Back to Journals » Infection and Drug Resistance » Volume 10
Post-influenza aspergillosis, do not underestimate influenza B
Authors Nulens EF ![]() , Bourgeois MJ, Reynders MB
, Bourgeois MJ, Reynders MB
Received 14 September 2016
Accepted for publication 12 January 2017
Published 21 February 2017 Volume 2017:10 Pages 61—67
DOI https://doi.org/10.2147/IDR.S122390
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Professor Suresh Antony
Eric FL Nulens,1 Marc JC Bourgeois,2 Marijke BML Reynders1
1Laboratory Medicine, Medical Microbiology, 2Department of Intensive Care, Algemeen Ziekenhuis Sint-Jan Brugge-Oostende AV, Brugge, Belgium
Abstract: Our objective is to highlight and focus on post-influenza aspergillosis, triggered by influenza B virus. This relatively new clinical entity is often associated with a fulminant course of respiratory decline and high mortality. A 51-year immunocompetent woman, without any medical history or risk factors for developing a complicated influenza infection, was admitted to the intensive care unit. During admission, she presented with an afebrile flu-like syndrome, myocarditis, rhabdomyolysis, multiple organ failure, and evolved to severe respiratory distress. The broncho-alveolar lavage contained influenza B RNA, and the culture revealed Aspergillus fumigatus. Despite maximal organ support, immunoglobulin, antiviral and antifungal therapy, the patient died. This case demonstrates that influenza B virus may be life threatening even to immunocompetent adults and may trigger an invasive Aspergillus superinfection.
Keywords: post-influenza aspergillosis, influenza B, invasive pulmonary aspergillosis, rhabdomyolysis, ARDS, antiviral therapy
Introduction
Every year, influenza A (IA) and B (IB) viruses co-circulate from late fall through to early spring, causing respiratory tract infections among persons of every age group with highest rates of illness in children. Severe infections often cause death due to bacterial superinfection. In immunocompromised patients, invasive pulmonary aspergillosis (IPA) is a well-known entity and is associated with a high mortality. Nevertheless, little is known about the association between invasive fungal superinfections and influenza. Since the introduction of multiparameter molecular assays for the detection of respiratory agents, it is clear that viral infections may trigger the development of fungal superinfections in both immunocompromised and non-immunocompromised patients. These superinfections occur predominantly after IA infections, whereas the exact role of an IB infection in the pathogenicity and its complications are less clear. In our opinion, this case proves that an IB virus infection may equal the severity of an IA infection. Critically ill patients are prone to develop disturbances in immunoregulation during their stay in the intensive care unit (ICU), which render them more vulnerable for fungal infections. Risk factors such as chronic obstructive pulmonary disease, prolonged use of steroids, preceding viral infection, advanced liver disease, chronic renal replacement therapy, near-drowning and diabetes mellitus have been described.
