Back to Journals » Drug Design, Development and Therapy » Volume 20
Population Pharmacokinetics and Model-Informed Precision Dosing of Lenalidomide Incorporating Total and Unbound Plasma Concentrations in Renally Impaired Patients with Multiple Myeloma
Authors Kim H ![]() , Park SS, Choi S
, Park SS, Choi S ![]() , Han S
, Han S ![]() , Han S
, Han S ![]() , Lee Y, Lee JH, Kim D, Kim H, Yang S, Shugg T, Chung EK
, Lee Y, Lee JH, Kim D, Kim H, Yang S, Shugg T, Chung EK ![]()
Received 27 February 2026
Accepted for publication 8 May 2026
Published 15 May 2026 Volume 2026:20 605741
DOI https://doi.org/10.2147/DDDT.S605741
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Anastasios Lymperopoulos
Hyeonji Kim,1– 3 Sung-Soo Park,4,* Suein Choi,5 Seunghoon Han,5 Sungpil Han,5 Yunbeom Lee,6 Ji Hyun Lee,6,7 Donghyun Kim,1– 3 Hyeonsu Kim,1– 3 Seungwon Yang,1– 3 Tyler Shugg,8 Eun Kyoung Chung1– 3,9,10,*
1Department of Pharmacy, College of Pharmacy, Kyung Hee University, Seoul, Republic of Korea; 2Department of Regulatory Science, Graduate School, Kyung Hee University, Seoul, Republic of Korea; 3Institute of Regulatory Innovation Through Science, Kyung Hee University, Seoul, Republic of Korea; 4Hematology Hospital, Seoul St. Mary’s Hospital, The Catholic University of Korea, Seoul, Republic of Korea; 5Department of Pharmacology, College of Medicine, The Catholic University of Korea, Seoul, Republic of Korea; 6Department of Biomedical Science and Technology, Kyung Hee University, Seoul, Republic of Korea; 7Department of Clinical Pharmacology and Therapeutics, College of Medicine, Kyung Hee University, Seoul, Republic of Korea; 8Division of Clinical Pharmacology, Department of Medicine, Indiana University School of Medicine, Indianapolis, IN, USA; 9Department of Pharmacy, Kyung Hee University Hospital at Gangdong, Seoul, Republic of Korea; 10Institute of Integrated Pharmaceutical Sciences, Kyung Hee University, Seoul, Republic of Korea
*These authors contributed equally to this work
Correspondence: Eun Kyoung Chung, Department of Pharmacy, College of Pharmacy, Kyung Hee University, 26 Kyungheedae-ro, Dongdaemun-gu, Seoul, Republic of Korea, Tel +82-2-961-2122, Fax +82-2-961-9580, Email [email protected]
Purpose: To develop a population pharmacokinetic (PK) model of lenalidomide and propose model-informed precision dosing based on total and unbound concentrations in multiple myeloma (MM) patients with moderate-to-severe renal impairment (RI).
Patients and Methods: Thirty MM patients orally receiving lenalidomide were prospectively enrolled with the following stratification based on renal function: 11 with an estimated glomerular filtration rate [eGFR] < 30 mL/min/1.73 m2, 11 with eGFR ≥ 30 to < 60 mL/min/1.73 m2, and 8 with eGFR ≥ 60 mL/min/1.73 m2. Serial plasma samples were collected at steady state. Total and unbound concentrations were analyzed simultaneously by population PK modeling using NONMEM. Monte Carlo simulations were performed to identify dosing regimens targeting the reference systemic exposure (area under the plasma concentration-time curve, AUC) for patients with normal renal function receiving 25 mg/day.
Results: A two-compartment model with linear protein binding best described both total and unbound plasma lenalidomide concentration-time data. Apparent unbound clearance (CL/F) increased with higher eGFR and body surface area (BSA), while apparent central volume of distribution (V1/F) increased with higher BSA (P < 0.05). No other PK parameters were significantly associated with patient characteristics including ABCB1 genotypes. Simulation results indicated that although labeled dosing was generally adequate, BSA-stratified adjustments offered improved precision. Specifically, dose reduction is recommended for patients with BSA < 1.6 m2 to maintain therapeutic exposure while minimizing toxicity; conversely, dose escalation is warranted for patients with BSA ≥ 2.0 m2 to prevent underexposure.
Conclusion: Our model identifies eGFR and BSA as critical covariates for precision dosing of lenalidomide to optimize therapeutic exposure in MM patients with RI. The infographic illustrates the population pharmacokinetics and model-informed precision dosing of lenalidomide for patients with multiple myeloma and renal impairment. It begins with a prospectively conducted clinical study in patients receiving lenalidomide, followed by simultaneous measurement of total and unbound plasma concentrations and genetic analysis. These data were used in population pharmacokinetic modeling and simulation. The lower section shows that apparent unbound clearance (CL/F) was significantly influenced by eGFR and body surface area (BSA), while apparent central volume of distribution (V₁/F) was associated with BSA, and the unbound fraction (Fu) was estimated at 61.4%. Based on the final model, virtual patient populations were generated across renal function and BSA strata, and Monte Carlo simulations were performed to evaluate drug exposure. The simulation results guide dosing adjustments based on BSA, indicating dose reduction for BSA less than 1.6 m² and dose escalation for BSA greater than or equal to 2.0 m².Lenalidomide dosing for renally impaired multiple myeloma patients using population pharmacokinetics.
Keywords: nonlinear mixed-effects modeling, personalized medicine, protein binding, Monte Carlo simulation, estimated glomerular filtration rate, body surface area
Introduction
Multiple myeloma (MM) is a hematologic malignancy primarily affecting elderly patients, and its incidence and prevalence are expected to increase with the aging population.1,2 The disease is characterized by malignant plasma cells that overproduce monoclonal immunoglobulins (M-proteins) detectable in serum or urine, leading to systemic organ damage. These clinical manifestations are commonly summarized by the CRAB features: hypercalcemia, renal impairment (RI), anemia, and bone lesions.3,4 Among these complications, RI is particularly significant for patient outcomes; up to 50% of MM patients present with RI at diagnosis, and RI represents one of the leading causes of death in MM.5 In patients with RI, excessive urinary loss of plasma proteins may occur, and elevated interleukin-6 (IL-6) levels in MM correlate with worsening renal function.6–8 These factors substantially increase the risk of hypoalbuminemia in MM patients, a condition associated with inferior clinical outcomes.9
Lenalidomide, a 4-amino-glutamyl analogue of thalidomide, is a second-generation immunomodulatory drug that retains potent anti-myeloma activity without neurologic toxicity.10 It is an oral anticancer agent rapidly absorbed (time to maximum concentration, Tmax 0.5–6 hours) with high bioavailability (>90%); the rate and extent of absorption can be reduced by high-fat food.11 Both the area under the plasma concentration-time curve (AUC) and maximum concentration (Cmax) increase proportionally with the administered dose, reflecting the linear pharmacokinetics (PK) of lenalidomide.12 It is widely used as monotherapy or combination therapy for newly diagnosed patients and those experiencing MM recurrence. In patients with RI, lenalidomide demonstrates effectiveness in treating myeloma and improving renal dysfunction within a few months. As lenalidomide is predominantly eliminated as unchanged drug in urine (~90% of dose), dosing adjustment is necessary based on renal function; however, limited data are available for patients with severe RI or receiving renal replacement therapy, necessitating careful hematologic monitoring.13,14 Even with renally adjusted dosing, considerable interindividual variability in systemic lenalidomide exposure has been reported in renally impaired patients with MM.15 This variability raises concerns about unexpected toxicities or treatment failure, highlighting the need for comprehensive population PK studies that consider various factors influencing PK parameters. In addition to renal function, genetic factors may substantially contribute to interpatient PK variability of lenalidomide. As lenalidomide is a known weak substrate for the P-glycoprotein (P-gp), several studies have linked genetic polymorphisms of ATP-binding cassette subfamily B member 1 (ABCB1) gene, encoding P-gp, to altered lenalidomide exposures.15–17 However, conflicting evidence suggests no significant association between ABCB1 variants and treatment outcomes; consequently, the clinical significance of these findings remains controversial.18 Furthermore, although body surface area (BSA) has been reported to influence PK of lenalidomide in a previous study, it is not reflected in current approved labeling.12,19 Given the high prevalence of elderly patients in MM and their substantial BSA variability, BSA should be considered alongside renal function for model-informed precision dosing.20 Additionally, lenalidomide PK may be further altered through changes in protein binding, resulting from hypoalbuminemia, which is frequently observed in MM patients with RI. Elevated IL-6 in MM suppresses hepatic albumin synthesis, while RI contributes additional reductions through impaired excretion.21 Beyond lower albumin levels, alterations in its binding properties may also occur. In MM, albumin can form complexes with dimeric immunoglobulin A and may undergo oxidative modifications, processes implicated in MM-related complications including nephropathy.22,23 Because albumin exhibits specific thermodynamic interactions with lenalidomide, such alterations could influence the unbound fraction of lenalidomide and thereby modify its PK behavior and therapeutic response.6,7,24 Despite these considerations, no population PK studies have comprehensively evaluated the effects of various patient factors on lenalidomide PK, particularly focusing on unbound lenalidomide concentrations in this clinical setting.
