthalmol. 2016;254(12):2307–2318. | ||
Malclès A, Dot C, Voirin N, et al. Safety of intravitreal dexamethasone implant (Ozurdex): the SAFODEX study. Incidence and risk factors of ocular hypertension. Retina. 2017;37(7):1352–1359. | ||
Capone A, Singer MA, Dodwell DG, et al. Efficacy and safety of two or more dexamethasone intravitreal implant injections for treatment of macular edema related to retinal vein occlusion (Shasta study). Retina. 2014;34(2):342–351. | ||
Lam WC, Albiani DA, Yoganathan P, et al. Real-world assessment of intravitreal dexamethasone implant (0.7 mg) in patients with macular edema: the CHROME study. Clin Ophthalmol. 2015;9:1255–1268. | ||
Kolar P. Risk factors for central and branch retinal vein occlusion: a meta-analysis of published clinical data. J Ophthalmol. 2014;2014:724780. | ||
Boyer DS, Yoon YH, Belfort R, et al. Three-year, randomized, sham-controlled trial of dexamethasone intravitreal implant in patients with diabetic macular edema. Ophthalmology. 2014;121(10):1904–1914. | ||
Malclès A, Dot C, Voirin N, et al. Real-life study in diabetic macular edema treated with dexamethasone implant: the RELDEX study. Retina. 2017;37(4):753–760. | ||
Khurana RN, Bansal AS, Chang LK, Palmer JD, Wu C, Wieland MR. Prospective evaluation of a sustained-release dexamethasone intravitreal implant for cystoid macular edema in quiescent uveitis. Retina. 2017;37(9):1692–1699. | ||
Tomkins-Netzer O, Taylor SR, Bar A, et al. Treatment with repeat dexamethasone implants results in long-term disease control in eyes with noninfectious uveitis. Ophthalmology. 2014;121(8):1649–1654. | ||