Written consent from the patient’s next of kin to publish this case report has been provided.
Case report
A previously healthy 51-year-old woman, without known comorbidity or risk factors for complicated influenza, was transferred from a regional hospital. She was admitted to the ICU with suspected myocarditis and rhabdomyolysis. Three days earlier, she had symptoms of a general malaise with myalgia, fatigue, nausea, dizziness, hyperventilation, no fever, but with diffuse severe pain. Amoxicillin 1 g IV q.i.d. treatment was started. A transthoracic echography showed a hypertrophic diffuse hypocontractile left ventricle, a left ventricle ejection fraction of 40%, a limited pericardial effusion without impairment of the myocardial contractility, and no evidence of a valvular disease. As she became hemodynamically unstable, acetylsalicylic acid 1 g IV q.i.d., methylprednisolone 40 mg/day and inotropic support were started. During ICU admission, C-reactive protein was 2.4 mg/dL, urea 101 mg/dL, creatinine 1.17 mg/dL, glomerular filtration rate 49 mL/min/1.73 m2, creatine kinase 166,752 U/L, platelets 115 × 106/µL, leukocytes 16.7 × 106/µL (85% neutrophils), D-dimer 1,268 ng/mL, potassium 5.76 mmol/L, calcium 3.1 mEq/L, troponin T 234 ng/L, aspartate transaminase 2,323 U/L, alanine transaminase 851 U/L, and myoglobin 91,180 µg/L. Arterial blood gas determination revealed a pH of 7.36, pO2 of 92.1 mmHg, O2 saturation of 96.0%, and a pCO2 of 32.4 mmHg. Initially, the chest X-ray showed no abnormalities (Figure 1 – day 1). She was admitted in severe organ failure (multiple organ failure), and shortly after developed severe respiratory distress (acute respiratory distress syndrome [ARDS]). After a failed trial of non-invasive ventilation, she became hypoxic and hypercapnic and was intubated. On day 3, a respiratory TaqMan™ microarray for the detection of 31 respiratory pathogens revealed a strong IB RNA signal in a nasopharynx sample. The influenza strain was typed by the National Reference Laboratory (Institute of Public Health, Brussels, Belgium) and belonged to the Yamagata lineage. Continuous venovenous hemofiltration (CVVH), meropenem 1 g IV t.i.d, and oseltamivir 75 mg orally b.i.d. were administered. On day 3, renal failure forced us to reduce the oseltamivir dose to 75 mg every other day. Eventually, oral oseltamivir dose was replaced by 40 mg IV b.i.d. in compassionate use for a total of 14 days. Zanamivir nebulization 10 mg b.i.d. was also added. For 3 days, methylprednisolone 