Back to Journals » OncoTargets and Therapy » Volume 13
Possible Oncogenic Viruses Associated with Lung Cancer
Authors Hu Y, Ren S, He Y ![]() , Wang L, Chen C
, Wang L, Chen C ![]() , Tang J, Liu W
, Tang J, Liu W ![]() , Yu F
, Yu F
Received 26 May 2020
Accepted for publication 2 September 2020
Published 20 October 2020 Volume 2020:13 Pages 10651—10666
DOI https://doi.org/10.2147/OTT.S263976
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Carlos E Vigil
Yan Hu,1 Siying Ren,2 Yu He,1 Li Wang,1 Chen Chen,1 Jingqun Tang,1 Wenliang Liu,1 Fenglei Yu1
1Department of Thoracic Surgery, The Thoracic Surgery Research Room, Second Xiangya Hospital, Central South University, Changsha 410011, People’s Republic of China; 2Department of Respiratory Medicine, Hunan Centre for Evidence-Based Medicine, Research Unit of Respiratory Diseases, Second Xiangya Hospital, Central South University, Changsha 410011, People’s Republic of China
Correspondence: Fenglei Yu
Department of Thoracic Surgery, The Thoracic Surgery Research Room, Second Xiangya Hospital, Central South University, No. 139 Renmin Road, Changsha 410011, People’s Republic of China
Email [email protected]
Abstract: Lung cancer is the most common cause of cancer death worldwide. Tobacco smoking is the most predominant etiology for lung cancer. However, only a small percentage of heavy smokers develop lung cancer, which suggests that other cofactors are required for lung carcinogenesis. Viruses have been central to modern cancer research and provide profound insights into cancer causes. Nevertheless, the role of virus in lung cancer is still unclear. In this article, we reviewed the possible oncogenic viruses associated with lung cancer.
Keywords: oncogenic virus, lung cancer, human papillomavirus, Merkel cell polyomavirus, Epstein–Barr virus, jaagsiekte sheep virus, John Cunningham virus
Introduction
Lung cancer is the most common cause of cancer death worldwide, with an estimated 1.8 million deaths each year.1 Lung cancer is divided into two main categories: non-small-cell lung cancer (NSCLC) that comprises approximately 85% of lung cancer cases and small-cell lung cancer (SCLC) that comprises about 15%.2 Tobacco smoking is the most predominant etiology for lung cancer, accounting for more than 80% of cases in the US and other countries where cigarette smoking is common.3 Lung cancer in nonsmokers is more common in women and in Asia and is a different disease with molecular characteristics that differ from lung cancer in smokers.4 This suggests that other factors are required for lung carcinogenesis, which include inherited genetic susceptibility and infectious agents, such as virus.3
Since Rous’s initial experiments suggesting virus as a possible transmissible agent for cancer, the virus-associated cancer research field has witnessed a roaring progress over the last century.5 Until now, seven human viruses have been discovered to cause 10–15% of human cancers worldwide, including Epstein–Barr virus (EBV), hepatitis B virus (HBV) or hepatitis C virus (HCV), human T-lymphotropic virus-1 (HTLV-1), human papillomavirus (HPV), Kaposi’s sarcoma herpesvirus (KSHV), and Merkel cell polyomavirus (MCPyV).5,6 Mounting evidences point to a potential role of several viruses for lung cancer (Figure 1.), such as HPV,7–9 MCPyV10–12 and EBV.13–15 It has been reported that Jaagsiekte sheep retrovirus (JSRV) spontaneously induces a transmissible lung adenocarcinoma through the viral envelope protein in sheep.16 However, human lung cancer shows no cogent evidence of being associated with oncogenic viruses so far.17 In this article, getting more insight into viral etiology for lung cancer, we thoroughly reviewed the possible oncogenic viruses associated with lung cancer through systematic literature searching.
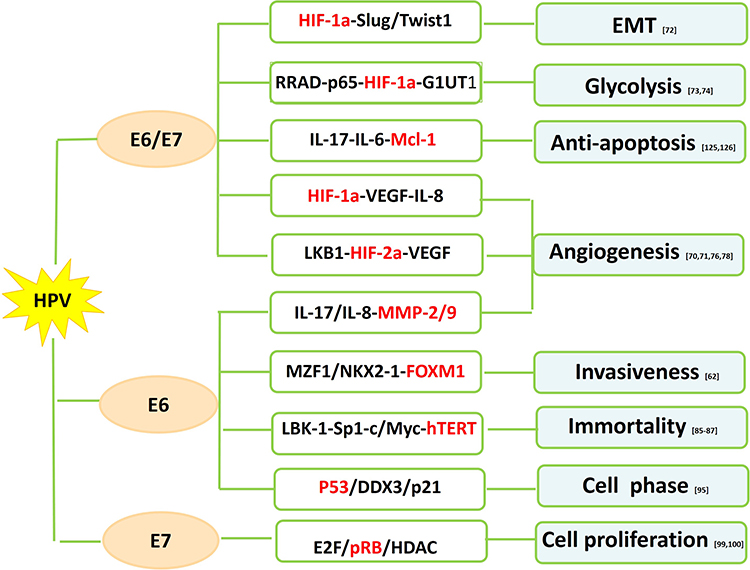 |
Figure 1 HPV-mediated oncogenic mechanisms in NSCLC. |
HPV
HPV Detection in Lung Cancer Tissues
Since Syrjanen’s suggestion of HPV involvement in bronchial squamous carcinoma reported in 1979,18,19 several studies have explored this relationship between HPV infection and lung cancer occurrence. However, the results of these studies have been inconsistent, with some authors in favor of this association20–36 and the others not37–43 (Table 1). HPV infection is the most common sexually transmitted infection, with estimates for the probability of infection with the virus exceeding 80% for women and 90% for men across their lifetime.44 The HPV DNA is detected in lung cancer tissue, but also detected in peripheral blood, bronchial brushing, and the exhaled breath condensate of patients with lung cancer.45–47 HPV 16 and 18 are the two most common genotypes detected in lung cancer worldwide.48 The other frequently detected high-risk subtypes are HPV 31 and 33 and the most prevalent low-risk subtypes are HPV 6 and 11.19 Recently, Xiong et al conducted a meta-analysis comparing HPV infection rates in lung cancer tissues (19.8% for HPV 16 and 18.59% for HPV 18) vs noncancer controls (5.84% for HPV 16 and 4.29% for HPV 18) and found that HPV infection was a risk factor of lung cancer.7
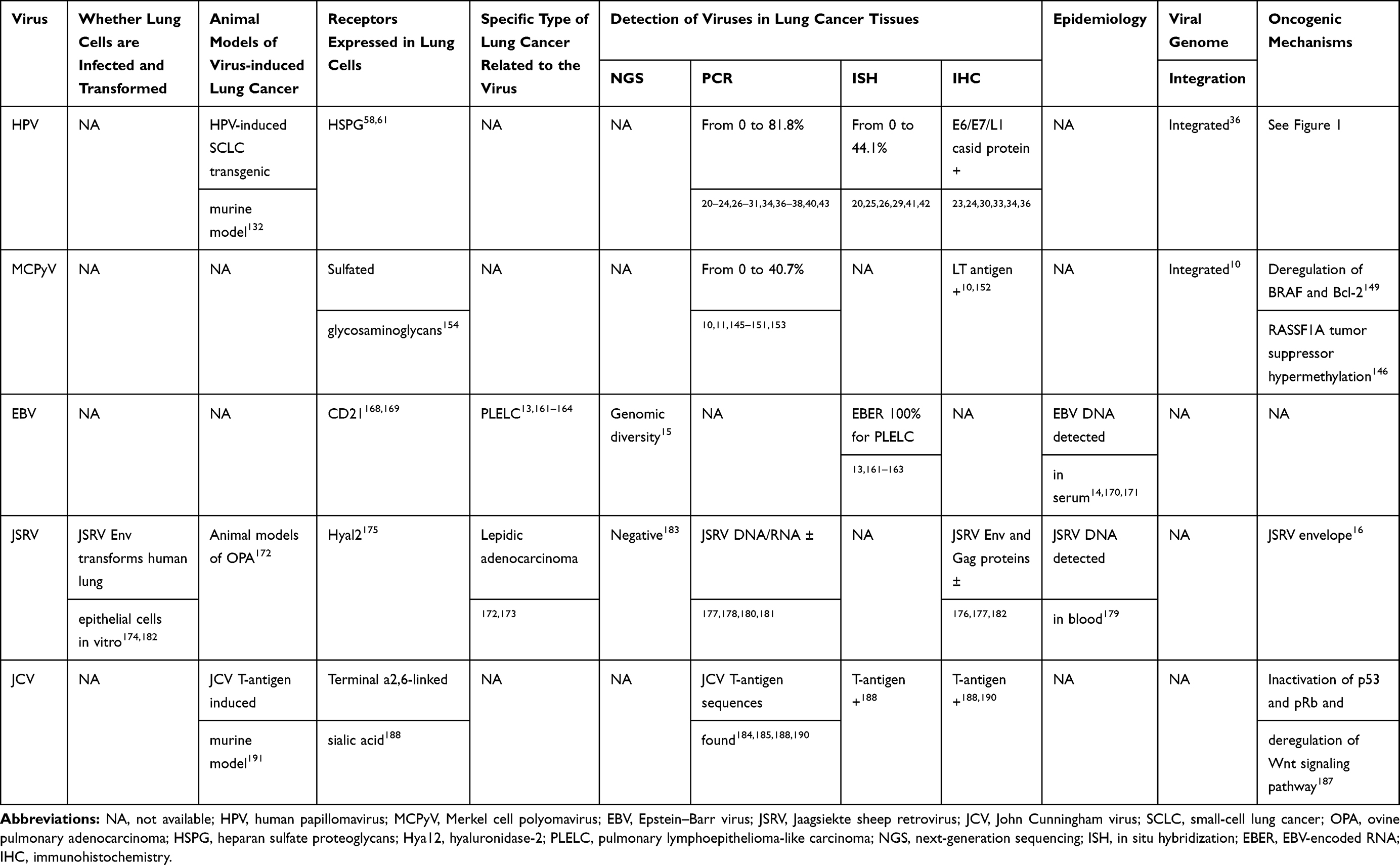 |
Table 1 Key Findings of Possible Oncogenic Viruses Associated with Lung Cancer |
Several studies have reported a higher prevalence of HPV infection in lung cancer tissues derived from patients in Asian than other continents.7,9,19,20,25,26,32 Syrjanen shows that the average HPV infection rate in lung cancer tissues worldwide is 26.5%, the highest in China (37.7%), the lowest in North America (12.5%), with Australia, Europe, South America, and other Asian regions (18.5%, 16.9%, 23.9%, and 17.2%), respectively.19 Moreover, HPV infection rate in squamous cell carcinoma (25.1%) is found to be higher than that in adenocarcinoma (15.1%). This result is consistent with that of the study by Xiong et al, which may be explained by the high affinity of HPV to squamous epithelial cells.7 They further explored that the reported wide variability in HPV infection rates of lung cancer tissues was not majorly owing to the HPV detection methods, but was better explained by the geographical origin of the study and the histological type of lung cancer.19
Transmission Routes of HPV into Lung
HPV can be transmitted through physical contact as well as vertically from the HPV-positive mother to her newborn and cause subclinical or clinical infection.49 How HPV is transmitted into the lung, however, remains unidentified. Several studies suggested that HPV might be transmitted to the lung from the aerodigestive tract, for instance, oral mucosa, esophagus, larynx, or sinonasal mucosa.50–52 In addition, the findings of higher risk of developing lung cancer in female patients with anogenital malignancies than those without53,54 and morphological resemblance of HPV-infected bronchial squamous cell carcinoma to HPV-infected genital warts18 indicate that HPV may be transmitted to the lung from the genital tract.55
Blood circulation may be another transmission route for HPV infection in the lung. Chiou et al found that HPV 16 DNA in blood circulation was significantly associated with that in lung cancer tissue.46 They proposed that peripheral lymphocytes could harbor HPV particles and might be involved in the spreading of HPV viral particles.