Previous population PK analyses of lenalidomide in MM have measured only total plasma concentrations.16,19,25,26 However, relying solely on total concentrations may be insufficient to adequately reflect the real-world clinical characteristics of this population. Since the unbound fraction exerts pharmacological and toxicological effects, evaluating unbound concentrations enables a more accurate assessment of clinically relevant exposure.27 Moreover, previous studies either did not include patients with severe RI or enrolled only a limited number of such patients. Although renal function was frequently identified as a significant covariate, applying established models to patients with severe RI relies on extrapolation beyond the observed data range. This lack of validation across the full spectrum of renal function precludes the development of model-informed precision dosing regimens. While several PK studies have demonstrated the safety and feasibility of lenalidomide in patients with RI, these investigations were not population PK-based analyses.28,29 Consequently, they could not quantitatively consider interindividual variability or support simulations that incorporate patient-specific covariates, thereby limiting their applicability for precision dosing. Therefore, the objective of this study was to characterize the population PK of both total and unbound lenalidomide concentrations in MM patients with moderate-to-severe RI. We investigated genetic polymorphisms and protein binding as potential contributors to interpatient variability and developed evidence-based dosing recommendations to optimize lenalidomide therapy in MM patients with varying degrees of RI.
Methods
Patients and Study Design
A prospective, open-label, population PK study was conducted in the Department of Hemato-oncology at St. Mary’s Hospital in Republic of Korea. The protocol was approved by the Institutional Review Board (approval date: 29 October 2024; approval number: KC24MISK0219). All procedures were performed in compliance with relevant laws and institutional guidelines. The written informed consent was obtained from all participants prior to initiating any study procedures.
Eligible patients were adults (aged >19 years) diagnosed with MM who had not received lenalidomide before participating in the study. All patients were anticipated to receive lenalidomide as a part of long-term maintenance treatment regimen for MM. Patients were excluded if they had smoldering MM, active plasma cell leukemia, monoclonal gammopathy of undetermined significance (MGUS), light chain amyloidosis, or central nervous system involvement. Pregnant or breastfeeding women, individuals with comorbid psychiatric conditions that could interfere with study compliance, and patients with confirmed diagnosis of HIV or viral hepatitis were also excluded. Patients were stratified based on their renal function: estimated glomerular filtration rate (eGFR) ≥ 60 mL/min/1.73 m2 (ie, control), 30 mL/min/1.73 m2 to < 60 mL/min/1.73 m2 (ie, moderate RI), and < 30 mL/min/1.73 m2 (ie, severe RI).
Lenalidomide was administered orally on days 1 to 28 of each treatment cycle. Dosing was adjusted based on renal function and the discretion of treating clinicians. Participants were instructed to fast for at least 10 hours before drug intake and to continue fasting for 4 hours post-dose during the PK study. Concomitant medications, including bisphosphonates and proton pump inhibitors, were allowed if clinically indicated. Prophylactic antibiotics and antivirals were used following routine clinical practice; however, their use was restricted on PK sampling days.
Serial blood samples were obtained on the third day of the first cycle of lenalidomide-based anti-cancer therapy. Sampling time points were at pre-dose (0 h) and 2, 4, 8, 12, and 24 hours after oral administration. At each sampling time, 4 mL of blood was collected into EDTA-K2 tubes to prevent coagulation. For the pre-dose sample, 3.5 mL was allocated for PK analysis and 0.5 mL for genotyping; subsequent samples were used exclusively for PK analysis. Samples for genetic analysis were stored at −70°C without centrifugation. For PK analysis, plasma was isolated by centrifuging whole blood at 20,000× g for 10 minutes at 4°C. Plasma aliquots were stored at −70°C and shipped on dry ice to the Kyung Hee Drug Analysis Center for quantification of total and unbound lenalidomide concentrations.
Determination of Total and Unbound Lenalidomide Concentrations
Quantification of total and unbound lenalidomide concentrations in plasma was conducted using a previously validated high-performance liquid chromatography-tandem mass spectrometry (HPLC-MS/MS) method.30 For the analysis of the unbound form, plasma ultrafiltrate was obtained by separating protein-bound fractions via centrifugal ultrafiltration using Amicon® Ultra-0.5 Centrifugal Filter Devices (Sigma-Aldrich; Steinheim, Germany). Chromatographic separation was achieved using a Halo® C18 column with lenalidomide-d5 as the internal standard and a mobile phase consisting of 0.1% formic acid and methanol in a 20:80 (v/v) ratio. The mass spectrometric detection was performed using positive electrospray ionization (ESI) in multiple reaction monitoring (MRM) mode. The lower limit of quantification (LLOQ) for both total and unbound lenalidomide was determined to be 5 ng/mL, with a signal-to-noise (S/N) ratio of ≥10, confirming adequate sensitivity and selectivity. Calibration curves were constructed over a range of 5–1,000 ng/mL using 1/x2-weighted linear regression (R2 ≥ 0.999). Matrix effects, evaluated using six different sources of human blank plasma, ranged from 99.69% to 106.82% for total and 95.81% to 100.04% for unbound lenalidomide. The corresponding coefficients of variation (CV) were 1.62% and 2.16%, respectively, indicating no significant ion suppression or enhancement. The analytical method demonstrated excellent accuracy and precision for both total and unbound lenalidomide: intra-assay bias (%) and precision (%CV) below 7.86% and 5.01%, and inter-assay bias and precision below 6.05% and 10.55%, respectively. The method was fully compliant with FDA and MFDS bioanalytical method validation guidance.
Non-Compartmental Analysis
Non-compartmental analysis (NCA) was performed to estimate initial PK parameters for building the population PK model of lenalidomide. NCA was conducted using Phoenix WinNonlin software (version 8.6.0, CertaraTM, Princeton, NJ, USA). From total and unbound concentration-time data, the following non-compartmental PK parameters were estimated: Cmax, Tmax, steady-state AUC over the 24-h dosing interval (AUC24; calculated by linear trapezoidal linear/log interpolation method), T1/2, apparent clearance (CL/F), and apparent volume of distribution during the terminal phase (Vz/F). The elimination rate constant (λz) was estimated from the terminal log-linear phase, with T1/2 = 0.693/λz. To evaluate concentration-dependent protein binding, linear regression analysis was performed to assess the relationship between total plasma lenalidomide concentration and unbound fraction.
Genetic Analysis
Genotypic analysis was performed to explore the association between genetic variations and lenalidomide PK, using residual pre-dose blood samples. Targeted next-generation sequencing (NGS) was employed as the genetic testing platform, utilizing a liquid-phase hybridization-based target capture panel (Celemics, Seoul, Republic of Korea). Detailed methods for genomic DNA library preparation, sequencing on the Illumina NextSeq 500 system, and the bioinformatics pipeline for variant calling to generate VCF files are provided in Supplementary Method 1. Specifically, genotyping focused on ABCB1 variants previously reported to affect drug response: 1236C>T (rs1128503), 2677G>T/A (rs2032582), and 3435C>T (rs1045642). Lenalidomide PK parameters calculated by NCA, ie, Cmax, Tmax, AUC24, and T1/2, were tested against each single nucleotide polymorphism (SNP) using the Mann–Whitney U-test. Genotypes were categorized into two or three groups according to their assumed effect on the phenotype; associations were evaluated under dominant, recessive, and additive genetic models. The Benjamini–Hochberg procedure, a multiple comparison correction method, was applied to control the False Discovery Rate (FDR). An FDR-adjusted p-value (q-value) < 0.05 was considered statistically significant.
Population Pharmacokinetic Analysis
Nonlinear mixed effect modeling (NONMEM) approach was used as a population PK analysis method to estimate PK parameters by assessing variability in lenalidomide concentrations in plasma.31 Population PK modeling for total and unbound concentrations of lenalidomide was simultaneously conducted with NONMEM software (version 7.5.1; ICON, Ellicott City, MD, USA) using second-order conditional estimation with interaction (Laplace).32 The interindividual variability (IIV, η) of the PK parameters was described with an exponential function, assuming the value of η following a log-normal distribution with a mean of zero and variance of ω2. The residual variability (ε) was evaluated using the additive, proportional, or combined additive-proportional error model for total and unbound concentrations, respectively, under the assumption of ε values following a normal distribution with a mean of zero and variance of σ2.