1 g daily was initiated owing to polymyositis suspicion. Five days after admission, Aspergillus fumigatus was cultured from bronchial aspirations. A loading dose of 6 mg/kg voriconazole followed by 4 mg/kg b.i.d. maintenance and caspofungin with a loading dose of 70 mg followed by 50 mg once a day was started. During the entire ICU stay, no bacteria were cultured from respiratory samples. On day 10, a muscle biopsy was performed. Neither histopathology nor a respiratory TaqMan microarray showed any evidence of infection. Histopathological examination revealed a necrotizing myopathy and a type IIB muscle fiber atrophy. Features of classic polymyositis, dermatomyositis, or inclusion body myositis were absent. An autoimmune necrotizing myopathy could not be excluded, and there were no arguments for a classic metabolic or mitochondrial myopathy. Autoimmune serology was negative, and there was no evidence of any recent infection due to cytomegalovirus, Epstein–Barr virus, Coxsackie virus, hepatitis C virus, parvo B19 virus, or Toxoplasma gondii. However, a repeat broncho-alveolar lavage (BAL) confirmed the strong IB RNA signal. Bronchoscopy showed white-yellow cauliflower-shaped budding as well as soft plate-shaped plaque-like elevations at the distal trachea, carina, and the lower airways. These findings are compatible with necrotizing tracheobronchitis.
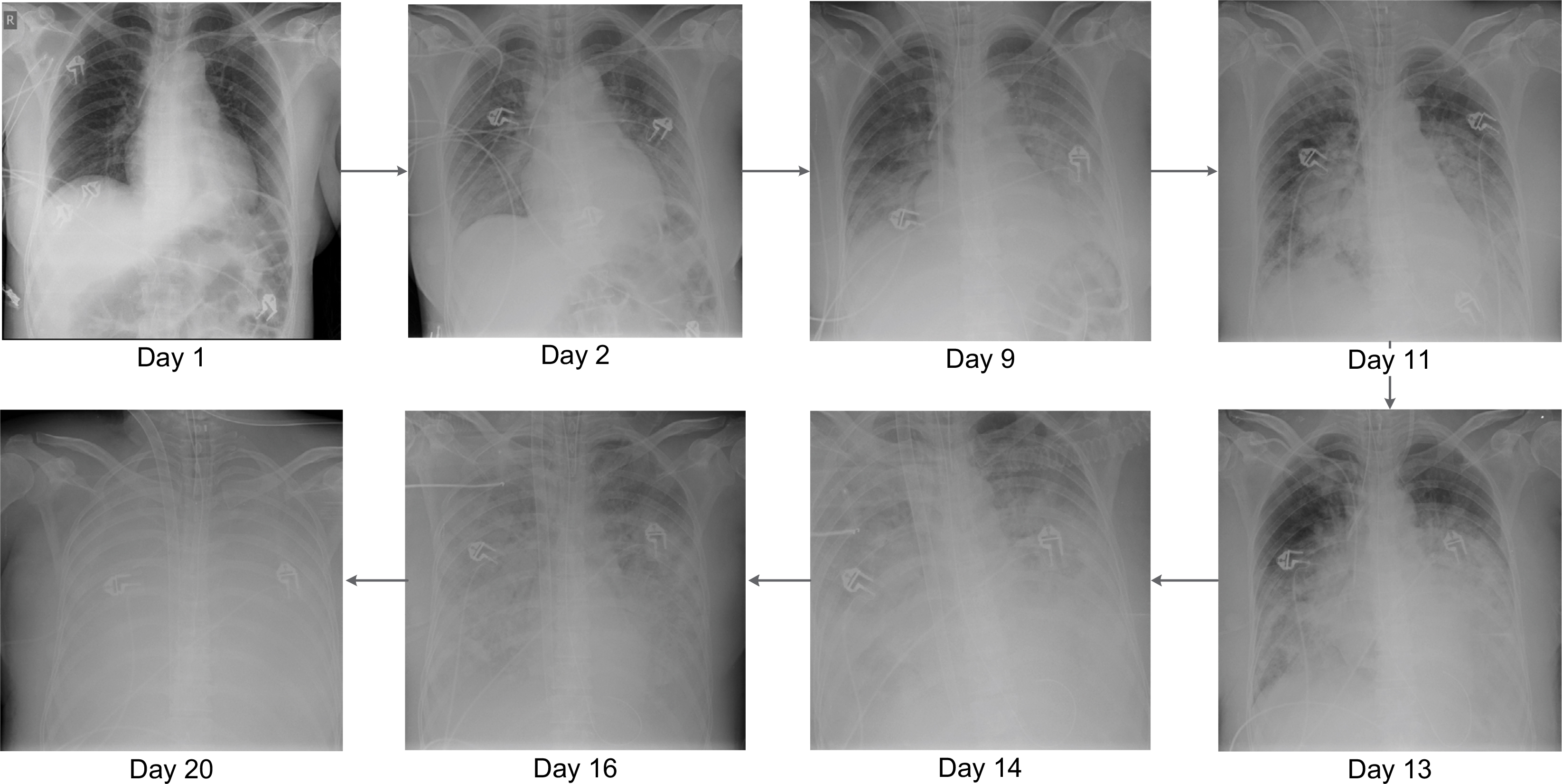 | Figure 1 Daily evolution of chest X-ray from admittance to the ICU to death of the patient. Abbreviation: ICU, intensive care unit. |
Histopathology of the biopsies showed necrosis, massive presence of hyphae, and very little remaining squamous endothelium. A. fumigatus grew from the biopsy, neither bacteria nor herpes viruses were detected. The serum galactomannan index determined on days 14 and 18 was <0.5. On the other hand, the index in BAL determined on days 11 and 16 was 7.8 and 10.9, respectively. A. fumigatus was repetitively grown from respiratory secretions. Serum voriconazole levels determined on days 14 and 20 were 2.9 and 8.7 µg/mL, respectively. From day 7, development of pleural effusion with bilateral basal compressive atelectasis of the lower lobes and diffuse alveolar infiltrates over both lungs were noted, suggestive for ARDS or overwhelming pneumonia. After 2 weeks, bilateral confluent parenchymal shadowing over the entire lungs ensued, with signs of lung edema (Figure 1).
Her condition worsened further. On day 15, poor oxygenation, refractory to nitrous oxide ventilation, and prone position led to extracorporeal membrane oxygenation (ECMO), and the administration of intravenous immunoglobulin (IVIG). Finally, after 22 days without any clinical improvement as well as the development of massive lung edema, all efforts were ceased and the patient died. No autopsy was performed.