Several studies have confirmed the presence of HPV DNA in surgical smoke and previous cases of treating gynecologist developing laryngeal HPV-associated neoplasm after performing laser therapy have been reported as well.56 In addition, Carpagnano et al found the presence of HPV in the exhaled breath condensate of patients with lung cancer.45 This evidence suggests another possible means of HPV transmission through inhalation.
Interplay of HPV with Lung Cell Receptors and Cell Entry
Cell entry is a fundamental process of the infectivity of any virus into host cell.57 Host cell entry of HPV is initiated by binding of the virus particle to cell surface receptors. Heparan sulfate proteoglycans (HSPG) is suggested as the initial attachment receptor for HPV and is ubiquitously expressed in the extracellular matrix and on the surface of most cells, including baseline membrane of type 1 alveolar epithelial cells and endothelial cell and endothelial cell surface.58 HPV can specifically attach to exposed basement membrane HSPG, followed by a series of conformational changes and, ultimately transfer of encapsidated plasmid DNA into the host cells.59 Although evidence of infectivity of human lung cell lines by HPV is lacking, the presence of HSPG in lung cells indicates that HPV may interact with these receptors and enter into lung cells. Moreover, mutation and modification in HSPG chains/sulfation patterns on a variety of solid tumors has been demonstrated.60 In particular, a recent study showed HPV capsids preferentially bind and infect lung cancer cells in vitro and in vivo, at least supporting HPV as a cofactor in the process of lung cancer carcinogenesis.61
HPV and Survival
Several studies have explored the impact of HPV infection on lung cancer prognosis.23,34,37,62–66 However, the results of these studies have been inconsistent, with some authors supporting the prognostic role of HPV infection34,64–66 and others supporting no association.23,37,62,63 Among them, Miyagi et al suggested that high intratumor infiltration of Langerhans cell might be responsible for better prognosis of HPV-infected lung cancer.66 Wang et al demonstrated the prognostic value of HPV status in lung adenocarcinoma, showing that patients with HPV-infected lung adenocarcinoma had a better prognosis than those without, with a 32% reduction in mortality.34 Guo et al further conducted a meta-analysis confirming the association between HPV infection and improved survival for lung adenocarcinoma patients, but not for squamous cell carcinoma patients.67 It seems mutually conflicting when it comes to HPV with pro-oncogenic potential seeming to improve survival of patients with lung cancer. We can only hypothesize that the presence of HPV may attract more immune cells including Langerhans cells in the tumor microenvironment and trigger stronger antitumor immune responses, leading to better prognosis. Without doubt, more evidence is needed to explain this phenomenon.
Oncogenic Mechanisms of HPV-associated NSCLC
HPV-16 E6/7 and HIF-1a/HIF-2a
HIF-1a is a transcription factor involved in the regulation of angiogenesis, which plays a vital role in tumor progression, metastasis, and drug resistance.68 VEGF, one of the key downstream targets of HIF pathway, regulates vessel formation through their effect on endothelial cell migration, proliferation, permeability and survival.69 HIF-1a expression tends to be higher in HPV-infected NSCLC than HPV-negative NSCLC. HPV-16 E6/7 oncoproteins significantly potentiate angiogenic phenotype of NSCLC cells in vitro and in vivo by upregulating the expression of HIF-1a, VEGF and IL-870,71 (Figure 1). PI3K/Akt and c-Jun signaling pathway might be responsible for HIF-1a/VEGF-mediated angiogenesis triggered by HPV-16 oncoproteins.70 HPV-16 E6/7 also promote NSCLC progression by facilitating epithelial-mesenchymal transition (EMT) process through their effect on EMT-related transcription factors including ZEB1, Snail1, Slug and Twist1.72 Moreover, upregulated expression of HPV-16 E6/7 enhanced GLUT1 expression in NSCLC cells through the inhibition of RRAD and translocation of p65,73 suggesting HPV oncoproteins involved in the regulation of the Warburg effect.74
HIF-2a, however, possesses similarly proangiogenic functions as to HIF-1a via the activation of downstream effectors including VEGF.75 HPV-16 E6/7 stimulates the expression of HIF-2a and subsequent VEGF in several NSCLC cell lines by suppressing LKB1.76
HPV E6 and MMPs/TIMP-3
MMPs are enzymes that degrade protein and collagen in the extracellular matrix (ECM) and are implicated in tumor invasion and metastasis.77 An in vitro study has shown that HPV-16 E6 enhances the expression of MMP-2 and MMP-9 by stimulating IL-8 expression in lung adenocarcinoma cells.78 Tissue inhibitor of metalloproteinase (TIMP), rather, acts as a MMP inhibitor to reduce proteolytic destruction of the cell matrix to decrease cancer metastasis and improve prognosis.79 Loss of heterozygosity of TIMP-3 has been involved in several cancer types.80–82 Frequency of TIMP-3 loss by LOH and/or promoter hypermethylation is higher in HPV-16/18 infected NSCLC than HPV-16/18 negative NSCLC.63 Loss of TIMP-3 potentiates malignant behaviors and poor survival of HPV-infected NSCLC by elevating IL-6 production via the tumor necrosis factor a/nuclear factor κB axis.63
HPV E6 and hTERT
The activation of human telomerase reverse transcriptase (hTERT), a catalytic subunit of the enzyme telomerase, is implicated in the process of human cell immortality and malignant transformation.83 The hTERT expression is found to be elevated in NSCLC including preneoplastic lesions, suggesting its role in the early stage of lung cancer development.84 HPV E6 seems to activate hTERT overexpression in HPV related lung cancer.85 Cheng et al further explored that Sp1 cooperated with c-Myc to activate hTERT transcription in HPV E6-positive lung cancer cells under the context of c-Myc induced by E6 promoting its binding onto hTERT promoter.85 However, p53 has been reported to inhibit hTERT expression by binding to Sp1 and preventing its access to the hTERT promoter.86 Moreover, LBK1 inhibition and subsequent Sp1 upregulation are required for the HPV E6-mediated hTERT upregulation in lung carcinogenesis.87
HPV E6 and p53
p53 has been described as “the guardian of the genome” because of its role in conserving the integrity of the genome by inducing cell cycle arrest or apoptosis on DNA damage. Inactivation of tumor suppressor p53 has been found to occur in most cancers including lung cancer. The classic function of oncogene protein E6 is to induce p53 degradation through its binding to the LxxLL motif of the cellular ubiquitin ligase E6AP.88,89 E6-mediated p53 inactivation results in chromosomal instability and increased potential of HPV-infected cells becoming malignant.89 Transcription of p21WAF1/CIP1 and mdm-2, two downstream targets of p53, are inhibited by E6 in lung tumors. p21WAF1/CIP1, a cyclin-dependent kinase (CDK) inhibitor, acts on cyclin E/cdk2 complexes and inhibits the phosphorylation of the pRb protein, thus preventing S phase entry.90 Induction of p21 is fulfilled through p53-dependent91 or p53-independent92 pathways. mdm-2, a cellular oncogene product, regulates the activity of p53 protein, which in turn modulates the transcription of mdm-2 gene.93 The human dead-box RNA helicase (DDX3), which plays a role in the regulation of gene expression via RNA metabolism,94 has been implicated in the development of viral-associated cancers.95 DDX3 transcription is directly regulated by p53 and DDX3 synergistically promotes p53-activated p21 transcription via increased Sp1 binding affinity onto the p21 promoter in NSCLC cells.95 p21 reduction by the E6-inactivated p53 pathway contributes to tumor progression and a poor relapse-free survival in lung cancer patients.95
HPV E7 and pRb
Oncoprotein E7 targets retinoblastoma suppressor protein (pRb) to induce its degradation, allowing the dissociation of the E2F/pRb/histone deacetylase (HDAC) complex and deregulation of cell proliferation.96 E7 also target and degrade the “pocket proteins” p107 and p130, both of which are E2F regulators.96 p16INK4A, an inhibitor cyclin-dependent kinase, is mapped to a critical region at chromosome 9p21 and hypermethylation of p16INK4A in the CpG-rich promoter regions occurs frequently in NSCLC.97,98 E7-mediated pRb degradation leads to the release of HDAC to enhance p16INK4 hypermethylation through chromatin remodeling by HDAC in HPV-infected tumors.99,100 Wu et al confirmed the potential correlation between p16INK4A hypermethylation and HPV infection in nonsmoking female patients with NSCLC with the finding that p16INK4A hypermethylation frequency was as high as 70% with HPV infection as compared to those without HPV infection.100 Reports from the same research group further indicated the linking of expression of DNA methyltransferase 3 (DNMT3) protein and HPV infection.101 They argued that, HPV infection upregulated DNMT3 protein expression, which subsequently increased p16INK4A hypermethylation.