The population PK model building proceeded in a stepwise manner:
- Development of the structural base model
- Identification of significant covariates
- Evaluation and validation of the final model
Structural base model building
A number of candidate models were assessed to establish a basic stochastic model structure for total and unbound lenalidomide concentration-time profiles. Initially, one- and two-compartment models with first-order elimination models were evaluated. Various absorption models were tested, including zero-order absorption, first-order absorption, and a fixed absorption rate constant, with additional evaluation of absorption delay using a transit compartment model and a lag time (Tlag). Unbound plasma concentrations of lenalidomide were used to estimate its PK parameters. The protein-binding characteristics of lenalidomide were assessed using both linear and nonlinear binding models to describe the relationship between total and unbound concentrations, as defined by the following equations:
Linear Protein Binding
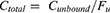
Non-Linear Protein Binding

where Ctotal represents total concentration (ng/mL), Cunbound does unbound concentration (ng/mL), Fu does unbound fraction, 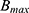 is maximum binding capacity (ng/mL), and
is maximum binding capacity (ng/mL), and 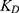 is dissociation constant (ng/mL). The best structural PK model was selected as the model with the lowest Akaike information criteria value based on the objective function value (OFV). Additionally, diagnostic goodness-of-fit (GOF) plots, relative standard error (RSE) and η-shrinkage values were evaluated during the model development process. Parameter estimates with an RSE of less than 30% were considered to be estimated with acceptable precision.
is dissociation constant (ng/mL). The best structural PK model was selected as the model with the lowest Akaike information criteria value based on the objective function value (OFV). Additionally, diagnostic goodness-of-fit (GOF) plots, relative standard error (RSE) and η-shrinkage values were evaluated during the model development process. Parameter estimates with an RSE of less than 30% were considered to be estimated with acceptable precision.
Covariate Analysis
Stepwise covariate modeling was used to identify significant covariates associated with unbound PK parameters of lenalidomide to develop the final model. Demographic data of our study patients were tested as candidate covariates for the PK parameters. Candidate covariates included sex; age; body size descriptor such as total body weight or BSA (m2) calculated using the Du-Bois method; platelet count (PLT, ×109/L); white blood cell count (WBC, ×109/L); absolute neutrophil count (ANC, ×109/L); total protein (TPN, g/dL); albumin (ALB, g/dL); C-reactive protein (CRP, mg/L); and genetic polymorphisms of ABCB1.33 For renal function, either creatinine clearance (CrCl, mL/min), estimated using the Cockcroft-Gault equation, or eGFR (mL/min/1.73 m2), calculated by the CKD-EPI 2021 equation, was evaluated as covariates in the population PK model.34,35 The covariate analysis followed stepwise forward inclusions and then, backward elimination steps. The statistical criteria for covariate selection were a decrease in OFV of > 3.84 (P < 0.05, χ2 distribution; 1 degree of freedom [df] for one additional parameter) for forward inclusion and an increase in OFV of > 6.63 (P < 0.01, χ2 distribution, 1df) for backward elimination. The effects of covariates on each PK parameter were explored using linear, power, or exponential functions for continuous covariates, and a linear function for categorical covariates. In addition to statistical criteria, the physiological plausibility of covariate effects on each PK parameter was considered throughout the final model building.
Final Model Evaluation
The robustness and accuracy of the final model were evaluated using a bootstrap resampling approach and visual predictive checks (VPCs). Bootstrap analysis was performed by generating 1,000 resampled datasets through random sampling with replacement from the original dataset, implemented in Wings for NONMEM version 7.5.1. The parameter estimates of the final model were compared to the 95% confidence intervals (CIs) derived from the bootstrap replicates. The 95% CIs were derived as the range between the 2.5th and 97.5th percentiles from 1,000 bootstrap-estimated PK parameters, employing a nonparametric approach. VPCs were conducted using Xpose with Perl Speaks NONMEM (PsN, version 5.3.1). A total of 1,000 simulations were executed based on the final model with the original dataset. The simulated 5th-, 50th-, and 95th-percentile concentrations and their corresponding 95% prediction intervals were plotted over time and overlaid with the observed lenalidomide concentrations.
Dosing Simulation
Systemic exposures of lenalidomide were simulated as AUC24 using different dosing regimens for comparison among severe RI (eGFR < 30 mL/min/1.73 m2), moderate RI (30 ≤ eGFR < 60 mL/min/1.73 m2), and control (ie, mildly impaired to normal renal function, eGFR ≥ 60 mL/min/1.73 m2) groups. Virtual patient populations for simulation were generated based on the distribution of baseline characteristics from the original dataset. Monte Carlo simulations were performed using the virtual patient population data through PsN based on the final population PK model developed for lenalidomide in MM patients. A total of 1,000 times simulations were performed using the simulation dataset for each lenalidomide dose of 5, 7.5, 10, 15, 20, and 25 mg once daily, generating 22,000 concentration-time profiles. Simulation results were stratified into the following two RI groups: severe and moderate RI. Simulated AUC24 profiles were further categorized based on any additional covariate included in the final population PK model of lenalidomide. Simulated AUC24 were compared with the predefined target AUC range to determine the probability of target attainment (PTA) for each patient category at each dosing regimen. The target AUC range was defined as 1,615.5–2,692.5 ng·h/mL, representing the reference value of 2,154 ng·h/mL ±25%. This reference corresponds to the average exposure reported in previous clinical studies29,36 for MM patients with normal to mildly impaired renal function (baseline serum creatinine ≤1.6 mg/dL) receiving the standard dose of 25 mg once daily.
Results
Patient Characteristics
A total of 30 patients with MM were enrolled in this study; among them, 11 patients were in the severe RI group (ie, eGFR < 30 mL/min/1.73 m2), 11 in the moderate RI group (ie, 30 ≤ eGFR < 60 mL/min/1.73 m2), and 8 in the control group (ie, eGFR ≥ 60 mL/min/1.73 m2). No patients received renal replacement therapy of any type throughout the study period. Treatment-emergent severe adverse events (grade ≥ 3) associated with lenalidomide dose reduction, specifically thrombocytopenia and neutropenia, were observed in 5 and 9 patients, respectively. In particular, severe adverse events were distributed as follows: 2 in the severe RI group, 6 in the moderate RI group, and 2 in the control group. Demographic and clinical characteristics of all enrolled patients are summarized in Table 1.
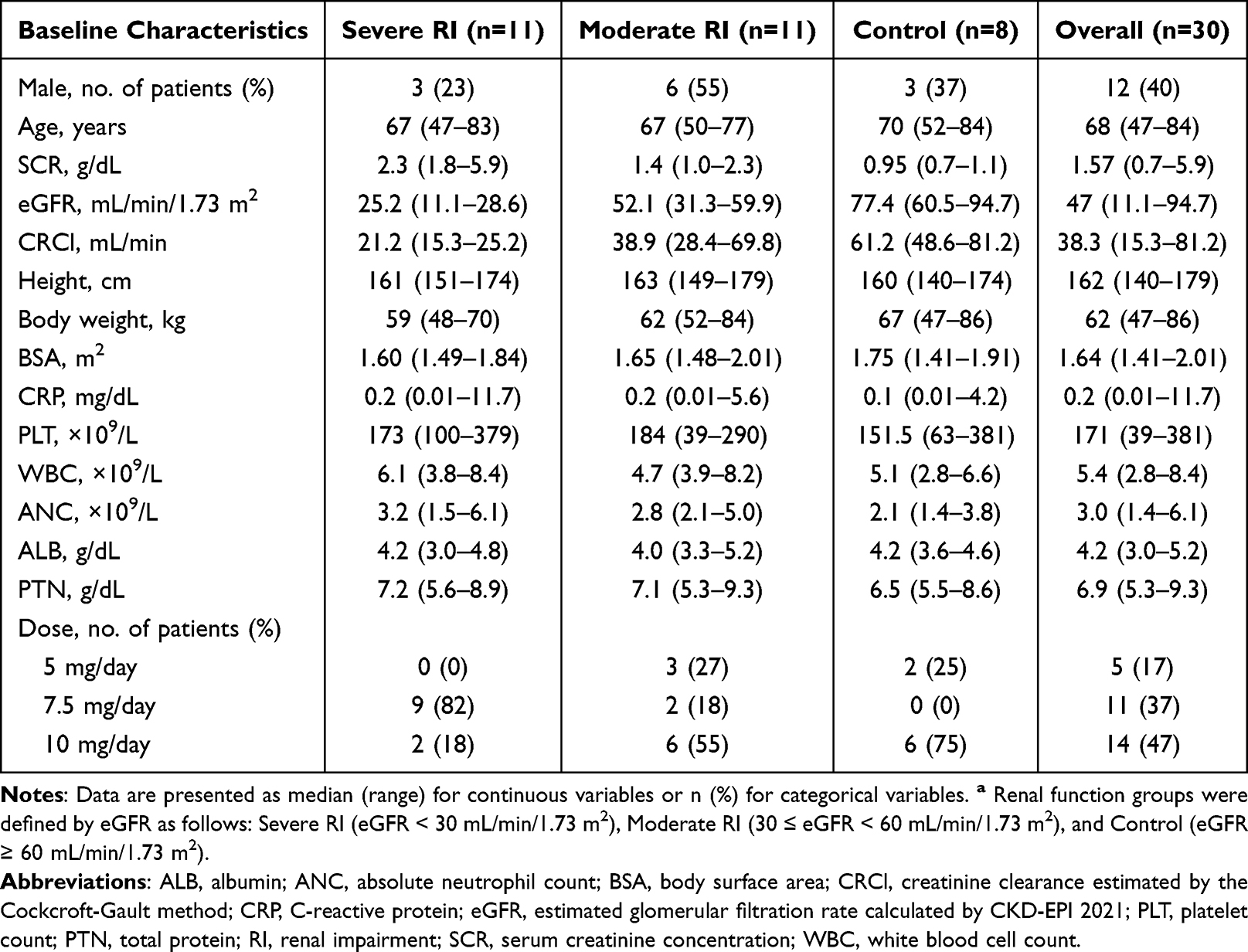 |
Table 1 Baseline Characteristics of Study Patients Grouped by Renal Function Statusa |
Non-Compartmental Analysis
Figure 1 illustrates individual plasma concentration-time profiles for total and unbound lenalidomide. The corresponding non-compartmental PK parameters are summarized in Table 2. The highest observed Cmax values were found in the severe RI group. Across all renal function groups, lenalidomide was absorbed with the median observed Tmax values ranging from 1.69 to 2.05 hours. As renal function worsened, systemic exposure (AUC24) and T1/2 increased, while CL/F decreased. Compared to the control group, the severe RI group showed an approximately two-fold higher AUC24, more than double the T1/2, and a reduction in CL/F less than half. In contrast, Vz/F was similar across all renal function groups, indicating that the extent of drug distribution was unaffected by RI.