Discussion
Discovered in 1940, IB like IA belongs to the family Orthomyxoviridae. Unlike IA, the antigenic drift of IB appears more slowly, does not undergo an antigenic shift, and therefore never causes pandemics. IB has had an evolution into two distinct circulating lineages: B/Victoria lineage and B/Yamagata lineage.1,2 Except for seals, humans are the only animal IB reservoir. IB causes infection of the tracheal mucous glands and respiratory bronchial epithelium, but not of the alveolar endothelium.1,2 The capability of an influenza virus to infect epithelial cells of the upper or lower respiratory tract is dependent on the virus hemagglutinin (HA) and the cellular receptors on the respiratory epithelium.1 The IB HA and neuraminidase are heavily glycosylated, bind collagenous lectins in lung surfactant, and are then readily cleared from the lungs. The respiratory tract cellular receptor preference of influenza virus may also play an important role in localization of the infection. The Yamagata virus lineage predominantly bind to α-2,6-linked sialic acids, which are mainly present in the upper respiratory tract. Although Victory lineage virus also cause mainly upper respiratory tract infection, some viruses are able to bind both α-2,6- and α-2,3-linked sialic acids.1,2 Therefore, IB is probably only involved in infection and inflammation of the upper respiratory tract, in contrast to IA which also destroys the alveolar endothelium of the lower respiratory tract.1–3 Consequently, during an IB infection, the bronchial ciliary epithelium is stripped, local hemorrhages and necrosis may occur, possibly leading to superinfections by bacteria or fungi.
Furthermore, IA infections are also responsible for cell-mediated defects and secondary leukopenia, which further may predispose infected patients to invasive fungal infections.4 IA viruses may cause mild to fulminant and fatal respiratory infections. For a long time, IB was considered to be less pathogenic than IA. However, there is now convincing evidence that IB may cause equally serious infections.1,3 This statement is supported by similar hospitalization rates, and comparable morbidity and mortality figures. It is currently assumed that infections caused by IB viruses are commonly milder than A/H3N2 infections, but more virulent than seasonal A/H1N1 infections.1–3,5
Epidemiologic surveillance indicates that, at the age of 7, every child has antibodies against at least one IA strain.1,6 Between 2004 and 2013, the average percentage of circulating IB viruses in the northern hemisphere was 21.4%. However, there is considerable variation among different epidemic seasons, ranging from <1% to >60%.7 Between 2004 and 2012, 27%–38% of US influenza-associated pediatric deaths were attributed to IB, while this only corresponded to 18%–26% of all circulating viruses.2,8,9 About half of these children did not have any known medical conditions.8 Therefore, children and adolescents may experience higher IB attack rate than adults, and infections may be more severe.1,2,8
An influenza virus infection typically presents as general malaise with fever, headache, cough, and rhinorrhea. Most cases present as a mild upper respiratory infection or a primary viral pneumonia/pneumonitis. Clinically, infections caused by the influenza viruses or other respiratory viruses are indistinguishable. Gastrointestinal complaints, including vomiting, diarrhea, and abdominal pain, usually occur in young children.6,9 Diffuse myalgia, occurring before or simultaneously with the systemic symptoms, may also be a common initial complaint. Influenza infection complications include sinusitis, otitis media, exacerbation of underlying medical conditions, secondary bacterial pneumonia, and co-infections with other viral or bacterial pathogens.
Non-pulmonary complications of influenza include myositis, myocarditis/pericarditis, encephalopathy, transverse myelitis, and Reye’s syndrome. The complications are observed in all age groups, but more frequently in school-aged children.6 Muscle involvement is greater with IB than IA virus. This phenomenon is not yet fully elucidated, but glycoprotein (NB) encoded by the viral RNA is suspected to be responsible for the muscle tropism. NB is an integral component of the IB virion membrane, which may have a role in viral entry and may render IB more myotropic than IA.1,5,6,9
Myositis differentiates from myalgia by its later and sudden onset, usually 1–5 days, more focal location, and greater intensity.6,9 Myositis occurs in, respectively, 6% and 34% of children with an IA and IB infection. The median age is 6–8 years, and boys are more affected than girls. The calf muscles are mostly affected. Tenderness on palpation and gait difficulties are present.10 The diagnosis is usually established by clinical history, presence of influenza in the community, and detecting influenza virus in respiratory samples. A muscle biopsy is not routinely recommended, and patients recover without the need for neuraminidase inhibitors.6,10