HPV and FHIT LOSS
The fragile histidine triad (FHIT) gene at chromosome 3p14.2 is altered by loss of heterozygosity (LOH) and occasional homozygous deletions in various human cancers including lung cancer.102,103 Allelic deletion of FHIT plays an important role in lung tumorigenesis104 and can be used as a negative prognostic marker.105 After HPV infection, HPV DNA integration into the fragile site FRA3B adjacent to FHIT occurs to cause allele loss of the gene.106 A study from Taiwan reported a high frequency of FHIT LOH in HPV-positive nonsmoking female lung cancer patients, suggesting its possible role in HPV-infected lung carcinogenesis.103 Carpagnano et al found microsatellite alterations (MA) at chromosome 3p existed in 100% of HPV-positive NSCLC patients enrolled in their study.107 Yu et al further suggested FHIT loss and p53 mutation might synergistically exerted on HPV-infected lung carcinogenesis.108 Verri et al found that different mechanisms as promoter methylation and LOH interplay to inactivate FHIT expression.109
HPV E6 and EGFR Mutation
EGFR somatic mutation is associated with HPV presence in NSCLC.110 A meta-analysis including four studies with a total of 498 Asian patients showed the presence of EGFR somatic mutation was significantly higher in HPV-positive patients compared with HPV-negative counterpart (P=0.012).110 Several studies reported that HPV infection in NSCLC denoted a better overall survival and better response to EGFR-TKI therapy.111,112 This observation could be explained that HPV-positive NSCLC patients are more likely to exhibit EGFR somatic mutation, thus having a better response to EGFR-TKI and better survival. However, the association between HPV infection and EGFR mutation or response to EGFR-TKI seems to be limited by geographical origin since Marquez-Medina et al reported the negative outcomes obtained from western patients.113 The underlying mechanism of this relationship remains unknown. Inhibitors of antiapoptosis proteins (IAP), including cIAP1, cIAP2 and XIAP, are a family of caspase inhibitors that block cell apoptosis and are considered as a therapeutic target in lung cancer.114 Wu et al indicated that HPV-16 E6 led to cIAP2 upregulation through phosphorylation of cAMP response element-binding protein (CREB) via EGFR/PI3K/AKT pathway and cIAP2 expression correlated with EGFR mutation.115 Inflammatory-induced oxidative stress is implicated in the development of lung adenocarcinoma and the level of 8-hydroxy-2ʹdeoxyguanosine (8-OH-dG), an oxidative stress biomarker, is closely associated with EGFR mutation in lung cancer.116 Tung et al found that HPV16/18 E6 elevated 8-OH-dG through increased ROS production, which in turn cooperated with HPV16/18 E6 to contribute to EGFR mutation in NSCLC.33
HPV and Smoking Exposures
Tobacco smoking is one of the well-known risk factors for developing lung cancer. However, only a small percentage of heavy smokers develop lung cancer. This phenomenon suggests that other cofactors are required for lung carcinogenesis. Whether HPV infection has a synergistic effect with smoking on lung carcinogenesis remains unknown. Munoz et al demonstrated that the proliferative rate and anchorage-independent growth of HPV16 E6/7 transfected lung epithelial cells were significantly elevated when exposed to cigarette smoke components (CSC), suggesting that the functional interaction between cigarette smoking and HPV infection promoted the possibility of lung carcinogenesis.117 Pena et al further showed that CSC activated HPV16 p97 promoter through their effects on the long control region (LCR) in lung epithelial cells.118 Moreover, HPV16 E6/7 was able to increase oxidative DNA damage induced by CSC.118
Benzo[a]pyrene (B[a]P), a major constituent of cigarette smoke, is associated with lung cancer development.119 B[a]P can increase the number of virions and genomes of HPV.120 B[a]P treatment contributes to gene promoter hypermethylation, which is the main pathway implicated in repair gene inactivation.121 In addition, combination of inactivation of repair genes and exposure to B[a]P facilitates DNA damage.122 Interestingly, HPV acts synergistically with B[a]P to induce DNA damage in NSCLC cells, promoting lung carcinogenesis, especially in female patients.123
Others
HPV E6/7 and Mcl-1
Myeloid cell leukemia (Mcl)-1 is an antiapoptotic member of the Bcl-2 family that contributes to the control of cancer development.124 The presence of Mcl-1 is implicated in cancer cell growth and evasion of apoptosis in various cancer types, including lung cancer.124 Concomitant expression of IL-6 or IL-17 and Mcl-1 is colocalized with HPV DNA in NSCLC.125 HPV E6/7 leads to upregulated expression of IL-6 or IL-17 and Mcl-1 through phosphatidylinositol-3-OH kinase pathway.126 The microenvironmental inflammation manifested by high level of IL-6 and IL-17 secreted by lung cancer cells in response to HPV stimuli is, therefore, likely to be responsible for HPV-infected lung tumorigenesis.125,126
HPV E6/7 and FOXM1
Increased expression of Foxhead box M1 (FOXM1) is associated with tumor progression and poor prognosis in various cancer types including NSCLC.127 FOXM1 is upregulated by E2F released by Rb phosphorylation through p53 inactivation and interacts with HPV-16 E7 to enhance the transformation potential of rat embryo fibroblasts.128 Chen et al failed to identify E7-triggered FOXM1 upregulation in HPV-positive cancer cells including lung cancer cells.62 However, elevated FOXM1 expression was triggered by E6 through the MZF1/NKX2-1 axis, which activated beta-catenin nuclear translocation and subsequently potentiated cell invasiveness and stemness in HPV-positive NSCLC.62
HPV E6 and miR-30c-2/MTA-1
miR-30c-2 is one of tumor suppressor microRNAs which are implicated in tumor development. The downregulation of miR-30c-2 promotes the invasion of NSCLC by targeting metastasis-associated protein-1 (MTA-1).129 Wu et al demonstrated that HPV-16/18 E6 negatively correlated with miR-30c-2 expression and positively with MTA-1 in NSCLC tissues and expression levels of miR-30c-2 and MTA-1 could predict prognosis and therapeutic response to chemotherapy of patients with NSCLC.130
Experimental Models of HPV-Associated Lung SCLC
In vitro and in vivo animal models are widely used in HPV research.131 No data are available concerning experimental models used to investigate how HPV infects and transforms lung cells. Importantly, Carraresi et al initially established a transgenic mouse model of SCLC induced by HPV-16 E6/E7 oncoproteins under the control of the cytokeratin 5 gene promoter.132 Furthermore, they developed two murine cell lines derived from transgenic lung SCLC, both of which showed absence of p53 and pRB and sustain tumor formation after subcutaneous injection in syngenic mice.133 These findings provide more direct evidence for ability of HPV to induce SCLC, possibly through the inactivation of p53 and pRB.
Possible Connection of HPV Vaccination and Lung Cancer
Prophylactic HPV vaccination, mainly covering girls and women under 25, is currently included into national vaccination programs in 60 countries worldwide.134 It aims at the formation of virus-neutralized antibodies and expected to protect from developing cervical cancer based on the established association between HPV and the progression of cervical neoplasia. The possible involvement of HPV in lung cancer may necessitate the introduction of prophylactic vaccination in both boys and girls. Undoubtedly, more lines of evidence are warranted to establish definitive evidence of HPV as an oncogenic factor of human lung cancer and to verify whether there is an impact on the lung cancer incidence of HPV-directed vaccine meant to prevent cervical cancer.
Merkel Cell Polyomavirus (MCPyV)
With the discovery by Feng et al in 2008 of Merkel cell polyomavirus (MCPyV) as a causative agent of Merkel cell carcinoma (MCC),135 several authors have investigated the association between the presence of MCPyV in several human tumors including lung cancer.136–139 MCPyV infections are widespread in the human population with MCPyV continuously shed from healthy skin.140 Moreover, MCPyV DNA fragments have been detected in a wide variety of anatomical locations, including lower respiratory tract.141 A recent study investigating MCPyV presence in 10 autopsies with an extensive organ sampling revealed a high prevalence of MCPyV was found in lung samples as well as in blood and brain samples.142 Persistent presence of MCPyV in the respiratory tract may facilitate the development of lung cancer.
MCPyV and Lung Cancer
Since SCLC harbors the histological similarities towards MCC,143,144 whether MCPyV leads to the development of SCLC has caught the attention of researchers. Studies have detected viral DNA sequences in SCLC tissues.145,146 However, there is evidence indicating no role of MCPyV in SCLC.147,148 Furthermore, the prevalence of MCPyV in NSCLC has been investigated as well. Researchers found varying degrees of MCPyV infection positivity in NSCLC at the DNA, RNA and protein levels.11,149–153 Hashida et al provided the first evidence of not only the detection of MCPyV DNA but also the expressions of both LT RNA transcripts and LT antigen in NSCLCs.10 They revealed that nine out of 32 SCC, nine out of 45 AC, one out of 32 large-cell carcinoma, and one out of three pleomorphic carcinomas were positive for MCPyV DNA.10 Another study showed statistically significant difference between stages of NSCLC and MCPyV LT-Ag DNA load, which suggested viral load may be increased with tumor progression.11
Possible NSCLC-Specific Oncogenic Mechanisms
Currently, neither human cell lines nor animal models are available to explore lung carcinogenesis by MCPyV. Like HPV, cell entry of MCPyV follows a two-step attachment-and-entry process154 and abundant expression of sulfated glycosaminoglycans in lung cells provides a basis for MCPyV to enter lung cells.
Integration of MCPyV DNA into the host cell genome is thought to cause MCC through the constitutive expression of the transforming large T (LT) and small T (ST) proteins with distinct mechanisms.155 However, the pathogenesis of MCPyV-induced NSCLC remains to be determined. It has been suggested that mutant BRAF drives the development of lung adenocarcinoma.156 Heterodimer of Bax/Bcl-2 induces a neutralization of Bax and a loss of apoptosis.157 Lasithiotaki et al revealed increased expression of BRAF as well as the downregulation of Bcl-2 in MCPyV-positive NSCLC samples as compared to MCPyV-negative NSCLC samples, suggesting a role of MCPyV in NSCLC through deregulation of BRAF and Bcl-2.149 They further demonstrated that the expression of NSCLC-associated microRNAs (miR-21, miR-376, and miR-145) and their corresponding target genes were influenced by the presence of MCPyV in lung cancer tissues, providing evidence of a MCPyV-associated epigenetic mechanism in NSCLC.12 Xu et al found a significant correlation between MCPyV infection and EGFR mutations by screening 189 NSCLC samples.152 Their finding suggested MCPyV infection might induce EGFR mutations in NSCLC. If this were the case, it would explain the phenomenon provided by Lasithiotaki et al of higher expression of BRAF in MCPyV-positive samples since BRAF is a downstream target of EGFR pathway.149 Hashida et al identified two MCPyV integration sites (5q23.1 and 11q25) in NSCLC patients.10 However, both integration sites were not close to the EGFR gene location (7p12).10 Therefore, whether MCPyV infection induces EGFR mutations in NSCLC warrants further investigations.
Epstein–Barr Virus (EBV)
EBV is a lymphotropic gamma herpes virus with oncogenic properties infecting more than 90% of adults worldwide. It is directly involved in the pathogenesis of a variety of lymphoproliferative and neoplastic disorders, including undifferentiated nasopharyngeal carcinoma and lymphoepithelioma-like carcinoma (LELC) at various sites.158,159 The association of EBV and lung cancer presents significant differences according to tumor histotype and geographical site.15,160 The EBV is often detected in pulmonary LELC occurring in patients from east and southeast Asia where nasopharyngeal carcinoma is highly prevalent,13,161–164 but rarely detected or even undetected in other types of lung cancer such as adenocarcinoma, squamous cell carcinoma, and SCLC.42,165,166 Recently, Wang et al explored the EBV genomic variations in lung carcinoma and reported four newly sequenced EBV genomes isolated from primary lung carcinomas with apparent genomic diversity among these EBV genomes.15 However, whether EBV genomic variations contribute to lung carcinogenesis remains unknown and deserves further investigation.
It is well-known that cell entry of EBV is initiated by the interaction of the viral envelope glycoprotein gp350 with the cellular surface receptor CD21 of B cells and epithelial cells.167 Several studies have demonstrated the expression of EBV receptor CD21 in human bronchial epithelium,168 and more specifically in type 2 alveolar epithelial cells,169 implying possibility of EBV to infect and possibly transform lung cells in a CD-21 dependent manner. Yet, it needs further investigations.