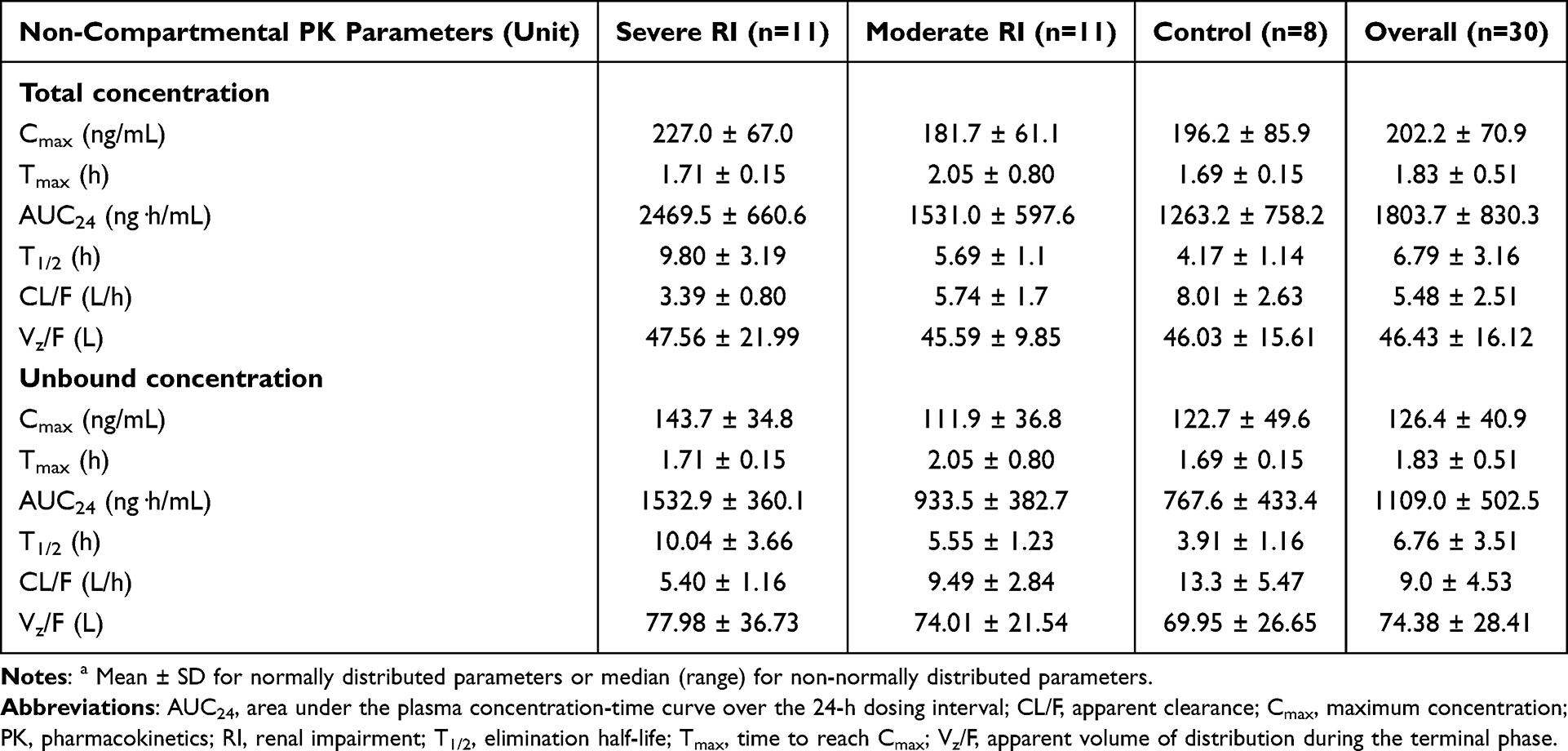 |
Table 2 Non-Compartmental PK Parameters of Lenalidomide Stratified by Renal Function Statusa |
 |
Figure 1 Plasma concentration-time profiles for (A) total and (B) unbound lenalidomide concentrations. Individual patients are distinguished by different markers. |
Figure 2 shows the relationship between total lenalidomide concentration and unbound fraction. The unbound fraction of lenalidomide remained constant across the observed concentration range, exhibiting a flat trend with increasing total concentrations. Linear regression analysis demonstrated no apparent concentration-dependent relationship, with a coefficient of determination (R2) of 0.0022.
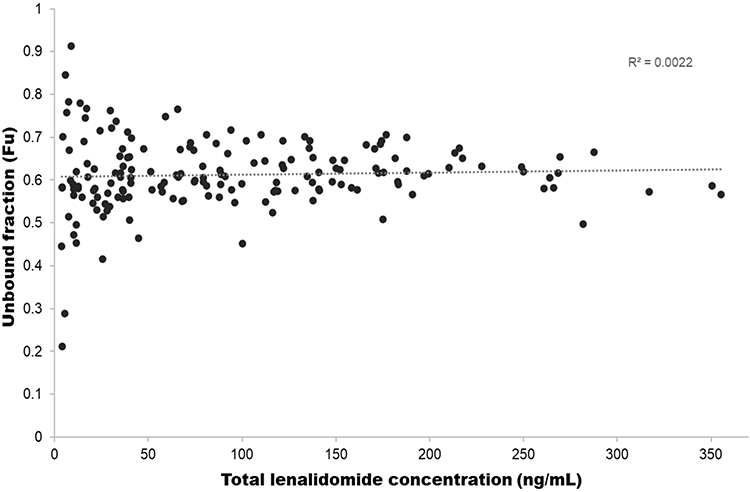 |
Figure 2 Relationship between total lenalidomide concentration and unbound fraction to assess concentration-dependent protein binding. |
Genotyping
Genotype distributions of SNPs of the ABCB1 in this study population and their associations with lenalidomide PK parameters, as assessed by Mann–Whitney U-test, are summarized in Supplementary Table 1. Several ABCB1 polymorphisms showed nominal associations with certain PK parameters; however, none remained statistically significant after adjusting for multiple comparisons with FDR methods (all adjusted P > 0.05).
Population Pharmacokinetic Modeling
Structural Base Model Building
The population PK analysis was conducted using 345 plasma lenalidomide concentrations (171 total, 174 unbound; a small number of samples with no detectable signal were excluded for total concentration quantification and treated as missing data in NONMEM dataset). The concentration-time profiles of lenalidomide were best described by a two-compartment model with first-order absorption, first-order elimination, and linear protein binding. Model-estimated PK parameters included unbound CL/F, apparent central volume of distribution (V1/F), apparent peripheral volume of distribution (V2/F), and apparent intercompartmental clearance (Q/F). The absorption rate constant (Ka) was fixed at 1.16 h−1 to improve the stability and precision of the model, based on our NCA analysis.37 Interindividual variability (IIV), assumed to be log-normally distributed, was estimated only for CL/F and V1/F. A combinational error model with both additive and proportional error terms was implemented to describe the residual variability for both total and unbound concentrations.
Covariate Analysis
Through the implementation of the stepwise covariate modeling (SCM) in PsN, the model OFV was significantly reduced with the following covariates in the forward inclusion process: eGFR added to CL/F (ΔOFV = −26.6), BSA to CL/F (ΔOFV = −14.8), BSA to V1/F (ΔOFV = −7.32), and eGFR to V1/F (ΔOFV = −3.89) (P < 0.05 for all). The ABCB1 SNPs did not significantly contribute to improvement in the model fit as an additional covariate to the unbound PK parameters of lenalidomide. Following backward elimination, eGFR was removed from V1/F (ΔOFV = 3.89); other covariates added to the PK parameters in the forward inclusion process remained in the model (P < 0.01). When testing CrCl as a covariate to account for renal function, the observed improvement in the model fit with the final CrCl-based model incorporating CrCl on CL/F and BSA on V1/F was less than that with the eGFR-based model (ΔOFV = −38.11 vs. −48.72).