Myocarditis is very rarely diagnosed in adults and is mostly associated with IA.11,12 There is a sudden onset of the disease. Echocardiography reveals normal ventricular dimensions, a strong left ventricle dysfunction, and a thickened left ventricular wall is noticed.11 In a recent publication on fatal IB virus infections, the mean age was 11 years, and 78% of them were <18 years old. Almost 70% of patients died within 4 days. Cardiac injury was seen in 20 of the 29 (69%) autopsies, predominantly in patients younger than 18 years. Significantly fewer patients <18 had a secondary bacterial infection, mostly with Staphylococcus aureus, compared with patients over 18, respectively, 24% and 82%. Therefore, these results indicate that in immunocompetent children, fatal viral infections may occur in the absence of a secondary bacterial infection.2 The treatment of influenza-associated myocarditis is supportive, with early initiation of ECMO.12
Rhabdomyolysis involves a rapid dissolution of damaged skeletal muscle, with intracellular muscle components release. The disease can range from an asymptomatic to a life-threatening condition, associated with acute renal failure (ARF), and disseminated intravascular coagulation.13 In severe rhabdomyolysis, the massive amounts of myoglobin released by necrotic myocytes exceed the binding capacity by albumin. Myoglobin is filtered by the glomeruli and reaches the tubules, where it can induce renal dysfunction.14 Rhabdomyolysis may be the consequence of both viral and bacterial infections and non-infectious causes.13 The classic triad of myalgia, weakness, and myoglobinuria is only observed in ~10% of patients. Over 50% of the patients have discolored urine as single clinical finding. In 10%–40%, patients develop ARF; in children, this may be up to 50%. So far, little is known about influenza-associated rhabdomyolysis (IAR).15 A study showed that influenza viruses account for 42% of all rhabdomyolysis cases. In 300 cases of influenza myositis, 3% was accompanied by rhabdomyolysis.6 In both children and adults, rhabdomyolysis was more frequently caused by influenza A. IAR may be complicated by the development of a compartment syndrome. While myositis appears more in boys, girls are more prone to develop rhabdomyolysis.6 A muscle biopsy may exclude alternative non-infectious causes, but usually influenza RNA is not recovered. Therefore, the cause of muscular inflammation or destruction can be derived from the identification of IB in respiratory aspirates during sickness.6,14 Rhabdomyolysis treatment is mainly supportive and aiming at ARF prevention, usually by using hemodialysis, IV fluid administration and, in severe cases, fasciotomy and ECMO.12,14 The effectiveness of a short course of very high doses of methylprednisolone in rhabdomyolysis treatment has not been established yet, but it was successful in a patient with post cytomegalovirus infection rhabdomyolysis.16
Finally, central nervous system (CNS) complications after an influenza virus infection are very rare, and a variety of CNS manifestations may be observed. Seizures (80%) and acute necrotizing encephalopathy (ANE) are the two most common influenza-related CNS complications.17 All age groups are affected. However, mostly children between 5 and 10 years tend to be a victim. Boys and girls are equally affected. Mortality is ~7% and is higher in adult patients. Complications usually appear in conjunction with a recent respiratory infection. In the presence of ANE symptoms occur early, and the clinical course progresses rapidly to coma and death within 2–3 days. Except for a slightly elevated protein concentration in cerebrospinal fluid, no abnormalities are found in 90% of the patients.17–19 Treatment of CNS complications is mainly supportive, with control of intracranial pressure, anti-epileptics for seizure control, management of brain edema, and the addition of antiviral treatment with oseltamivir.18
Treatment of severe IB infection is essentially the same as for an IA infection, with neuraminidase inhibitors oseltamivir, peramivir, laninamivir, and zanamivir.20 In Europe, oseltamivir and zanamivir are the only neuraminidase inhibitors available. Regarding the disappearance of fever and shedding of the virus from the respiratory tract, oseltamivir is believed to be less active against IB and influenza A/N1 subtypes, while isolates of the influenza A/N2 subtype were more sensitive.21,22 The amino acid homology between IA and IB neuraminidase is only 30%. The emergence of viral resistance is a serious concern, as far less is known regarding the IB resistance markers.2 Zanamivir, on the other hand, seems to be more active on IB but is only available for inhalation therapy or IV administration in compassionate use and is not used as first-line treatment.2,22,23 The general recommendation is to start oseltamivir within 48 h of symptom appearance. However, some recent observational studies reported that oral oseltamivir treatment started even 4–5 days after onset in hospitalized patients with suspected or confirmed influenza is also associated with a lower risk for severe outcomes.20,23 Furthermore, viral shedding in IA infection peaks during the first 2 days of illness, whereas viral shedding already increased 2 days before the onset of symptoms in IB infection and is more prolonged in contrast to