Previous studies have measured circulating EBV DNA in the plasma of patients with pulmonary LELC and suggested its role for monitoring response to therapy.170,171 Recently, Xie et al performed a prospective multicenter study in Southern China investigating the association between baseline EBV DNA and OS and disease-free survival (DFS) in a total of 429 patients with pulmonary LELC and showed that baseline EBV DNA copy of at least 4000 copies/mL predicted disease recurrence and poorer survival among patients with early- or advanced-stage pulmonary LELC.14 Through sequential blood draw, they found that plasma EBV DNA frequently preceded disease progression during posttherapy follow-up. Moreover, patients with persistently detectable plasma EBV DNA after radial resection had significantly worse OS and DFS than did those with EBV DNA after surgery.14 The above findings further supported an oncogenic role of EBV in a fraction of Asian patients of pulmonary LELC. Nevertheless, neither in vitro cellular nor animal models exist currently verifying EBV’s ability to infect and transform malignantly human lung cells. Therefore, the possible oncogenic mechanism in EBV-associated lung cancer remains unknown.
Jaagsiekte Sheep Retrovirus (JSRV)
JSRV is a known betaretrovirus capable of inducing the formation of transmissible lung cancer in sheep called ovine pulmonary adenocarcinoma (OPA).172 OPA is characterized by the multifocal mixed presentation of adenocarcinoma, with its early lesions having similarity to lepidic-predominant adenocarcinoma and its advanced lesions resembling adenocarcinoma with papillary or acinar characteristics.173 JSRV infects and transforms bronchiolar and alveolar epithelial cells, namely type II pneumocytes and club cells.174 The cellular receptor for JSRV is hyaluronidase-2 (Hyal2) and Hyal2 has been shown to mediate human cell entry of JSRV Env pseudotyped retroviral particles.175 The envelope (Env) protein of JSRV is an oncogenic protein and its carcinogenic property has been demonstrated in sheep and mice in vivo and in various cell lines in vitro, including human bronchial epithelial cells.16
Given that the capacity of JSRV to induce OPA and histological similarities between OPA and human adenocarcinoma, as well as due to the findings that human bronchial epithelial cells express Hyal2 for JSRV entry and JSRV Env protein transforms human lung epithelial cells in vitro, numerous studies have explored the role of JSRV in the induction of human lung cancer.176–183 By immunohistochemical analysis on human lung tissues using an antiserum to JSRV capsid protein, De las Heras et al revealed a positive reaction in 30% of 129 human lepidic adenocarcinomas, 26% of 65 other adenocarcinomas, and two of seven large cell carcinomas.176 By human lung cancer tissue arrays, Linnerth-Petrik et al further found the presence of JSRV Env protein by immunostaining and JSRV Env and Gag sequences by PCR in a subset of human adenocarcinomas.177 In addition, JSRV related DNA sequences have been detected in paraffin sections of lepidic adenocarcinoma specimens from lung cancer patients in Sardinia178 and in blood of Africans from Nigeria and Cameroon.179 Nonetheless, other groups failed to find evidence for JSRV Env and Gag proteins by immunostaining182 or for JSRV DNA or RNA by PCR in human adenocarcinoma.180,181 Recently, a more definitive method using a high-throughput sequencing approach was also unable to find evidence for JSRV sequences in five lepidic adenocarcinomas.183 Until now, no conclusive evidence exists regarding the link between JSRV and the development of lung adenocarcinomas in humans and much needs to be done.
John Cunningham Virus (JCV)
JCV is a member of polyomavirus family infecting a large proportion of the population worldwide and 80% to 90% of adults are seropositive.184 Recent literature reports the presence of JCV in various types of human neoplasm, including lung cancer.185 The JCV T-antigen is considered to play an important role in JCV-associated carcinogenesis.186 The T-antigen can inactivate tumor suppressor proteins, p53 and pRb, and deregulate the Wnt signaling pathway through promoting the stability and accumulation of beta-catenin via direct binding, which culminating in uncontrolled proliferation and immortal survival.187 Giuliani et al, for the first time, suggested the presence of JCV in lung tumors by showing JCV sequences were amplified in one lung carcinoma only.184 Zheng et al examined the JCV by targeting JCV T-antigen and expression of Ki-67, caspase-3, beta-catenin, p53, and pRb in 103 lung carcinomas and 18 normal lung tissues.188 In their study, the detection rate and copy number of JCV was higher in lung carcinoma than in normal lung tissue. JCV copies correlated positively with expression of Ki-67 and negatively with membrane beta-catenin expression, which suggested that lung carcinoma with high JCV copies exhibited high proliferation and downregulation of cell adhesion mediated by membrane beta-catenin. Moreover, JCV T-antigen was found in the nuclei of lung carcinoma cells and adjacent alveolar epithelial cells.188 These above findings along with previous report indicating that terminal a2,6-linked sialic acid is a critical component of the JCV receptor, which is abundantly expressed in normal lungs,189 suggested that JCV might be implicated in the malignant transformation of pulmonary epithelial cells and supported the notion that the respiratory tract might be a portal of entry for JCV infection.189 Abdel-Aziz et al explored the presence of JCV genome in 62 lung cancer and 23 normal lung tissue by targeting the T-antigen, VP, and agnoprotein.190 The JCV genome was detected in approximately half of lung cancer cases and JCV T-antigen correlated significantly with p53 and nuclear staining of beta-catenin. Sinagra et al recently investigated JCV gene sequences by targeting T-antigen in lung adenocarcinoma and its surrounding normal lung tissue.185 JCV positivity was observed in seven of 13 lung cancer tissues and none were JCV-positive for surrounding normal lung tissues. Noguchi et al generated transgenic (TG) mice with a transgene including the K-19 promoter, which was specific to bronchial and digestive epithelium and the JCV T-antigen and found pulmonary tumors in two out of 15 TG mice (13.3%) without any metastasis, suggesting possible association of JCV with bronchial tumorigenesis in experimental animals.191
Challenges in Virus-associated Cancer Research and Future Perspectives
The global health burden of viral infection in cancer is high but underappreciated. Infectious agents are estimated to be blamed for 15.4% of cancers worldwide, with most being viruses.192 Viruses have had a chequered history in cancer biology throughout the past century.5 There are several inherent characteristics of viral biology making it difficult to identify viral agents as causative factors for human cancers.193 First, most of the viruses are ubiquitous in nature, but only a small percentage of infected individuals develop cancers. Second, no human cancer arises as the acute consequence of infection. The latency period between infection and the development of a cancer make exposure markers difficult to assess along the carcinogenic process. Third, viral agents might act as indirect carcinogens, without persistence of their genes within the respective cancer cells. It remains unclear whether these viruses cause cancer solely through sustaining mature tumor cells by viral products or chronic infection and inflammation or directly through contributing to cancer cell transformation by viral oncogenes. Viral etiology research may be influenced by “hit and run” hypothesis, where the viral genes are lost as the tumor begins to mature. Fourth, species-specific barriers often limits the use of animals as surrogate hosts to study human tumor viruses.
Even so, the exploitation for viral etiology for selected solid cancers should not cease. We are now entering a more mature phase of research with the realization that a considerable proportion of cancers are indeed caused by viruses. With the advent of new sequencing technologies, it is highly probable that this proportion will increase. The discovery of cancers with an infectious origin is critical to develop antilatent viral drugs and immunological therapies. The recognition of the importance of viral cancers has already resulted in vaccines against HBV and high-risk HPV and targeted therapies against HCV and HIV, and will create more opportunities in cancer control.
Conclusion
This article highlights several important findings on lung cancer-associated oncogenic viruses. First, HPV is detected in a substantial fraction of human lung cancer tissues worldwide, with widely variable infection rates in lung cancer tissues depending on the geographical origin of the study. The association seems to be stronger in squamous cell carcinoma than in other lung cancer subtypes. Multiple oncogenic mechanisms have been proposed to play a part in HPV-infected NSCLC carcinogenesis, such as transcription of oncogenes that contribute to lung cancer cell transformation, induction of EGFR mutation, and clonal integration into the cellular genome. Furthermore, HPV might act as a cofactor of smoking exposures to facilitate lung cancer carcinogenesis. MCPyV has been put under the spotlight of searching oncogenic virus associated with lung cancer since the discovery of MCPyV being as a causative agent of MCC. Limited evidence indicates a role of MCPyV in lung cancer, especially NSCLC. Like HPV, MCPyV might also induce EGFR mutation in NSCLC, but through unknown mechanism. The association between MCPyV and lung cancer warrants further investigation. The association of EBV and lung cancer presents significant differences according to tumor histotype and geographical site. EBV is often detected in pulmonary LELC occurring in patients from east and southeast Asian and circulating EBV DNA in the plasma of patients with pulmonary LELC predicts disease recurrence. EBV may be related to pulmonary LELC, especially in east and southeast Asia, while a more general role of EBV in lung carcinogenesis seems unlikely. Although JSRV has been known to induce ovine lung adenocarcinoma through the viral envelope protein, no conclusive evidence exists related to the possible link between JSRV and the development of lung adenocarcinomas in humans so far. Compared with the above four viruses, JCV are less-studied. Obviously, more research in the future is needed to get more insight into their role in lung carcinogenesis.
Abbreviations
NSCLC, non-small-cell lung cancer; SCLC, small-cell lung cancer; EBV, Epstein–Barr virus; HBV, hepatitis B virus; HCV, hepatitis C virus; HTLV-1, human T-lymphotropic virus-1; HPV, human papillomavirus; KSHV, Kaposi’s sarcoma herpesvirus; MCPyV, Merkel cell polyomavirus; JSRV, Jaagsiekte sheep retrovirus; HSPG, heparan sulfate proteoglycans; FHIT, fragile histidine triad; LOH, loss of heterozygosity; MA, microsatellite alterations; EMT, epithelial-mesenchymal transition; ECM, extracellular matrix; TIMP, tissue inhibitor of metalloproteinase; Mcl-1, myeloid cell leukemia-1; hTERT, human telomerase reverse transcriptase; IAP, antiapoptosis proteins; CREB, cAMP response element-binding protein; 8-OH-dG, 8-hydroxy-2ʹdeoxyguanosine; CSC, cigarette smoke components; LCR, long control region; B[a]P, benzo[a]pyrene; FOXM1, foxhead box M1; MTA-1, metastasis-associated protein-1; CDK, cyclin-dependent kinase; DDX3, dead-box RNA helicase; pRb, retinoblastoma suppressor protein; HDAC, histone deacetylase; DNMT3, DNA methyltransferase; MCC, Merkel cell carcinoma; SCC, squamous cell carcinoma; AC, adenocarcinoma; LELC, lymphoepithelioma-like carcinoma; DFS, disease-free survival; OPA, ovine pulmonary adenocarcinoma; Hyal2, hyaluronidase-2; Env, envelope; JCV: John Cunningham virus.
Acknowledgments
The review was supported by National Natural Science Foundation of China (81700070 to SR), Natural Science Foundation of Hunan Province, China (2019JJ30038 to SR) and Health Department Research Fund of Hunan Province, China (B2018-0541 to SR).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68(6):394–424.
2. Molina JR, Yang P, Cassivi SD, Schild SE, Adjei AA. Non-small cell lung cancer: epidemiology, risk factors, treatment, and survivorship. Mayo Clinic Proceedings. 2008;83(5):584–594.
3. Alberg AJ, Brock MV, Ford JG, Samet JM, Spivack SD. Epidemiology of lung cancer: diagnosis and management of lung cancer, 3rd ed: american College of Chest Physicians evidence-based clinical practice guidelines. Chest. 2013;143(5Suppl):e1S–e29S.