Overall, based on the OFV values and other pre-defined model selection criteria as aforementioned in Methods Population Pharmacokinetic Analysis, the best-fit final population PK model for lenalidomide was significantly improved from the structural stochastic model (OFV reduced to 1842.34 from 1891.06, ΔOFV = −48.72) and described by the following equations:
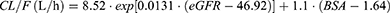
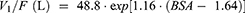



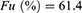
The IIV of CL/F and V1/F decreased from 38.6% to 18.9% and 36.6% to 30.8%, respectively, in the final model.
Final Model Evaluation
The PK parameter estimates for the final population PK model, along with results from the bootstrap, are listed in Table 3. A 1,000 replicate bootstrap analysis (99.4% success rate) showed all estimates within the 95% CIs with minimal bias (< 10%) compared to the bootstrap medians. Figures 3 and 4 present the goodness-of-fit (GOF) plots and visual predictive checks (VPCs) for both total and unbound lenalidomide concentrations. The VPC results showed that observed concentration percentiles (5th, 50th, and 95th) were well captured within the model-predicted 95% prediction intervals.
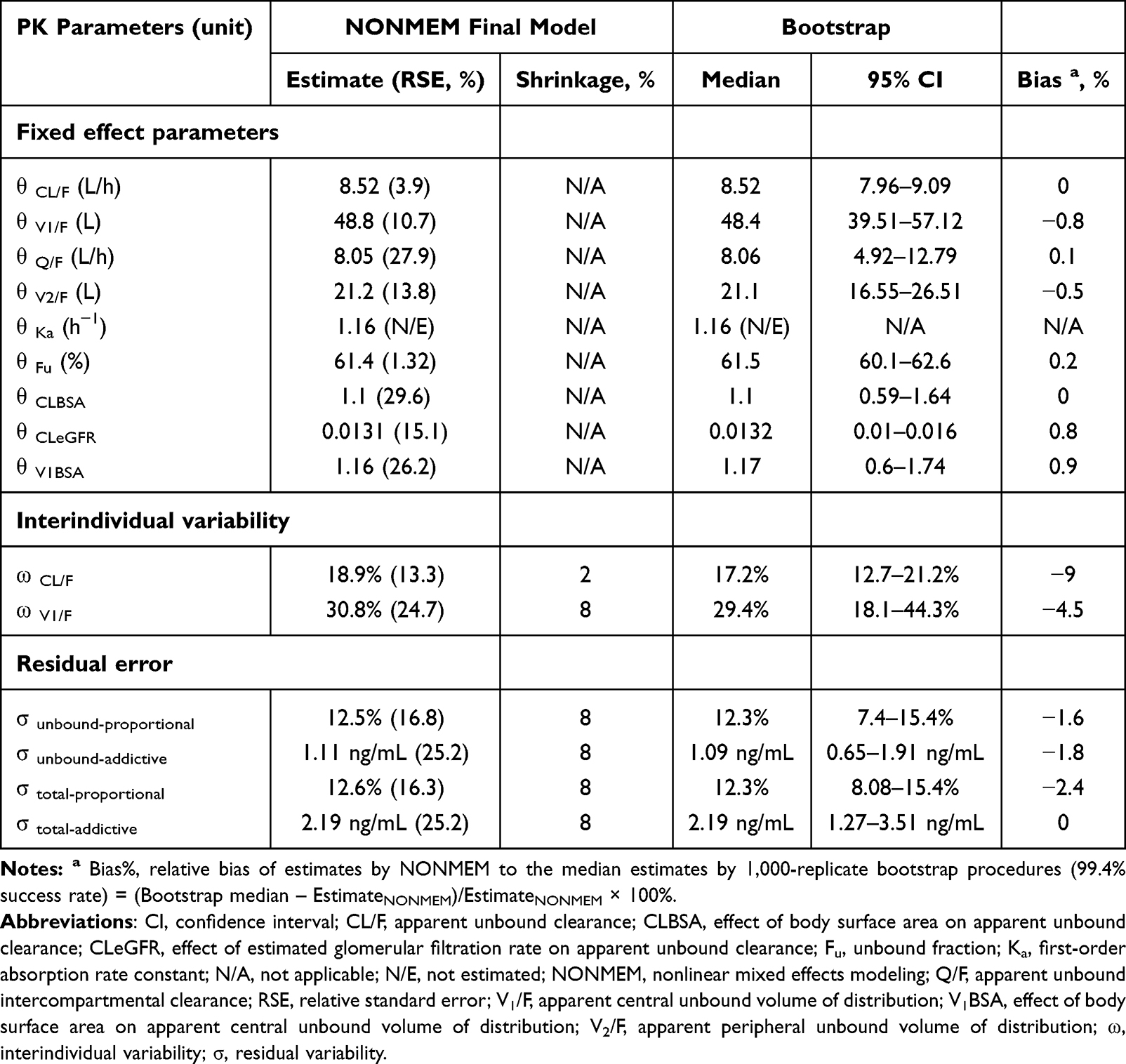 |
Table 3 Final Population PK Model Parameter Estimates and the Results of Bootstrap Validation of Lenalidomide |
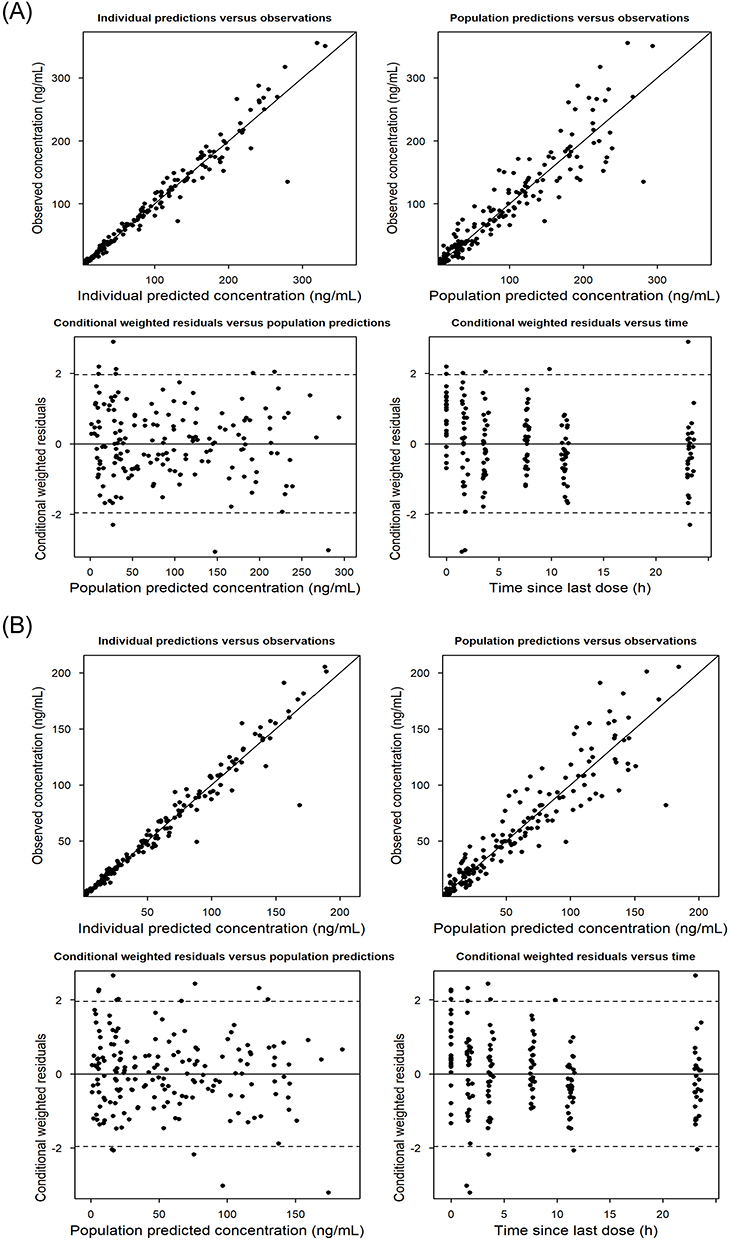 |
Figure 3 Goodness-of-fit plots of the final population PK model for (A) total and (B) unbound lenalidomide concentrations. |
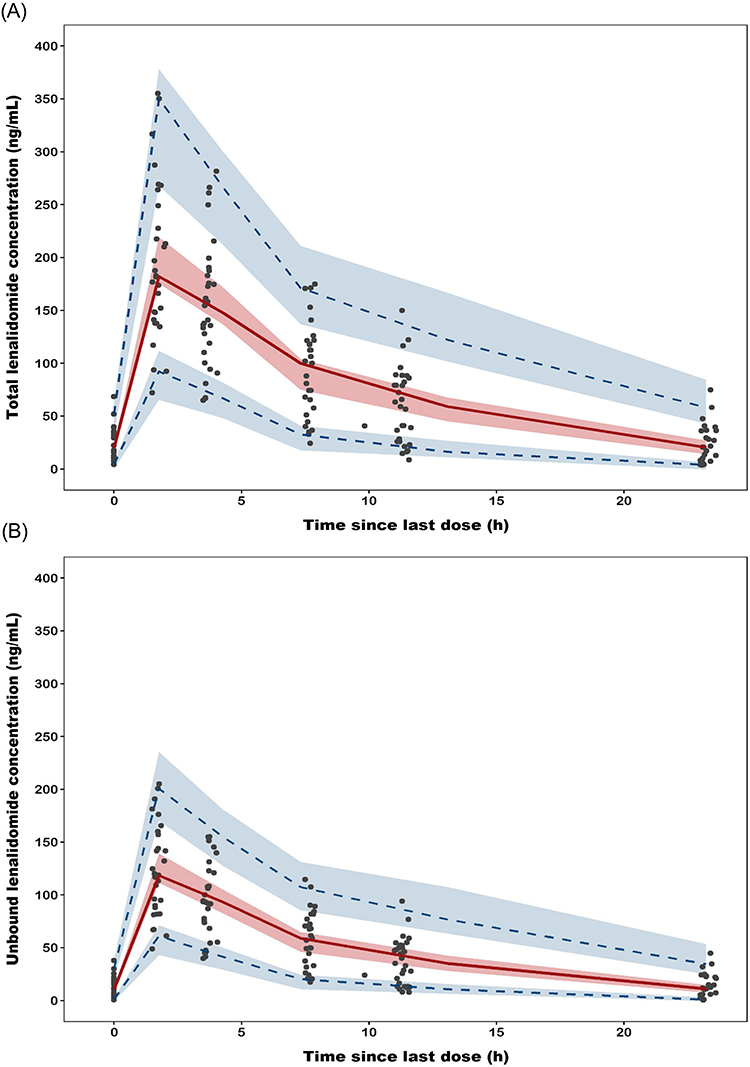 |
Figure 4 Visual predictive checks (n = 1,000 simulations) of the final population PK model for (A) total and (B) unbound lenalidomide concentrations. The solid red lines represent the median of the observed data, while the dashed blue lines indicate the 5th- and 95th-percentile observations. The shaded areas represent the 95% confidence intervals for corresponding percentiles of simulated concentrations. |
Simulation
To explore optimal lenalidomide dosing, simulations were conducted across clinically relevant dose levels of 5, 7.5, 10, 15, 20, and 25 mg to generate 22,000 virtual patient profiles at each dose level. Virtual patient population was stratified into severe RI and moderate RI, and further categorized by BSA values ranging from 1.4 to 2.0 m2.