IA.24 Consequently, IB-infected patients are contagious before symptoms appear, so more subsequent cases may develop. It remains to be elucidated whether early shedding influences the outcome of patients with severe IB infections under antiviral therapy. The shedding time may partially explain why IB virus infections respond more slowly to oseltamivir. However, IB viral clearance and clinical response are faster with zanamivir, and in our opinion, treatment regimens should match the clinical circumstances and may need to be altered.25 The duration of the antiviral treatment is determined by the severity of the illness, immune status, viral clearance, and often lasts longer than 5 days.20 The standard dose of oseltamivir is 75 mg b.i.d., but up to 150 mg b.i.d. may be administered for severe infections or immunocompromised hosts. A doubled dose is usually well tolerated and might be beneficial.26,27 In patients with end-stage renal disease, 75 mg once a day is given orally. In critically ill multiple organ failure patients, physicians may want to avoid overdosing oseltamivir and, therefore, the risk of not reaching adequate plasma drug levels may exist.28 A recent review comparing studies of critically ill patients shows that the oseltamivir absorption is not influenced by the presence of vasopressors or enteral feeding. Also, the serum level of oseltamivir carboxylate after a standard dose exceeds by far the pharmacodynamic threshold.23 However, evidence-based data identifying the optimal antiviral dose for severe infections with MOF are lacking in patients with ECMO and with continuous hemofiltration. Some studies demonstrated adequate drug concentrations with conventional oral dose therapy, while others noted inadequate drug exposure.28 However, dose reduction may be required for patients receiving continuous renal replacement therapy. Despite the fact that compared to healthy patients, ECMO patients have a larger distribution volume, dose adjustment is apparently not necessary.18 In a recent pharmacokinetic study, after only 100 mg oseltamivir IV b.i.d. to a patient under CVVH and ECMO, adequate plasma levels were obtained, resulting in declining viral loads and clinical improvement.23,28 It is reasonable to assume that the orally and IV administered oseltamivir in our patient was insufficient to decrease viral shedding rapidly. IV neuraminidase inhibitors for compassionate use are only available in some countries. Therefore, a collaboration between specialists and therapeutic drug monitoring of orally administered oseltamivir is necessary to establish the optimal personalized antiviral therapy.26 Also, children shed higher viral loads for a longer period than adult patients.8 The administration of IVIG shows significant cross-reactive-specific antibody concentrations against influenza and may serve as an adjuvant in severe infections, particularly in immunocompromised patients and in patients with drug-resistant influenza infections. Although promising, the isolated effect of IVIG in severe influenza infections is hard to assess, as most patients are simultaneously treated with high-dose oseltamivir.29
Respiratory viruses and influenza viruses are easily detected by sampling nasopharynx and throat. However, a recent publication of critically ill ICU patients with laboratory-confirmed influenza showed that sampling the lower respiratory tract may be more predictive of clinical outcome, duration of respiratory support, and length of ICU stay than sampling the nasopharynx.30
The association of bacterial superinfections after respiratory virus infections and influenza infections, in particular, is well described. Publications of post-influenza fungal superinfections are less numerous and often stem from before the availability of diagnostics tools for fungal infections such as galactomannan.4,31,32 Also, there is no doubt about an association between primary viral and fungal superinfections.
With the introduction of molecular assays for the detection of viral respiratory nucleic acids, virus infections are more rapidly detected, improving the clinical outcome of patients with more accurate patient management.
IPA is often described in severely immunocompromised patients and has a high mortality in this group. In apparently immunocompetent critical ill patients without classical immunosuppression but with potential aggravating factors related to ICU admittance, IPA should also be considered when respiratory viruses have been detected.3,4,33–37 In our case report, serum galactomannan remained undetectable during the entire course of the infection. This is not surprising as the patient was relatively young, immunocompetent, had no medical history, and did not take any medication. The most probable explanation is a transient methylprednisolone and viral-induced depression of the cell-mediated immunity. Loss of ciliary function of the bronchial tree during acute IB infection may have predisposed the patient to the development of an invasive fungal infection.1,34 A. fumigatus culture turned positive only after 5 days of intubation. Despite adequate treatment with voriconazole and association with caspofungin, our patient died.