4. Sun S, Schiller JH, Gazdar AF. Lung cancer in never smokers–a different disease. Nat Rev Cancer. 2007;7(10):778–790.
5. Moore PS, Chang Y. Why do viruses cause cancer? Highlights of the first century of human tumour virology. Nat Rev Cancer. 2010;10(12):878–889.
6. Pagano JS, Blaser M, Buendia MA, et al. Infectious agents and cancer: criteria for a causal relation. Semin Cancer Biol. 2004;14(6):453–471.
7. Xiong WM, Xu QP, Li X, Xiao RD, Cai L, He F. The association between human papillomavirus infection and lung cancer: a system review and meta-analysis. Oncotarget. 2017;8(56):96419–96432.
8. Robinson LA, Jaing CJ, Pierce Campbell C, et al. Molecular evidence of viral DNA in non-small cell lung cancer and non-neoplastic lung. Br J Cancer. 2016;115(4):497–504.
9. Hasegawa Y, Ando M, Kubo A, et al. Human papilloma virus in non-small cell lung cancer in never smokers: a systematic review of the literature. Lung Cancer. 2014;83(1):8–13.
10. Hashida Y, Imajoh M, Nemoto Y, et al. Detection of Merkel cell polyomavirus with a tumour-specific signature in non-small cell lung cancer. Br J Cancer. 2013;108(3):629–637. doi:10.1038/bjc.2012.567
11. Behdarvand A, Zamani MS, Sadeghi F, et al. Evaluation of Merkel cell polyomavirus in non-small cell lung cancer and adjacent normal cells. Microb Pathog. 2017;108:21–26. doi:10.1016/j.micpath.2017.04.033
12. Lasithiotaki I, Tsitoura E, Koutsopoulos A, et al. Aberrant expression of miR-21, miR-376c and miR-145 and their target host genes in Merkel cell polyomavirus-positive non-small cell lung cancer. Oncotarget. 2017;8(68):112371–112383. doi:10.18632/oncotarget.11222
13. Yeh Y-C, Kao H-L, Lee K-L, Wu M-H, Ho H-L, Chou T-Y. Epstein-Barr Virus–Associated Pulmonary Carcinoma. Am J Surg Pathol. 2019;43(2):211–219. doi:10.1097/PAS.0000000000001173
14. Xie M, Wu X, Wang F, et al. Clinical Significance of Plasma Epstein-Barr Virus DNA in Pulmonary Lymphoepithelioma-like Carcinoma (LELC) Patients. J Thoracic Oncol. 2018;13(2):218–227. doi:10.1016/j.jtho.2017.10.031
15. Wang S, Xiong H, Yan S, Wu N, Lu Z. Identification and Characterization of Epstein-Barr Virus Genomes in Lung Carcinoma Biopsy Samples by Next-Generation Sequencing Technology. Sci Rep. 2016;6(1):26156. doi:10.1038/srep26156
16. Monot M, Archer F, Gomes M, Mornex J-F, Leroux C. Advances in the study of transmissible respiratory tumours in small ruminants. Vet Microbiol. 2015;181(1–2):170–177. doi:10.1016/j.vetmic.2015.08.008
17. De Paoli P, Carbone A. Carcinogenic viruses and solid cancers without sufficient evidence of causal association. Int j Cancer. 2013;133(7):1517–1529.
18. Syrjanen KJ. Condylomatous changes in neoplastic bronchial epithelium. Rep Case Respiration. 1979;38(5):299–304.
19. Syrjanen K. Detection of human papillomavirus in lung cancer: systematic review and meta-analysis. Anticancer Res. 2012;32(8):3235–3250.
20. Cheng YW, Chiou HL, Sheu GT, et al. The association of human papillomavirus 16/18 infection with lung cancer among nonsmoking Taiwanese women. Cancer Res. 2001;61(7):2799–2803.
21. Nadji SA, Mokhtari-Azad T, Mahmoodi M, et al. Relationship between lung cancer and human papillomavirus in north of Iran, Mazandaran province. Cancer Lett. 2007;248(1):41–46.
22. Sarchianaki E, Derdas SP, Ntaoukakis M, et al. Detection and genotype analysis of human papillomavirus in non-small cell lung cancer patients. Tumour Biol. 2014;35(4):3203–3209.
23. Chen SP, Hsu NY, Wu JY, et al. Association of p53 codon 72 genotypes and clinical outcome in human papillomavirus-infected lung cancer patients. Ann Thorac Surg. 2013;95(4):1196–1203.
24. de Oliveira THA, do Amaral CM, de Franca Sao Marcos B, Nascimento KCG, de Miranda Rios AC, Quixabeira DCA, et al. Presence and activity of HPV in primary lung cancer. J Cancer Res Clin Oncol. 2018;144(12):2367–2376.
25. Fei Y, Yang J, Hsieh WC, et al. Different human papillomavirus 16/18 infection in Chinese non-small cell lung cancer patients living in Wuhan, China. Jpn J Clin Oncol. 2006;36(5):274–279.
26. Wang Y, Wang A, Jiang R, et al. Human papillomavirus type 16 and 18 infection is associated with lung cancer patients from the central part of China. Oncol Rep. 2008;20(2):333–339.
27. Wang YH, Chen DJ, Yi TN, Liu XH. The relationship among human papilloma virus infection, survivin, and p53 gene in lung squamous carcinoma tissue. Saudi Med J. 2010;31(12):1331–1336.
28. Yu Y, Yang A, Hu S, Zhang J, Yan H. Significance of human papillomavirus 16/18 infection in association with p53 mutation in lung carcinomas. Clin Respir J. 2013;7(1):27–33.
29. Badillo-Almaraz I, Zapata-Benavides P, Saavedra-Alonso S, et al. Human papillomavirus 16/18 infections in lung cancer patients in Mexico. Intervirology. 2013;56(5):310–315.
30. Cheng YW, Wu MF, Wang J, et al. Human papillomavirus 16/18 E6 oncoprotein is expressed in lung cancer and related with p53 inactivation. Cancer Res. 2007;67(22):10686–10693.
31. Kato T, Koriyama C, Khan N, et al. EGFR mutations and human papillomavirus in lung cancer. Lung Cancer. 2012;78(2):144–147.
32. Lin FC, Huang JY, Tsai SC, et al. The association between human papillomavirus infection and female lung cancer: A population-based cohort study. Medicine. 2016;95(23):e3856.
33. Tung MC, Wu HH, Cheng YW, et al. Association of epidermal growth factor receptor mutations with human papillomavirus 16/18 E6 oncoprotein expression in non-small cell lung cancer. Cancer. 2013;119(18):3367–3376.
34. Wang JL, Fang CL, Wang M, et al. Human papillomavirus infections as a marker to predict overall survival in lung adenocarcinoma. Int j Cancer. 2014;134(1):65–71.
35. Storey R, Joh J, Kwon A, Jenson AB, Ghim SJ, Kloecker GH. Detection of Immunoglobulin G against E7 of Human Papillomavirus in Non-Small-Cell Lung Cancer. J Oncol. 2013;2013:240164.
36. Aguayo F, Castillo A, Koriyama C, et al. Human papillomavirus-16 is integrated in lung carcinomas: a study in Chile. Br J Cancer. 2007;97(1):85–91.
37. Anantharaman D, Gheit T, Waterboer T, et al. No causal association identified for human papillomavirus infections in lung cancer. Cancer Res. 2014;74(13):3525–3534.
38. Argyri E, Tsimplaki E, Marketos C, Politis G, Panotopoulou E. Investigating the role of human papillomavirus in lung cancer. Papillomavirus Res. 2017;3:7–10.
39. van Boerdonk RA, Daniels JM, Bloemena E, et al. High-risk human papillomavirus-positive lung cancer: molecular evidence for a pattern of pulmonary metastasis. J Thoracic Oncol. 2013;8(6):711–718.
40. Galvan A, Noci S, Taverna F, et al. Testing of human papillomavirus in lung cancer and non-tumor lung tissue. BMC Cancer. 2012;12:512.
41. Chang SY, Keeney M, Law M, Donovan J, Aubry MC, Garcia J. Detection of human papillomavirus in non-small cell carcinoma of the lung. Hum Pathol. 2015;46(11):1592–1597.
42. Lim WT, Chuah KL, Leong SS, Tan EH, Toh CK. Assessment of human papillomavirus and Epstein-Barr virus in lung adenocarcinoma. Oncol Rep. 2009;21(4):971–975.
43. Silva EM, Mariano VS, Pastrez PRA, et al. Human papillomavirus is not associated to non-small cell lung cancer: data from a prospective cross-sectional study. Infect Agent Cancer. 2019;14:18.
44. Chesson HW, Dunne EF, Hariri S, Markowitz LE. The estimated lifetime probability of acquiring human papillomavirus in the United States. Sex Transm Dis. 2014;41(11):660–664.
45. Carpagnano GE, Koutelou A, Natalicchio MI, et al. HPV in exhaled breath condensate of lung cancer patients. Br J Cancer. 2011;105(8):1183–1190.
46. Chiou HL, Wu MF, Liaw YC, et al. The presence of human papillomavirus type 16/18 DNA in blood circulation may act as a risk marker of lung cancer in Taiwan. Cancer. 2003;97(6):1558–1563.
47. Bodaghi S, Wood LV, Roby G, Ryder C, Steinberg SM, Zheng ZM. Could human papillomaviruses be spread through blood? J Clin Microbiol. 2005;43(11):5428–5434.
48. Zhai K, Ding J, Shi HZ. HPV and lung cancer risk: a meta-analysis. J Clin Virology. 2015;63:84–90.
49. Mammas IN, Sourvinos G, Spandidos DA. The paediatric story of human papillomavirus (Review). Oncol Lett. 2014;8(2):502–506.
50. Gillison ML, Shah KV. Chapter 9: role of mucosal human papillomavirus in nongenital cancers. J Natl Cancer Inst Monogr. 2003;31:57–65.
51. Chen HB, Chen L, Zhang JK, et al. Human papillomavirus 16 E6 is associated with the nuclear matrix of esophageal carcinoma cells. World j Gastroenterol. 2001;7(6):788–791.
52. Syrjanen KJ. HPV infections and lung cancer. J Clin Pathol. 2002;55(12):885–891.
53. Rabkin CS, Biggar RJ, Melbye M, Curtis RE. Second primary cancers following anal and cervical carcinoma: evidence of shared etiologic factors. Am J Epidemiol. 1992;136(1):54–58.
54. Frisch M, Melbye M. Risk of lung cancer in pre- and post-menopausal women with ano-genital malignancies. Int j Cancer. 1995;62(5):508–511.
55. Li YJ, Tsai YC, Chen YC, Christiani DC. Human papilloma virus and female lung adenocarcinoma. Semin Oncol. 2009;36(6):542–552.
56. Zhou Q, Hu X, Zhou J, Zhao M, Zhu X, Zhu X. Human papillomavirus DNA in surgical smoke during cervical loop electrosurgical excision procedures and its impact on the surgeon. Cancer Manag Res. 2019;11:3643–3654.