Figure 5 illustrates the distribution of simulated AUCs stratified by simulated doses and BSA values for the severe and moderate RI groups, allowing visual assessment of simulated systemic exposures relative to the pre-defined therapeutic target. Based on these simulated AUC ranges, Figure 6 shows the probability of attaining the target AUC range (ie, 1,615.5–2,692.5 ng·h/mL) for the severe and moderate RI groups as heatmaps; each cell of the heatmap represents PTA at the specific dose of lenalidomide for the virtual patient with a specific BSA in the corresponding RI group.
 |
Figure 5 Simulated AUC24 distributions of lenalidomide across varying daily doses and body surface areas. (A) Severe renal impairment group (ie, eGFR < 30 mL/min/1.73 m2) and (B) moderate renal impairment group (ie, 30 ≤ eGFR < 60 mL/min/1.73 m2). Boxplots represent the distribution of simulated AUC24 values. Red dashed lines indicate the predefined target AUC range (1,615.5–2,692.5 ng·h/mL). a Dose approved by regulatory agencies (MFDS and US FDA) for patients with severe renal impairment. b Dose approved by regulatory agencies (MFDS and US FDA) for patients with moderate renal impairment. Abbreviations: AUC24, area under the plasma concentration-time curve over the 24-h dosing interval; BSA, body surface area; eGFR, estimated glomerular filtration rate; MFDS, Ministry of Food and Drug Safety; FDA, Food and Drug Administration. |
 |
Figure 6 Heatmaps of probability of target attainment (PTA) for simulated lenalidomide doses based on body surface area range. (A) Severe renal impairment group (ie, eGFR < 30 mL/min/1.73 m2) and (B) moderate renal impairment group (ie, 30 ≤ eGFR < 60 mL/min/1.73 m2). Each cell indicates PTA of virtual patients whose AUC24 values were within the predefined target range (1,615.5–2,692.5 ng·h/mL). a Dose approved by regulatory agencies (MFDS and US FDA) for patients with severe renal impairment. b Dose approved by regulatory agencies (MFDS and US FDA) for patients with moderate renal impairment. Abbreviations: AUC24, area under the plasma concentration-time curve over the 24-h dosing interval; BSA, body surface area; eGFR, estimated glomerular filtration rate; PTA, probability of target attainment; MFDS, Ministry of Food and Drug Safety; FDA, Food and Drug Administration. |
Discussion
In this study, we developed a population PK model for lenalidomide that represents one of the first few studies to encompass the full spectrum of renal insufficiency, specifically including patients with severe RI (ie, eGFR < 30 mL/min/1.73 m2). Given the pathophysiological alterations in MM patients with concomitant RI that can significantly affect protein binding, accurately assessing Fu is crucial for predicting clinically relevant drug distribution. Therefore, we performed simultaneous modeling using total and unbound lenalidomide concentration-time profiles. The developed models were robust and precise in describing both total and unbound concentrations and enabled estimation of Fu for characterizing protein binding. Importantly, Fu of lenalidomide was not influenced by patient factors including renal function. Rather, the PK profile of lenalidomide was significantly affected by eGFR and BSA; however, we observed no impact of ABCB1 gene polymorphisms on lenalidomide PK. To evaluate therapeutically effective exposures of lenalidomide, model-informed simulations were conducted to identify precision dosing regimens based on renal function as well as BSA for MM patients with moderate-to-severe RI.
The PK parameters from NCA in our study (Table 2), including AUC24, T1/2, and CL/F, were comparable to those reported by Bridoux et al across different stages of RI.29 Minor discrepancies observed in absorption parameters, such as Cmax and Tmax, were attributable to variations in sampling schedules between the studies. Notably, VZ/F remained constant across renal function categories, further supporting that protein binding is unaffected by RI. In the final population PK model (Table 3), the typical value of CL/F was estimated at 8.52 L/h (95% CI 7.96–9.09) with distribution characterized by a two-compartment model: V1/F of 48.8 L (95% CI 39.51–57.12) and V2/F of 21.2 L (95% CI 16.55–26.51), resulting in a total volume of approximately 70 L. Variations in CL/F estimates across previous studies were primarily driven by differences in renal function inclusion criteria. For instance, Connarn et al, Guglieri-López et al, and Hughes et al reported higher typical CL/F estimates of 14.2 L/h, 14 L/h and 12.4 L/h, respectively, compared to the present study.19,25,26 These prior studies excluded patients with severe RI and predominantly enrolled those with normal to mildly impaired renal function. Given that lenalidomide is excreted mainly via the kidneys, our inclusion of patients with significantly reduced GFR consequently resulted in a lower population CL/F estimate. Indeed, applying the median CrCl of the present cohort to the Connarn et al model yielded a CL/F of 8.43 L/h. This value is highly comparable to the estimate (8.52 L/h) in the current analysis. Regarding the apparent volume of distribution (V/F), our estimate demonstrated consistency with values reported by Connarn et al (62.4 L), Guglieri-López et al (57 L), and Hughes et al (75.4 L) In contrast, Liang et al reported lower estimates for both CL/F (7.25 L/h) and V/F (29.1 L) compared to our study and other previous reports.16 Even when accounting for their specific model parameterization involving ABCB1 genotype and dietary status, these values remain notably low, especially given the exclusion of severe RI. A key distinction in study design lies in the concomitant medications, where our study and previous reports primarily included patients treated with lenalidomide alone or in combination with dexamethasone—which is known to have no clinically significant PK interaction—whereas Liang et al analyzed a cohort receiving various lenalidomide-based combination therapies.38 To further explore the factors influencing distribution characteristics, we evaluated plasma protein binding. Fu was estimated at 0.614, corresponding to a plasma protein binding of approximately 38.6%. This value differs notably from in vitro data referenced in EMA and FDA labels, which report lower binding rates of 23% to 30% using [14C]-lenalidomide.12,39 However, our results align with previous ex vivo studies, which reported binding rates ranging from 35% to 44% with no significant differences between normal and RI groups.36 This suggests that the discrepancy between in vitro and ex vivo values can be attributed to methodological differences and physiological factors of the study population. To further elucidate the factors contributing to this variability within MM patients, we conducted a subgroup analysis based on the presence of serum electrophoresis M-Peak (n=29). The analysis revealed that protein binding was significantly higher in patients with detectable serum M-peak compared to those without (P < 0.05, data not shown). These findings suggest that excessive M-protein may serve as an additional binding reservoir for lenalidomide, thereby increasing the ex vivo protein binding. Further research is warranted to clarify the specific binding interactions between lenalidomide and paraproteins.