Even so, our patient survived for 3 weeks. For one, she was young and immunocompetent, and further early admittance to ICU, adequate mechanical support, and respiratory stabilization certainly influenced the course of her stay.
Unfortunately, we lacked existent information regarding zanamivir/oseltamivir plasma concentrations in this severely infected patient under CVVH and ECMO.
Disclosure
The authors report no conflicts of interest in this work.
References
van de Sandt CE, Bodewes R, Rimmelzwaan GF, de Vries RD. Influenza B viruses: not to be discounted. Future Microbiol. 2015;10(9):1447–1465. | ||
Paddock CD, Liu L, Denison AM, et al. Myocardial injury and bacterial pneumonia contribute to the pathogenesis of fatal influenza B virus infection. J Infect Dis. 2012;205(6):895–905. | ||
McCullers JA, Hayden FG. Fatal influenza B infections: time to reexamine influenza research priorities. J Infect Dis. 2012;15;205(6):870–872. | ||
Lat A, Bhadelia N, Miko B, Furuya EY, Thompson GR 3rd. Invasive aspergillosis after pandemic (H1N1) 2009. Emerg Infect Dis. 2010;16(6):971–973. | ||
van der Vries E, Ip DK, Cowling BJ, et al. Outcomes and susceptibility to neuraminidase inhibitors in individuals infected with different influenza B lineages: the influenza resistance information study. J Infect Dis. 2016;213(2):183–190. | ||
Crum-Cianflone NF. Bacterial, fungal, parasitic, and viral myositis. Clin Microbiol Rev. 2008;21(3):473–494. | ||
Burnham AJ, Baranovich T, Govorkova EA. Neuraminidase inhibitors for influenza B virus infection: efficacy and resistance. Antiviral Res. 2013;100(2):520–534. | ||
Wong KK, Jain S, Blanton L, Dhara R, Brammer L, Fry AM, Finelli L. Influenza-associated pediatric deaths in the United States, 2004-2012. Pediatrics. 2013;132(5):796–804. | ||
Hu JJ, Kao CL, Lee PI, et al. Clinical features of influenza A and B in children and association with myositis. J Microbiol Immunol Infect. 2004;37(2):95–98. | ||
Sham CO, Tse K. Review of children diagnosed with acute myositis of calves admitted to a regional hospital in Hong Kong in the period 2003-2012. HK J Paediatr (new series). 2015;20:145–150. | ||
Taremi M1, Amoroso A, Nace HL, Gilliam BL. Influenza B-induced refractory cardiogenic shock: a case report. BMC Infect Dis. 2013;13:452. | ||
Chang HL, Hsu JF, Tsai YM, Lin SY, Kuo HF, Yang CJ. Acute respiratory distress syndrome and acute myocarditis developed in a previously healthy adult with influenza B. BMC Pulm Med. 2016;16:1. | ||
Torres PA, Helmstetter JA, Kaye AM, Kaye AD. Rhabdomyolysis: pathogenesis, diagnosis, and treatment. Ochsner J. 2015;15(1):58–69. | ||
Naderi AS, Palmer BF. Rhabdomyolysis and acute renal failure associated with influenza virus type B infection. Am J Med Sci. 2006;332(2):88–89. | ||
Hung PL, Lin PC, Tseng PL. Influenza-B associated rhabdomyolysis and acute renal failure. Indian Pediatr. 2013;50(6):595–596. | ||
Ciarambino T, Adinolfi LE, Giordano M. Acute rhabdomyolysis in healthy woman. Am J Emerg Med. 2016;34(1):113. | ||
Mizuguchi M. Influenza encephalopathy and related neuropsychiatric syndromes. Influenza Other Respir Viruses. 2013;(7 Suppl 3):67–71. | ||
Studahl M. Influenza virus and CNS manifestations. J Clin Virol. 2003;28(3):225–232. | ||
Ak Ö, Biteker F, Cag Y, et al. Influenza B-associated encephalopathy in two adults. J Infect Chemother. 2012;18(6):961–964. | ||