57. Schiller JT, Day PM, Kines RC. Current understanding of the mechanism of HPV infection. Gynecol Oncol. 2010;118(1 Suppl):S12–S27.
58. Smits NC, Shworak NW, Dekhuijzen PN, van Kuppevelt TH. Heparan sulfates in the lung: structure, diversity, and role in pulmonary emphysema. Anatomical Record. 2010;293(6):955–967.
59. Kines RC, Thompson CD, Lowy DR, Schiller JT, Day PM. The initial steps leading to papillomavirus infection occur on the basement membrane prior to cell surface binding. Proc Natl Acad Sci U S A. 2009;106(48):20458–20463.
60. Hammond E, Khurana A, Shridhar V, Dredge K. The Role of Heparanase and Sulfatases in the Modification of Heparan Sulfate Proteoglycans within the Tumor Microenvironment and Opportunities for Novel Cancer Therapeutics. Front Oncol. 2014;4:195.
61. Kines RC, Cerio RJ, Roberts JN, et al. Human papillomavirus capsids preferentially bind and infect tumor cells. Int j Cancer. 2016;138(4):901–911.
62. Chen PM, Cheng YW, Wang YC, Wu TC, Chen CY, Lee H. Up-regulation of FOXM1 by E6 oncoprotein through the MZF1/NKX2-1 axis is required for human papillomavirus-associated tumorigenesis. Neoplasia. 2014;16(11):961–971.
63. Wu DW, Tsai LH, Chen PM, et al. Loss of TIMP-3 promotes tumor invasion via elevated IL-6 production and predicts poor survival and relapse in HPV-infected non-small cell lung cancer. Am J Pathol. 2012;181(5):1796–1806.
64. Hsu NY, Cheng YW, Chan IP, et al. Association between expression of human papillomavirus 16/18 E6 oncoprotein and survival in patients with stage I non-small cell lung cancer. Oncol Rep. 2009;21(1):81–87.
65. Iwamasa T, Miyagi J, Tsuhako K, et al. Prognostic implication of human papillomavirus infection in squamous cell carcinoma of the lung. Pathol Res Pract. 2000;196(4):209–218.
66. Miyagi J, Kinjo T, Tsuhako K, et al. Extremely high Langerhans cell infiltration contributes to the favourable prognosis of HPV-infected squamous cell carcinoma and adenocarcinoma of the lung. Histopathology. 2001;38(4):355–367.
67. Guo L, Liu S, Zhang S, et al. Human papillomavirus infection as a prognostic marker for lung adenocarcinoma: a systematic review and meta-analysis. Oncotarget. 2017;8(21):34507–34515.
68. Jackson AL, Zhou B, Kim WY. HIF, hypoxia and the role of angiogenesis in non-small cell lung cancer. Expert Opin Ther Targets. 2010;14(10):1047–1057.
69. Ellis LM, Hicklin DJ. VEGF-targeted therapy: mechanisms of anti-tumour activity. Nat Rev Cancer. 2008;8(8):579–591.
70. Zhang E, Feng X, Liu F, Zhang P, Liang J, Tang X. Roles of PI3K/Akt and c-Jun signaling pathways in human papillomavirus type 16 oncoprotein-induced HIF-1alpha, VEGF, and IL-8 expression and in vitro angiogenesis in non-small cell lung cancer cells. PLoS One. 2014;9(7):e103440.
71. Li G, He L, Zhang E, et al. Overexpression of human papillomavirus (HPV) type 16 oncoproteins promotes angiogenesis via enhancing HIF-1alpha and VEGF expression in non-small cell lung cancer cells. Cancer Lett. 2011;311(2):160–170.
72. Liu J, Huang B, Xiu Z, et al. PI3K/Akt/HIF-1alpha signaling pathway mediates HPV-16 oncoprotein-induced expression of EMT-related transcription factors in non-small cell lung cancer cells. J Cancer. 2018;9(19):3456–3466.
73. Gu NJ, Wu MZ, He L, et al. HPV 16 E6/E7 up-regulate the expression of both HIF-1alpha and GLUT1 by inhibition of RRAD and activation of NF-kappaB in lung cancer cells. J Cancer. 2019;10(27):6903–6909.
74. Fan R, Hou WJ, Zhao YJ, et al. Overexpression of HPV16 E6/E7 mediated HIF-1alpha upregulation of GLUT1 expression in lung cancer cells. Tumour Biol. 2016;37(4):4655–4663.
75. Joshi S, Singh AR, Zulcic M, Durden DL. A macrophage-dominant PI3K isoform controls hypoxia-induced HIF1alpha and HIF2alpha stability and tumor growth, angiogenesis, and metastasis. Mol Cancer Res. 2014;12(10):1520–1531.
76. Shao JS, Sun J, Wang S, et al. HPV16 E6/E7 upregulates HIF-2alpha and VEGF by inhibiting LKB1 in lung cancer cells. Tumour Biol. 2017;39(7):1010428317717137.
77. Kessenbrock K, Plaks V, Werb Z. Matrix metalloproteinases: regulators of the tumor microenvironment. Cell. 2010;141(1):52–67.
78. Shiau MY, Fan LC, Yang SC, et al. Human papillomavirus up-regulates MMP-2 and MMP-9 expression and activity by inducing interleukin-8 in lung adenocarcinomas. PLoS One. 2013;8(1):e54423.
79. Chetty C, Lakka SS, Bhoopathi P, Kunigal S, Geiss R, Rao JS. Tissue inhibitor of metalloproteinase 3 suppresses tumor angiogenesis in matrix metalloproteinase 2-down-regulated lung cancer. Cancer Res. 2008;68(12):4736–4745.
80. Barski D, Wolter M, Reifenberger G, Riemenschneider MJ. Hypermethylation and transcriptional downregulation of the TIMP3 gene is associated with allelic loss on 22q12.3 and malignancy in meningiomas. Brain Pathol. 2010;20(3):623–631.
81. Wild A, Langer P, Celik I, Chaloupka B, Bartsch DK. Chromosome 22q in pancreatic endocrine tumors: identification of a homozygous deletion and potential prognostic associations of allelic deletions. Eur j Endocrinol. 2002;147(4):507–513.
82. Nakamura M, Ishida E, Shimada K, et al. Frequent LOH on 22q12.3 and TIMP-3 inactivation occur in the progression to secondary glioblastomas. Lab Invest. 2005;85(2):165–175.
83. Horikawa I, Barrett JC. Transcriptional regulation of the telomerase hTERT gene as a target for cellular and viral oncogenic mechanisms. Carcinogenesis. 2003;24(7):1167–1176.
84. Lantuejoul S, Soria JC, Morat L, et al. Telomere shortening and telomerase reverse transcriptase expression in preinvasive bronchial lesions. Clin Cancer Res. 2005;11(5):2074–2082.
85. Cheng YW, Wu TC, Chen CY, Chou MC, Ko JL, Lee H. Human telomerase reverse transcriptase activated by E6 oncoprotein is required for human papillomavirus-16/18-infected lung tumorigenesis. Clin Cancer Res. 2008;14(22):7173–7179.
86. Xu D, Wang Q, Gruber A, et al. Downregulation of telomerase reverse transcriptase mRNA expression by wild type p53 in human tumor cells. Oncogene. 2000;19(45):5123–5133.
87. Yang JH, Li XY, Wang X, et al. Long-term persistent infection of HPV 16 E6 up-regulate SP1 and hTERT by inhibiting LKB1 in lung cancer cells. PLoS One. 2017;12(8):e0182775.
88. Martinez-Zapien D, Ruiz FX, Poirson J, et al. Structure of the E6/E6AP/p53 complex required for HPV-mediated degradation of p53. Nature. 2016;529(7587):541–545.
89. Zur Hausen H. Papillomaviruses and cancer: from basic studies to clinical application. Nat Rev Cancer. 2002;2(5):342–350.
90. Sherr CJ, Roberts JM. CDK inhibitors: positive and negative regulators of G1-phase progression. Genes Dev. 1999;13(12):1501–1512.
91. el-Deiry WS, Tokino T, Velculescu VE, et al. WAF1, a potential mediator of p53 tumor suppression. Cell. 1993;75(4):817–825.
92. Marchetti A, Doglioni C, Barbareschi M, et al. p21 RNA and protein expression in non-small cell lung carcinomas: evidence of p53-independent expression and association with tumoral differentiation. Oncogene. 1996;12(6):1319–1324.
93. Wu X, Bayle JH, Olson D, Levine AJ. The p53-mdm-2 autoregulatory feedback loop. Genes Dev. 1993;7(7A):1126–1132.
94. Rosner A, Rinkevich B. The DDX3 subfamily of the DEAD box helicases: divergent roles as unveiled by studying different organisms and in vitro assays. Curr Med Chem. 2007;14(23):2517–2525.
95. Wu DW, Liu WS, Wang J, Chen CY, Cheng YW, Lee H. Reduced p21(WAF1/CIP1) via alteration of p53-DDX3 pathway is associated with poor relapse-free survival in early-stage human papillomavirus-associated lung cancer. Clin Cancer Res. 2011;17(7):1895–1905.
96. Sherr CJ, McCormick F. The RB and p53 pathways in cancer. Cancer Cell. 2002;2(2):103–112.
97. Ulivi P, Zoli W, Calistri D, et al. p16INK4A and CDH13 hypermethylation in tumor and serum of non-small cell lung cancer patients. J Cell Physiol. 2006;206(3):611–615.
98. Esteller M, Sanchez-Cespedes M, Rosell R, Sidransky D, Baylin SB, Herman JG. Detection of aberrant promoter hypermethylation of tumor suppressor genes in serum DNA from non-small cell lung cancer patients. Cancer Res. 1999;59(1):67–70.
99. Finzer P, Kuntzen C, Soto U, et al. Inhibitors of histone deacetylase arrest cell cycle and induce apoptosis in cervical carcinoma cells circumventing human papillomavirus oncogene expression. Oncogene. 2001;20(35):4768–4776.
100. Wu M-F, Cheng Y-W, Lai J-C, et al. Frequent p16INK4a promoter hypermethylation in human papillomavirus-infected female lung cancer in Taiwan. Int j Cancer. 2005;113(3):440–445. doi:10.1002/ijc.20597
101. Lin T-S, Lee H, Chen R-A, et al. An association of DNMT3b protein expression with P16INK4a promoter hypermethylation in non-smoking female lung cancer with human papillomavirus infection. Cancer Lett. 2005;226(1):77–84. doi:10.1016/j.canlet.2004.12.031
102. Sozzi G, Veronese ML, Negrini M, et al. The FHIT Gene at 3p14.2 Is Abnormal in Lung Cancer. Cell. 1996;85(1):17–26. doi:10.1016/S0092-8674(00)81078-8
103. Wang J, Cheng Y-W, Wu D-W, et al. Frequent FHIT gene loss of heterozygosity in human papillomavirus-infected non-smoking female lung cancer in Taiwan. Cancer Lett. 2006;235(1):18–25. doi:10.1016/j.canlet.2005.03.058
104. Sozzi G, Pastorino U, Moiraghi L, et al. Loss of FHIT function in lung cancer and preinvasive bronchial lesions. Cancer Res. 1998;58(22):5032–5037.