The final population PK model was established as a two-compartment model with first-order absorption and elimination, distinguishing it from the one-compartment structure frequently reported in prior studies.16,19,25,26 This structural characterization is closely linked to the altered PK profile in the study population. Unlike patients with normal renal function, the RI population exhibits significantly delayed elimination.36 Our study employed a sufficiently dense sampling schedule that was appropriate for capturing both the initial distribution and the prolonged elimination phases, thereby enabling the precise identification of two distinct compartments. For the absorption phase, Ka was fixed at 1.16 h−1 due to sparse data points during the initial absorption period. Given the rapid absorption characteristics of lenalidomide, the drug transitions quickly into the distribution and elimination phases; thus, fixing this parameter was considered a reasonable modeling strategy that did not compromise the overall model robustness. In the covariate analysis, BSA and eGFR were identified as significant covariates explaining interindividual variability in lenalidomide PK. Specifically, larger BSA was associated with higher CL/F and V1/F, while higher eGFR correlated with increased CL/F. Currently, prescribing information approved by regulatory agencies (eg., MFDS and US FDA) recommends dosing adjustments primarily based on CrCl. However, our analysis demonstrated that the PK profile of lenalidomide was better characterized when eGFR was incorporated as a covariate. This finding aligns with the KDIGO clinical practice guidelines, which support the use of validated eGFR equations rather than CrCl for drug dosing adjustments, because CrCl is highly dependent on body weight. In particular, CKD-EPI eGFR is advised for optimizing antineoplastic therapy as it reflects renal function more accurately than CrCl.40 Ultimately, our final population PK model confirms that these most recent evidence-based guidelines are indeed applicable and consistent for the specific case of lenalidomide. Nevertheless, the application of eGFR requires careful consideration regarding body size, as it is indexed to a standard BSA of 1.73 m2. Since MM patients with RI are typically older and prone to significant changes in body weight, relying on normalized eGFR may obscure actual renal function; thus, employing non-indexed eGFR individualized to the patient’s actual BSA could be considered for dosing decisions.41–44 In our study, we also developed a model incorporating non-indexed eGFR; however, the model that included BSA as a linear covariate on CL/F described the total and unbound concentration data more accurately in MM patients with RI. Consequently, we propose that a dosing strategy incorporating BSA in addition to eGFR would provide a more accurate and precise model-informed dosing regimen. We also evaluated the variability of protein binding. Unbound fraction remained consistent across all renal function groups, and no significant covariates associated with Fu were identified. Although RI affects approximately 50% of MM patients and is frequently associated with hypoalbuminemia, participants in our study — even those with severe RI — maintained serum albumin concentrations above 3 g/dL.45,46 Within this albumin range, protein binding of lenalidomide appeared unaffected by the severity of RI, which is further supported by the relatively low protein binding of lenalidomide (approximately 30–40%), suggesting that typical albumin fluctuations would have a minimal impact on Fu. Nevertheless, caution is warranted when extrapolating these findings to patients with profound hypoalbuminemia, as such patients were not represented in our study population. Furthermore, ABCB1 polymorphisms were not significantly associated with lenalidomide PK in our analysis, as presented in Supplementary Table 1. The allele frequencies of the three ABCB1 variants (1236C>T, 2677G>T/A, and 3435C>T) in our cohort were broadly comparable to publicly available East Asian/Korean reference data.47–49 Because rs2032582 is a triallelic variant, interpretation of its frequency distribution requires caution, particularly in a modest-sized cohort. To further explore possible combined effects of the three ABCB1 loci, we performed an additional multi-locus analysis using an unweighted zygosity score and the Jonckheere–Terpstra trend test; no statistically significant association with any lenalidomide PK parameter was detected after multiple-comparison correction (Supplementary Table 2).
Based on the final population PK model, we performed stratified Monte Carlo simulations across renal function categories and body size strata to provide evidence-based dosing recommendations. Current dosing recommendations include a substantial dose reduction at a CrCl threshold of 60 mL/min (eg., from 25 mg to 10 mg); however, given the steep exposure–response relationship characteristic of anticancer agents, such stepwise reductions may result in subtherapeutic exposure in some patients, particularly those near the dose-adjustment boundary.50 At the same time, maintaining effective lenalidomide treatment in renally impaired MM patients remains crucial based on clinical evidence. Across several studies, including Phase 3 trials, lenalidomide-based therapy was associated with renal function improvement in a considerable proportion of patients (up to ~70%).28,51,52 Additionally, a PK study demonstrated that effective myeloma control is a prerequisite for renal recovery, and conversely, restored renal function serves as a prognostic indicator for improved overall prognosis.52 Collectively, these findings underscore the need to optimize dosing in this population to achieve appropriate therapeutic exposure. Although definitive PK endpoints for lenalidomide efficacy have not been established, prior studies have used AUC observed in patients receiving the standard 25 mg dose originally established as the maximum tolerated dose.19,29,53 Chen et al and Bridoux et al showed that AUC-matched dose reductions in moderate and severe RI maintain comparable systemic exposure. Furthermore, Bridoux et al reported that clinical responses (very good partial response [VGPR] and partial response [PR]) were observed across a wide Cmax range (101–728 ng/mL), indicating that Cmax may not be the sole determinative factor for lenalidomide efficacy. Renal impairment reduces lenalidomide clearance, leading to increased systemic exposure through drug accumulation; therefore, when adjusting maintenance dosing regimens in renally impaired patients, targeting AUC may be more clinically relevant than preserving other PK parameters such as Cmax. To better reflect the distribution of renal function observed in real-world clinical practice, we used eGFR data from our patient cohort. We also generated virtual patient profiles incorporating the BSA distribution representative of our cohort, with values ranging from 1.4 to 2.0 m2. The simulation results highlight the necessity of BSA-stratified dosing alongside renal function monitoring. In the severe RI group, the approved dose of 7.5 mg provided reasonable target attainment rates across the majority of BSA ranges. For patients with BSA < 1.6 m2 (the median BSA in our cohort), a reduced dose of 5 mg maintained a comparable PTA of 58.9–69.6%. However, for patients with a BSA of 2.0 m2 (classified as overweight), the PTA for the 7.5 mg dose declined to 42.5%. Simulating a dose increase to 10 mg improved the PTA to 70.0%, suggesting that standard dose might lead to under-dosing for overweight patients.54 Similarly, in the moderate RI group, while the standard dose of 10 mg is generally recommended, reducing the dose to 7.5 mg for patients with BSA < 1.6 m2 achieved a PTA of 56.0–63.8%. This suggests that dose reduction is feasible to minimize excess exposure without compromising efficacy. Conversely, for patients with a BSA of 2.0 m2, the standard 10 mg dose resulted in a suboptimal PTA of 30.6%. Increasing the dose to 15 mg in these patients significantly improved the PTA to 65.9%, ensuring adequate therapeutic exposure. These findings demonstrate that simultaneously incorporating both eGFR and BSA as covariates can account for the multiple sources of PK variability encountered in clinical practice. This enables individualized dosing recommendations that maintain consistent and predictable systemic exposure across the full spectrum of patient characteristics in this population. Optimizing dosing regimens necessitates a careful balance between safety and efficacy. A prior study established a correlation between lenalidomide minimum concentration (Cmin) and hematological toxicity, proposing a safety threshold of 10.95 ng/mL.55 However, our simulations (Supplementary Figure 1) revealed that predicted Cmin values in RI patients frequently exceeded this threshold. This elevation is a mechanistic consequence of impaired renal elimination; urinary recovery of unchanged lenalidomide decreases from approximately 84% in normal renal function to 43% in severe RI, prolonging the elimination half-life and flattening the concentration-time profile.36 Importantly, a flatter profile with an equivalent AUC inherently yields a higher Cmin, reflecting altered drug elimination rather than excessive total drug exposure. The clinical relevance of this Cmin elevation warrants careful interpretation. First, the threshold proposed by Song et al was derived from a cohort excluding patients with significant RI, and its applicability to this population remains unvalidated; moreover, Cmin was adopted in that study as a practical metric due to the logistical constraints of multi-point sampling required for AUC estimation in routine clinical practice, rather than as a pharmacologically superior toxicity endpoint.55 Second, a randomized Phase 2 study demonstrated that patients receiving lenalidomide twice daily exhibited lower Cmin values yet experienced greater and earlier myelosuppression compared with once daily dosing, directly challenging the utility of Cmin as a hematotoxicity predictor in this setting.56 Consequently, further investigation is required to validate the clinical relevance of Cmin regarding hematologic toxicity specifically in RI patients.