Fiore AE, Fry A, Shay D, Gubareva L, Bresee JS, Uyeki TM; Centers for Disease Control and Prevention (CDC). Antiviral agents for the treatment and chemoprophylaxis of influenza recommendations of the Advisory Committee on Immunization Practices (ACIP). MMWR Recomm Rep. 2011;60(1):1–24. | ||
McKimm-Breschkin JL. Influenza neuraminidase inhibitors: antiviral action and mechanisms of resistance. Influenza Other Respir Viruses. 2013;(7 Suppl 1):25–36. | ||
McKimm-Breschkin J, Trivedi T, Hampson A, et al. Neuraminidase sequence analysis and susceptibilities of influenza virus clinical isolates to zanamivir and oseltamivir. Antimicrob Agents Chemother. 2003;47(7):2264–2272. | ||
Flannery AH, Thompson Bastin ML. Oseltamivir dosing in critically ill patients with severe influenza. Ann Pharmacother. 2014;48(8):1011–1018. | ||
Ip DK, Lau LL, Chan KH, et al. The dynamic relationship between clinical symptomatology and viral shedding in naturally acquired seasonal and pandemic influenza virus infections. Clin Infect Dis. 2016;62(4):431–437. | ||
Li TC, Chan MC, Lee N. Clinical implications of antiviral resistance in influenza. Viruses. 2015;7(9):4929–4944. | ||
He G, Massarella J, Ward P. Clinical pharmacokinetics of the prodrug oseltamivir and its active metabolite Ro 64-0802. Clin Pharmacokinet. 1999;37(6):471–484. | ||
Abdel-Ghafar AN, Chotpitayasunondh T, Gao Z, et al; Writing Committee of the Second World Health Organization Consultation on Clinical Aspects of Human Infection with Avian Influenza A (H5N1) Virus. Update on avian influenza A (H5N1) virus infection in humans. N Engl J Med. 2008;358(3):261–273. | ||
Karsch K, Chen X, Miera O, et al. Pharmacokinetics of oral and intravenous oseltamivir treatment of severe influenza B virus infection requiring organ replacement therapy. Eur J Drug Metab Pharmacokinet. 2016. Epub ahead of print. | ||
Hui DS, Lee N. Adjunctive therapies and immunomodulating agents for severe influenza. Influenza Other Respir Viruses. 2013;(7 Suppl 3):52–59. | ||
Reddy KP, Bajwa EK, Parker RA, Onderdonk AB, Walensky RP. Relationship between upper respiratory tract influenza test result and clinical outcomes among critically ill influenza patients. Open Forum Infect Dis. 2016;3(1):1–3. | ||
Dong Won Park DW, Yhi JY, et al. Fatal clinical course of probable invasive pulmonary aspergillosis with influenza B infection in an immunocompetent patient. Tuberc Respir Dis. 2014;77(3):141–144. | ||
Hasejima N, Yamato K, Takezama S, Kobayashi H, Kadoyama C. Invasive pulmonary aspergillosis associated with influenza B. Respirology. 2005;10(1):116–119. | ||
Taccone FS, Van den Abeele AM, Bulpa P, et al; AspICU Study Investigators. Epidemiology of invasive aspergillosis in critically ill patients: clinical presentation, underlying conditions, and outcomes. Crit Care. 2015;19:7. | ||
Pietsch U, Müller-Höcker C, Enzler-Tschudy A, Filipovic M. Severe ARDS in a critically ill influenza patient with invasive pulmonary aspergillosis. Intensive Care Med. 2016;42(10):1632–1633. | ||
Wauters J, Baar I, Meersseman P, et al. Invasive pulmonary aspergillosis is a frequent complication of critically ill H1N1 patients: a retrospective study. Intensive Care Med. 2012; 389(11):1761–1768. | ||
Stevens DA, Melikian GL. Aspergillosis in the “nonimmunocompromised” host. Immunol Invest. 2011;40(7–8):751–766. | ||
Alshabani K1, Haq A, Miyakawa R, Palla M, Soubani AO. Invasive pulmonary aspergillosis in patients with influenza infection: report of two cases and systematic review of the literature. Expert Rev Respir Med. 2015;9(1):89–96. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.