105. Burke L, Khan MA, Freedman AN, et al. Allelic deletion analysis of the FHIT gene predicts poor survival in non-small cell lung cancer. Cancer Res. 1998;58(12):2533–2536.
106. Wilke CM, Hall BK, Hoge A, Paradee W, Smith DI, Glover TW. FRA3B extends over a broad region and contains a spontaneous HPV16 integration site: direct evidence for the coincidence of viral integration sites and fragile sites. Hum Mol Genet. 1996;5(2):187–195. doi:10.1093/hmg/5.2.187
107. Carpagnano GE, Lacedonia D, Crisetti E, et al. Exhaled HPV infection in lung cancer: role of MA at 3p. Arch Med Res. 2014;45(5):383–387. doi:10.1016/j.arcmed.2014.06.006
108. Yu Y, Liu X, Yang Y, et al. Effect of FHIT loss and p53 mutation on HPV-infected lung carcinoma development. Oncol Lett. 2015;10(1):392–398. doi:10.3892/ol.2015.3213
109. Verri C, Roz L, Conte D, et al. Fragile histidine triad gene inactivation in lung cancer: the European Early Lung Cancer project. Am J Respir Crit Care Med. 2009;179(5):396–401. doi:10.1164/rccm.200807-1153OC
110. Liang H, Pan Z, Cai X, et al. The association between human papillomavirus presence and epidermal growth factor receptor mutations in Asian patients with non-small cell lung cancer. Translational Lung Cancer Res. 2018;7(3):397–403. doi:10.21037/tlcr.2018.03.16
111. Baba M, Castillo A, Koriyama C, et al. Human papillomavirus is frequently detected in gefitinib-responsive lung adenocarcinomas. Oncol Rep. 2010;23(4):1085–1092.
112. Li M, Deng F, Qian L-T, et al. Association between human papillomavirus and EGFR mutations in advanced lung adenocarcinoma. Oncol Lett. 2016;12(3):1953–1958. doi:10.3892/ol.2016.4847
113. Marquez-Medina D, Gasol-Cudos A, Taberner-Bonastre MT, Samame Perez-Vargas JC, Salud-Salvia A, Llombart-Cussac A. Human papillomavirus in non-small-cell lung cancer: the impact of EGFR mutations and the response to erlotinib. Archivos de Bronconeumología (English Edition). 2013;49(2):79–81. doi:10.1016/j.arbr.2012.04.006
114. Schimmer AD. Inhibitor of apoptosis proteins: translating basic knowledge into clinical practice. Cancer Res. 2004;64(20):7183–7190. doi:10.1158/0008-5472.CAN-04-1918
115. Wu -H-H, Wu J-Y, Cheng Y-W, et al. cIAP2 Upregulated by E6 Oncoprotein via Epidermal Growth Factor Receptor/Phosphatidylinositol 3-Kinase/AKT Pathway Confers Resistance to Cisplatin in Human Papillomavirus 16/18–Infected Lung Cancer. Clin Cancer Res. 2010;16(21):5200–5210. doi:10.1158/1078-0432.CCR-10-0020
116. Kawahara A, Azuma K, Hattori S, et al. The close correlation between 8-hydroxy-2ʹ-deoxyguanosine and epidermal growth factor receptor activating mutation in non-small cell lung cancer. Hum Pathol. 2010;41(7):951–959.
117. Munoz JP, Gonzalez C, Parra B, et al. Functional interaction between human papillomavirus type 16 E6 and E7 oncoproteins and cigarette smoke components in lung epithelial cells. PLoS One. 2012;7(5):e38178. doi:10.1371/journal.pone.0038178
118. Pena N, Carrillo D, Munoz JP, et al. Tobacco smoke activates human papillomavirus 16 p97 promoter and cooperates with high-risk E6/E7 for oxidative DNA damage in lung cells. PLoS One. 2015;10(4):e0123029. doi:10.1371/journal.pone.0123029
119. Rubin H. Synergistic mechanisms in carcinogenesis by polycyclic aromatic hydrocarbons and by tobacco smoke: a bio-historical perspective with updates. Carcinogenesis. 2001;22(12):1903–1930. doi:10.1093/carcin/22.12.1903
120. Alam S, Conway MJ, Chen H-S, Meyers C. The cigarette smoke carcinogen benzo[a]pyrene enhances human papillomavirus synthesis. J Virol. 2008;82(2):1053–1058. doi:10.1128/JVI.01813-07
121. Lee M-N, Tseng R-C, Hsu H-S, et al. Epigenetic inactivation of the chromosomal stability control genes BRCA1, BRCA2, and XRCC5 in non-small cell lung cancer. Clin Cancer Res. 2007;13(3):832–838. doi:10.1158/1078-0432.CCR-05-2694
122. Johansson F. A method to monitor replication fork progression in mammalian cells: nucleotide excision repair enhances and homologous recombination delays elongation along damaged DNA. Nucleic Acids Res. 2004;32(20):e157. doi:10.1093/nar/gnh154
123. Cheng Y-W, Lin FC-F, Chen C-Y, Hsu N-Y. Environmental exposure and HPV infection may act synergistically to induce lung tumorigenesis in nonsmokers. Oncotarget. 2016;7(15):19850–19862. doi:10.18632/oncotarget.7628
124. De Blasio A, Vento R, Di Fiore R. Mcl-1 targeting could be an intriguing perspective to cure cancer. J Cell Physiol. 2018;233(11):8482–8498. doi:10.1002/jcp.26786
125. Chang Y-H, Yu C-W, Lai L-C, et al. Up-regulation of interleukin-17 expression by human papillomavirus type 16 E6 in nonsmall cell lung cancer. Cancer. 2010;116(20):4800–4809. doi:10.1002/cncr.25224
126. Cheng Y-W, Lee H, Shiau M-Y, Wu T-C, Huang -T-T, Chang Y-H. Human papillomavirus type 16/18 up-regulates the expression of interleukin-6 and antiapoptotic Mcl-1 in non-small cell lung cancer. Clin Cancer Res. 2008;14(15):4705–4712. doi:10.1158/1078-0432.CCR-07-4675
127. Zhang J, Zhang J, Cui X, et al. FoxM1: a novel tumor biomarker of lung cancer. Int J Clin Exp Med. 2015;8(3):3136–3140.
128. Luscher-Firzlaff JM, Westendorf JM, Zwicker J, et al. Interaction of the fork head domain transcription factor MPP2 with the human papilloma virus 16 E7 protein: enhancement of transformation and transactivation. Oncogene. 1999;18(41):5620–5630. doi:10.1038/sj.onc.1202967
129. Xia Y, Chen Q, Zhong Z, et al. Down-regulation of miR-30c promotes the invasion of non-small cell lung cancer by targeting MTA1. Cell Physiol Biochem. 2013;32(2):476–485. doi:10.1159/000354452
130. Wu Y-L, Hsu N-Y, Cheau-Feng Lin F, Lee H, Cheng Y-W. MiR-30c-2* negative regulated MTA-1 expression involved in metastasis and drug resistance of HPV-infected non-small cell lung cancer. Surgery. 2016;160(6):1591–1598. doi:10.1016/j.surg.2016.06.025
131. Christensen ND, Budgeon LR, Cladel NM, Hu J. Recent advances in preclinical model systems for papillomaviruses. Virus Res. 2017;231:108–118. doi:10.1016/j.virusres.2016.12.004
132. Carraresi L, Tripodi SA, Mulder LC, et al. Thymic hyperplasia and lung carcinomas in a line of mice transgenic for keratin 5-driven HPV16 E6/E7 oncogenes. Oncogene. 2001;20(56):8148–8153. doi:10.1038/sj.onc.1205007
133. Carraresi L, Martinelli R, Vannoni A, et al. Establishment and characterization of murine small cell lung carcinoma cell lines derived from HPV-16 E6/E7 transgenic mice. Cancer Lett. 2006;231(1):65–73. doi:10.1016/j.canlet.2005.01.027
134. Vonsky MS, Runov AL, Gordeychuk IV, Isaguliants MG. Therapeutic Vaccines Against Human Papilloma Viruses: achievements and Prospects. Biochemistry (Moscow). 2019;84(7):800–816. doi:10.1134/S0006297919070101
135. Feng H, Shuda M, Chang Y, Moore PS. Clonal integration of a polyomavirus in human Merkel cell carcinoma. Science. 2008;319(5866):1096–1100.
136. Pantulu ND, Pallasch CP, Kurz AK, et al. Detection of a novel truncating Merkel cell polyomavirus large T antigen deletion in chronic lymphocytic leukemia cells. Blood. 2010;116(24):5280–5284. doi:10.1182/blood-2010-02-269829
137. Dworkin AM, Tseng SY, Allain DC, Iwenofu OH, Peters SB, Toland AE. Merkel cell polyomavirus in cutaneous squamous cell carcinoma of immunocompetent individuals. J Invest Dermatol. 2009;129(12):2868–2874. doi:10.1038/jid.2009.183
138. Imajoh M, Hashida Y, Nemoto Y, et al. Detection of Merkel cell polyomavirus in cervical squamous cell carcinomas and adenocarcinomas from Japanese patients. Virol J. 2012;9(1):154. doi:10.1186/1743-422X-9-154
139. Sadeghi F, Salehi-Vaziri M, Alizadeh A, et al. Detection of Merkel cell polyomavirus large T-antigen sequences in human central nervous system tumors. J Med Virol. 2015;87(7):1241–1247. doi:10.1002/jmv.24178
140. Schowalter RM, Pastrana DV, Pumphrey KA, Moyer AL, Buck CB. Merkel cell polyomavirus and two previously unknown polyomaviruses are chronically shed from human skin. Cell Host Microbe. 2010;7(6):509–515. doi:10.1016/j.chom.2010.05.006
141. Babakir-Mina M, Ciccozzi M, Lo Presti A, Greco F, Perno CF, Ciotti M. Identification of Merkel cell polyomavirus in the lower respiratory tract of Italian patients. J Med Virol. 2010;82(3):505–509. doi:10.1002/jmv.21711
142. Mancuso G, Antona J, Sirini C, et al. Frequent detection of Merkel cell polyomavirus DNA in tissues from 10 consecutive autopsies. J Gen Virol. 2017;98(6):1372–1376. doi:10.1099/jgv.0.000778
143. van der Heijden HFM, Heijdra YF. Extrapulmonary small cell carcinoma. South Med J. 2005;98(3):345–349. doi:10.1097/01.SMJ.0000145724.40477.50
144. Pulitzer MP, Amin BD, Busam KJ. Merkel cell carcinoma: review. Adv Anat Pathol. 2009;16(3):135–144. doi:10.1097/PAP.0b013e3181a12f5a
145. Andres C, Ihrler S, Puchta U, Flaig MJ. Merkel cell polyomavirus is prevalent in a subset of small cell lung cancer: a study of 31 patients. Thorax. 2009;64(11):1007–1008. doi:10.1136/thx.2009.117911
146. Helmbold P, Lahtz C, Herpel E, Schnabel PA, Dammann RH. Frequent hypermethylation of RASSF1A tumour suppressor gene promoter and presence of Merkel cell polyomavirus in small cell lung cancer. Eur J Cancer. 2009;45(12):2207–2211. doi:10.1016/j.ejca.2009.04.038
147. Wetzels CTAH, Hoefnagel JGM, Bakkers JMJE, Dijkman HBPM, Blokx WAM, Melchers WJG. Ultrastructural proof of polyomavirus in Merkel cell carcinoma tumour cells and its absence in small cell carcinoma of the lung. PLoS One. 2009;4(3):e4958. doi:10.1371/journal.pone.0004958
148. Karimi S, Yousefi F, Seifi S, Khosravi A, Nadji SA. No evidence for a role of Merkel cell polyomavirus in small cell lung cancer among Iranian subjects. Pathology - Research and Practice. 2014;210(12):836–839. doi:10.1016/j.prp.2014.08.011
149. Lasithiotaki I, Antoniou KM, Derdas SP, et al. The presence of Merkel cell polyomavirus is associated with deregulated expression of BRAF and Bcl-2 genes in non-small cell lung cancer. Int j Cancer. 2013;133(3):604–611. doi:10.1002/ijc.28062
150. Joh J, Jenson AB, Moore GD, et al. Human papillomavirus (HPV) and Merkel cell polyomavirus (MCPyV) in non small cell lung cancer. Exp Mol Pathol. 2010;89(3):222–226. doi:10.1016/j.yexmp.2010.08.001
151. Gheit T, Munoz JP, Levican J, et al. Merkel cell polyomavirus in non-small cell lung carcinomas from Chile. Exp Mol Pathol. 2012;93(1):162–166. doi:10.1016/j.yexmp.2012.04.008
152. Xu S, Jiang J, Yu X, Sheng D, Zhu T, Jin M. Association of Merkel cell polyomavirus infection with EGFR mutation status in Chinese non-small cell lung cancer patients. Lung Cancer. 2014;83(3):341–346.