Pleyer U, Klamann M, Laurent TJ, et al. Fast and successful management of intraocular inflammation with a single intravitreal dexamethasone implant. Ophthalmologica. 2014;232:223–229. | ||
Rezkallah A, Kodjikian L, Malclès A, Dot C. DEX implant intravitreal injection, sustained intraocular hypertension, and steroid-induced glaucoma in patients with no risk factors. Graefes Arch Clin Exp Ophthalmol. 2018;256(1):219–220. | ||
Maturi RK, Pollack A, Uy HS, et al. Intraocular pressure in patients with diabetic macular edema treated with dexamethasone intravitreal implant in the 3-year MEAD study. Retina. 2016;36(6):1143–1152. | ||
Meyer CH, Michels S, Rodrigues EB, et al. Incidence of rhegmatogenous retinal detachments after intravitreal antivascular endothelial factor injections. Acta Ophthalmol. 2011;89(1):70–75. | ||
Bezatis A, Spital G, Höhn F, et al. Functional and anatomical results after a single intravitreal Ozurdex injection in retinal vein occlusion: a 6-month follow-up – the SOLO study. Acta Ophthalmol. 2013;91(5):e340–e347. | ||
Fassbender Adeniran JM, Jusufbegovic D, Schaal S. Common and rare ocular side-effects of the dexamethasone implant. Ocul Immunol Inflamm. 2017;25(6):834–840. |
Supplementary materials
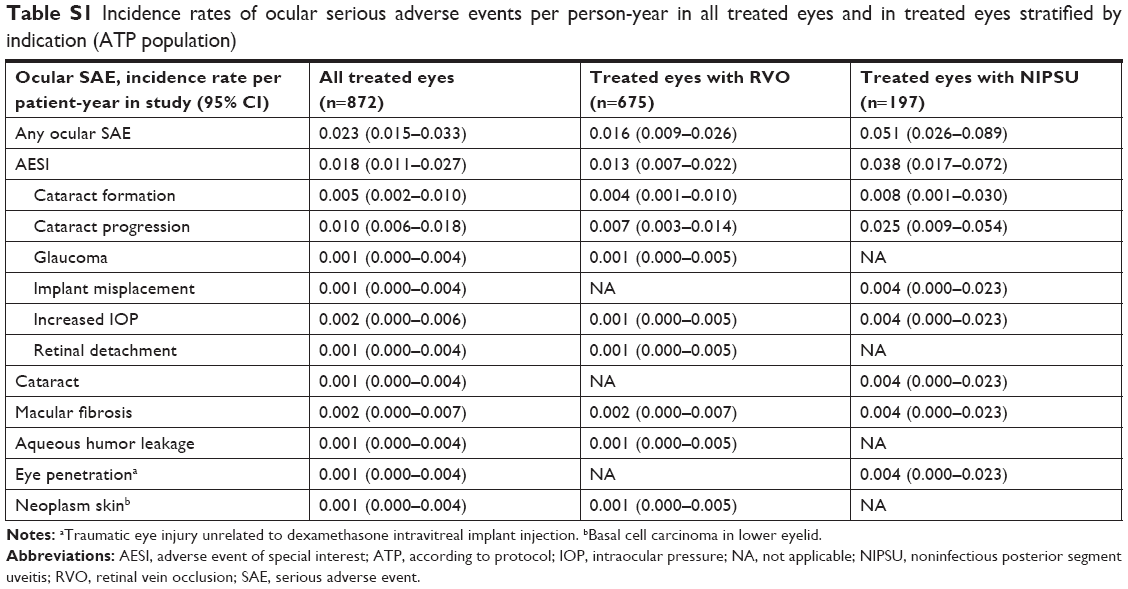 | Table S1 Incidence rates of ocular serious adverse events per person-year in all treated eyes and in treated eyes stratified by indication (ATP population) |
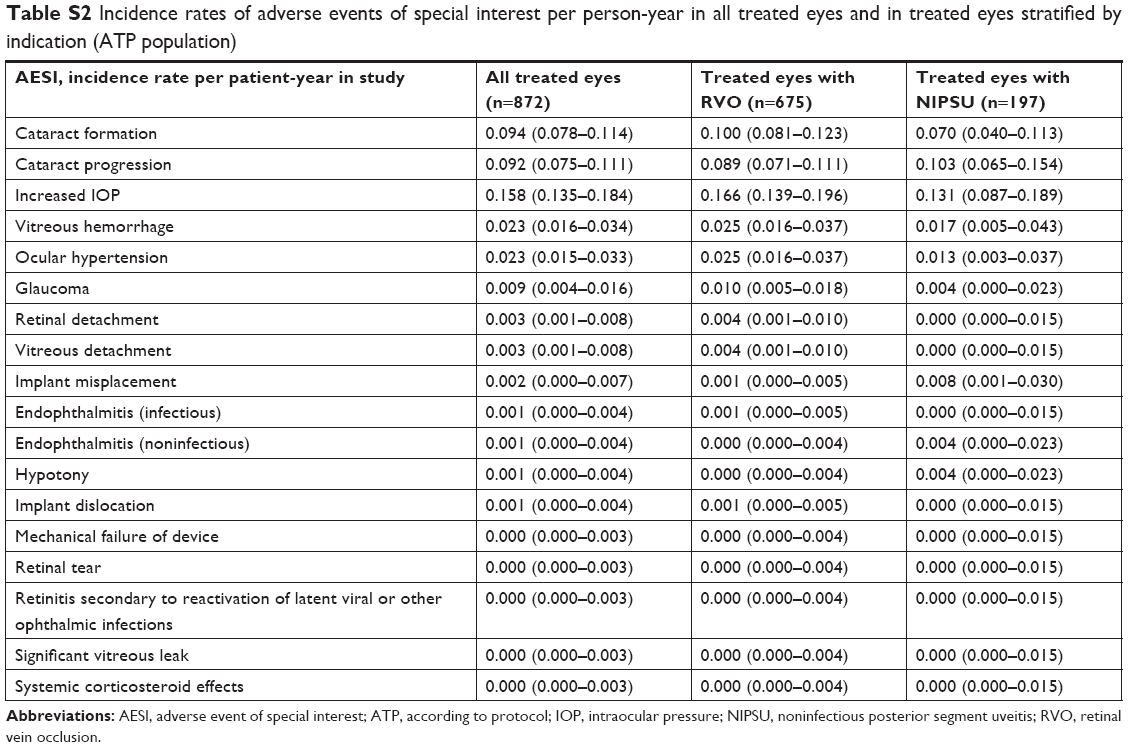 | Table S2 Incidence rates of adverse events of special interest per person-year in all treated eyes and in treated eyes stratified by indication (ATP population) |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.