153. Kim G-J, Lee J-H, Lee DH. Clinical and prognostic significance of Merkel cell polyomavirus in nonsmall cell lung cancer. Medicine. 2017;96(3):e5413. doi:10.1097/MD.0000000000005413
154. Becker M, Dominguez M, Greune L, et al. Infectious Entry of Merkel Cell Polyomavirus. J Virol. 2019;93(6):6. doi:10.1128/JVI.02004-18
155. Samimi M, Kervarrec T, Touze A. Immunobiology of Merkel cell carcinoma. Curr Opin Oncol. 2020;32(2):114–121. doi:10.1097/CCO.0000000000000608
156. Paik PK, Arcila ME, Fara M, et al. Clinical characteristics of patients with lung adenocarcinomas harboring BRAF mutations. J clin oncol. 2011;29(15):2046–2051.
157. Brambilla E, Negoescu A, Gazzeri S, et al. Apoptosis-related factors p53, Bcl2, and Bax in neuroendocrine lung tumors. Am J Pathol. 1996;149(6):1941–1952.
158. Chang CM, Yu KJ, Mbulaiteye SM, Hildesheim A, Bhatia K. The extent of genetic diversity of Epstein-Barr virus and its geographic and disease patterns: a need for reappraisal. Virus Res. 2009;143(2):209–221. doi:10.1016/j.virusres.2009.07.005
159. Deyrup AT. Epstein-Barr virus–associated epithelial and mesenchymal neoplasms. Hum Pathol. 2008;39(4):473–483. doi:10.1016/j.humpath.2007.10.030
160. Kheir F, Zhao M, Strong MJ, et al. Detection of Epstein-Barr Virus Infection in Non-Small Cell Lung Cancer. Cancers. 2019;11(6):6. doi:10.3390/cancers11060759
161. Lin Z, Situ D, Chang X, et al. Surgical treatment for primary pulmonary lymphoepithelioma-like carcinoma. Interact Cardiovasc Thorac Surg. 2016;23(1):41–46.
162. Liang Y, Wang L, Zhu Y, et al. Primary pulmonary lymphoepithelioma-like carcinoma: fifty-two patients with long-term follow-up. Cancer. 2012;118(19):4748–4758. doi:10.1002/cncr.27452
163. Chang Y-L, Wu C-T, Shih J-Y, Lee Y-C. Unique p53 and epidermal growth factor receptor gene mutation status in 46 pulmonary lymphoepithelioma-like carcinomas. Cancer Sci. 2011;102(1):282–287. doi:10.1111/j.1349-7006.2010.01768.x
164. Chen Y-P, Chan ATC, Le Q-T, Blanchard P, Sun Y, Ma J. Nasopharyngeal carcinoma. Lancet. 2019;394(10192):64–80. doi:10.1016/S0140-6736(19)30956-0
165. Chu PG, Cerilli L, Chen -Y-Y, Mills SE, Weiss LM. Epstein–Barr virus plays no role in the tumorigenesis of small-cell carcinoma of the lung. Modern Pathol. 2004;17(2):158–164. doi:10.1038/modpathol.3800024
166. Gomez-Roman JJ, Martinez MN, Fernandez SL, Val-Bernal JF. Epstein–Barr virus-associated adenocarcinomas and squamous-cell lung carcinomas. Modern Pathol. 2009;22(4):530–537. doi:10.1038/modpathol.2009.7
167. Tanner J, Weis J, Fearon D, Whang Y, Kieff E. Epstein-Barr virus gp350/220 binding to the B lymphocyte C3d receptor mediates adsorption, capping, and endocytosis. Cell. 1987;50(2):203–213.
168. Timens W, Boes A, Vos H, Poppema S. Tissue distribution of the C3d/EBV-receptor: CD21 monoclonal antibodies reactive with a variety of epithelial cells, medullary thymocytes, and peripheral T-cells. Histochemistry. 1991;95(6):605–611.
169. Malizia AP, Egan JJ, Doran PPIL-4. increases CD21-dependent infection of pulmonary alveolar epithelial type II cells by EBV. Mol Immunol. 2009;46(8–9):1905–1910.
170. Ngan RK, Yip TT, Cheng WW, et al. Clinical role of circulating Epstein-Barr virus DNA as a tumor marker in lymphoepithelioma-like carcinoma of the lung. Ann N Y Acad Sci. 2004;1022:263–270.
171. Ngan RK, Yip TT, Cheng WW, et al. Circulating Epstein-Barr virus DNA in serum of patients with lymphoepithelioma-like carcinoma of the lung: a potential surrogate marker for monitoring disease. Clin Cancer Res. 2002;8(4):986–994.
172. Gray ME, Meehan J, Sullivan P, et al. Ovine Pulmonary Adenocarcinoma: A Unique Model to Improve Lung Cancer Research. Front Oncol. 2019;9:335.
173. Youssef G, Wallace WA, Dagleish MP, Cousens C, Griffiths DJ. Ovine pulmonary adenocarcinoma: a large animal model for human lung cancer. ILAR j. 2015;56(1):99–115.
174. Martineau HM, Cousens C, Imlach S, Dagleish MP, Griffiths DJ. Jaagsiekte sheep retrovirus infects multiple cell types in the ovine lung. J Virol. 2011;85(7):3341–3355.
175. Rai SK, Duh FM, Vigdorovich V, Danilkovitch-Miagkova A, Lerman MI, Miller AD. Candidate tumor suppressor HYAL2 is a glycosylphosphatidylinositol (GPI)-anchored cell-surface receptor for jaagsiekte sheep retrovirus, the envelope protein of which mediates oncogenic transformation. Proc Natl Acad Sci U S A. 2001;98(8):4443–4448.
176. De Las Heras M, Barsky SH, Hasleton P, et al. Evidence for a protein related immunologically to the jaagsiekte sheep retrovirus in some human lung tumours. Eur Respir J. 2000;16(2):330–332.
177. Linnerth-Petrik NM, Walsh SR, Bogner PN, Morrison C, Wootton SK. Jaagsiekte sheep retrovirus detected in human lung cancer tissue arrays. BMC Res Notes. 2014;7:160.
178. Rocca S, Sanna MP, Leoni A, et al. Presence of Jaagsiekte sheep retrovirus in tissue sections from human bronchioloalveolar carcinoma depends on patients’ geographical origin. Hum Pathol. 2008;39(2):303–304.
179. Morozov VA, Lagaye S, Lower J, Lower R. Detection and characterization of betaretroviral sequences, related to sheep Jaagsiekte virus, in Africans from Nigeria and Cameroon.. Virology. 2004;327(2):162–168.
180. Yousem SA, Finkelstein SD, Swalsky PA, Bakker A, Ohori NP. Absence of jaagsiekte sheep retrovirus DNA and RNA in bronchioloalveolar and conventional human pulmonary adenocarcinoma by PCR and RT-PCR analysis. Hum Pathol. 2001;32(10):1039–1042.
181. Hiatt KM, Highsmith WE. Lack of DNA evidence for jaagsiekte sheep retrovirus in human bronchioloalveolar carcinoma. Hum Pathol. 2002;33(6):680.
182. Miller AD, De Las Heras M, Yu J, et al. Evidence against a role for jaagsiekte sheep retrovirus in human lung cancer. Retrovirology. 2017;14(1):3.
183. Berthet N, Frangeul L, Olaussen KA, et al. No evidence for viral sequences in five lepidic adenocarcinomas (former “BAC”) by a high-throughput sequencing approach. BMC Res Notes. 2015;8:782.
184. Giuliani L, Jaxmar T, Casadio C, et al. Detection of oncogenic viruses SV40, BKV, JCV, HCMV, HPV and p53 codon 72 polymorphism in lung carcinoma. Lung Cancer. 2007;57(3):273–281.
185. Sinagra E, Raimondo D, Gallo E, et al. JC Virus and Lung Adenocarcinoma: fact or Myth? Anticancer Res. 2017;37(6):3311–3314.
186. White MK, Khalili K. Expression of JC virus regulatory proteins in human cancer: potential mechanisms for tumourigenesis. Eur J Cancer. 2005;41(16):2537–2548.
187. Khalili K, Del Valle L, Otte J, Weaver M, Gordon J. Human neurotropic polyomavirus, JCV, and its role in carcinogenesis. Oncogene. 2003;22(33):5181–5191.
188. Zheng H, Abdel Aziz HO, Nakanishi Y, et al. Oncogenic role of JC virus in lung cancer. J Pathol. 2007;212(3):306–315.
189. Eash S, Tavares R, Stopa EG, Robbins SH, Brossay L, Atwood WJ. Differential distribution of the JC virus receptor-type sialic acid in normal human tissues. Am J Pathol. 2004;164(2):419–428.
190. Abdel-Aziz HO, Murai Y, Hong M, et al. Detection of the JC virus genome in lung cancers: possible role of the T-antigen in lung oncogenesis. Applied Immunohistochemistry Mol Morphol. 2007;15(4):394–400.
191. Noguchi A, Kikuchi K, Ohtsu T, et al. Pulmonary tumors associated with the JC virus T-antigen in a transgenic mouse model. Oncol Rep. 2013;30(6):2603–2608.
192. Plummer M, de Martel C, Vignat J, Ferlay J, Bray F, Franceschi S. Global burden of cancers attributable to infections in 2012: a synthetic analysis. Lancet Glob Health. 2016;4(9):E609–E16.
193. Zur Hausen H. The search for infectious causes of human cancers: where and why. Virology. 2009;392(1):1–10.
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.