Despite these meaningful findings, several limitations should be acknowledged. First, since blood samples were collected starting at 2 hours post-dose, limited data were available to characterize the absorption phase, necessitating the use of a fixed Ka value derived from NCA rather than estimating the best-fit Ka value within the final model. Second, although patients with moderate-to-severe RI were included, no patients receiving renal replacement therapy were included, thus limiting application of our study findings to MM patients undergoing such treatment. Third, body size descriptors tested with BSA selected as a significant covariate; however, the potential effect of body composition on lenalidomide PK was not considered, which might be more physiologically plausible. Fourth, none of our study patients had clinically significant hypoalbuminemia; all patients had serum albumin concentrations of 3 g/dL or greater. Considering hypoalbuminemia as a frequent complication in MM patients with RI, the generalizability of our study findings might be limited to patients without hypoalbuminemia. However, our present study confirmed the fact that not all of MM patients with RI had hypoalbuminemia. Finally, prospective clinical validation evaluating treatment outcomes was not conducted in our current study. While our internal validation supports the PK reliability of the predictions, there remains a lack of direct effectiveness or safety evidence for these model-informed dosing recommendations. Therefore, future studies are warranted with larger sample sizes, evaluation of diverse covariates such as body composition, inclusion of more complicated patients (eg., those receiving renal replacement therapy or with clinically significant hypoalbuminemia), and a more dense-scheduled sampling, particularly during the initial absorption-dominant phase.
Conclusions
In conclusion, renal function (eGFR) and body size (BSA) are the key factors influencing PK variability of lenalidomide in renally impaired MM patients. While protein binding remained constant regardless of renal status, our simulations using the model reveal that current dosing protocols based solely on renal function may result in suboptimal drug exposure, particularly in patients at the extremes of the BSA distribution. Notably, for patients with BSA below the cohort median (< 1.6 m2), dose reduction is recommended to minimize excess exposure without compromising efficacy, whereas for large body-size patients (BSA ≥ 2.0 m2), dose escalation is warranted to achieve therapeutic exposures comparable to those in patients with normal renal function. These findings support a model-informed precision dosing approach that incorporates both eGFR and BSA to optimize lenalidomide exposure, potentially enhancing treatment response and minimizing toxicity in MM patients with moderate-to-severe RI.
Ethics Statement
The study was conducted in accordance with the Declaration of Helsinki, and the protocol was approved by the Institutional Review Board of Seoul St. Mary’s Hospital, The Catholic University of Korea (Approval Date: October 29, 2024; Approval No. KC24MISK0219). Written informed consent was obtained from all patients prior to enrollment.
Acknowledgments
The authors thank Seungmin Jun and Soyoung Lee for their assistance in project administration and data management.
Funding
This research was partially supported by the BK21 FOUR program of Graduate School, Kyung Hee University (GS-1-JO-NON-info2120250482) and by the Ministry of Food and Drug Safety (No. 23212MFDS228, No. 21153MFDS601-1, RS-2024-00331810) in 2024.
Disclosure
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
1. Hou Q, Li X, Ma H, Fu D, Liao A. A systematic epidemiological trends analysis study in global burden of multiple myeloma and 29 years forecast. Sci Rep. 2025;15(1). doi:10.1038/s41598-024-83630-x
2. Wildes TM, Rosko A, Tuchman SA. Multiple myeloma in the older adult: better prospects, more challenges. J Clin Oncol. 2014;32(24):2531–20. doi:10.1200/jco.2014.55.1028
3. Palumbo A, Anderson K. Multiple Myeloma. N Engl J Med. 2011;364(11):1046–1060. doi:10.1056/nejmra1011442
4. Kumar V, Abbas AK, Fausto N, Aster JC. Robbins and Cotran Pathologic Basis of Disease.
5. Bridoux F, Leung N, Hutchison CA, et al. Diagnosis of monoclonal gammopathy of renal significance. Kidney Int. 2015;87(4):698–711. doi:10.1038/ki.2014.408
6. Tanaka T, Kishimoto T. The biology and medical implications of Interleukin-6. Cancer Immunol Res. 2014;2(4):288–294. doi:10.1158/2326-6066.cir-14-0022
7. Shevra C, Singh S, Singh N, Kumar S, Rai M, Singh U. Interleukin-6 and interleukin-4 levels in multiple myeloma and correlation of interleukin-6 with β2 microglobulin and serum creatinine. Clin Cancer Invest J. 2015;4(2):211. doi:10.4103/2278-0513.148963
8. Walther CP, Gutiérrez OM, Cushman M, et al. Serum albumin concentration and risk of end-stage renal disease: the REGARDS study. Nephrol Dial Transplant. 2018;33(10):1770–1777. doi:10.1093/ndt/gfx331
9. Komrokji RS, Corrales-Yepez M, Kharfan-Dabaja MA, et al. Hypoalbuminemia is an independent prognostic factor for overall survival in myelodysplastic syndromes. Am J Hematol. 2012;87(11):1006–1009. doi:10.1002/ajh.23303
10. Kotla V, Goel S, Nischal S, et al. Mechanism of action of lenalidomide in hematological malignancies. J Hematol Oncol. 2009;2(1):36. doi:10.1186/1756-8722-2-36
11. Chen N, Kasserra C, Reyes J, Liu L, Lau H. Single-dose pharmacokinetics of lenalidomide in healthy volunteers: dose proportionality, food effect, and racial sensitivity. Cancer Chemother Pharmacol. 2012;70(5):717–725. doi:10.1007/s00280-012-1966-z
12. U.S. FaDA. Prescribing information for REVLIMID® (lenalidomide) tablets, for oral use. 2025. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/label/2019/021880s057lbl.pdf.
13. Dimopoulos MA, Merlini G, Bridoux F, et al. Management of multiple myeloma-related renal impairment: recommendations from the International Myeloma Working Group. Lancet Oncol. 2023;24(7):e293–e311. doi:10.1016/s1470-2045(23)00223-1
14. Dimopoulos MA, Sonneveld P, Leung N, et al. International myeloma working group recommendations for the diagnosis and management of myeloma-related renal impairment. J Clin Oncol. 2016;34(13):1544–1557. doi:10.1200/jco.2015.65.0044
15. Kobayashi T, Miura M, Abumiya M, Akamine Y, Ito F, Takahashi N. Influence of ABCB1 polymorphisms on the pharmacokinetics and toxicity of lenalidomide in patients with multiple myeloma. Med Oncol. 2019;36(6). doi:10.1007/s12032-019-1280-2
16. Liang X, Shi H, Bi K, et al. Population pharmacokinetics of lenalidomide in Chinese patients with influence of genetic polymorphisms of ABCB1. Sci Rep. 2024;14(1). doi:10.1038/s41598-024-52460-2
17. Tong Z, Yerramilli U, Surapaneni S, Kumar G. The interactions of lenalidomide with human uptake and efflux transporters and UDP-glucuronosyltransferase 1A1: lack of potential for drug–drug interactions. Cancer Chemother Pharmacol. 2014;73(4):869–874. doi:10.1007/s00280-014-2415-y
18. Jakobsen Falk I, Lund J, Gréen H, et al. Pharmacogenetic study of the impact of ABCB1 single-nucleotide polymorphisms on lenalidomide treatment outcomes in patients with multiple myeloma: results from a Phase IV observational study and subsequent Phase II clinical trial. Cancer Chemother Pharmacol. 2018;81(1):183–193. doi:10.1007/s00280-017-3481-8
19. Guglieri-López B, Pérez-Pitarch A, Moes DJAR, et al. Population pharmacokinetics of lenalidomide in multiple myeloma patients. Cancer Chemother Pharmacol. 2017;79(1):189–200. doi:10.1007/s00280-016-3228-y
20. Abdallah NH, Nagayama H, Takahashi N, et al. Muscle and fat composition in patients with newly diagnosed multiple myeloma. Blood Cancer J. 2023;13(1). doi:10.1038/s41408-023-00934-3
21. Don BR, Kaysen G. POOR NUTRITIONAL STATUS AND INFLAMMATION: serum albumin: relationship to inflammation and nutrition. Semin Dial. 2004;17(6):432–437. doi:10.1111/j.0894-0959.2004.17603.x
22. Alanazi AM, Abdelhameed AS, Bakheit AH, Darwish IA. Exploring the interaction forces involved in the binding of the multiple myeloma drug lenalidomide to bovine serum albumin. J Mol Liq. 2017;238:3–10. doi:10.1016/j.molliq.2017.04.110
23. Nagumo K, Tanaka M, Chuang VTG, et al. Cys34-cysteinylated human serum albumin is a sensitive plasma marker in oxidative stress-related chronic diseases. PLoS One. 2014;9(1):e85216. doi:10.1371/journal.pone.0085216
24. Kawata Y, Hirano H, Takahashi R, et al. Detailed structure and pathophysiological roles of the IgA-Albumin complex in multiple myeloma. Int J Mol Sci. 2021;22(4):1766. doi:10.3390/ijms22041766
25. Connarn JN, Hwang R, Gao Y, Palmisano M, Chen N. Population pharmacokinetics of lenalidomide in healthy volunteers and patients with hematologic malignancies. Clin Pharmacol Drug Dev. 2018;7(5):465–473. doi:10.1002/cpdd.372
26. Hughes JH, Phelps MA, Upton RN, et al. Population pharmacokinetics of lenalidomide in patients with B-cell malignancies. Br J Clin Pharmacol. 2019;85(5):924–934. doi:10.1111/bcp.13873
27. Van Steeg TJ, Boralli VB, Krekels EHJ, et al. Influence of plasma protein binding on pharmacodynamics: estimation of in vivo receptor affinities of β blockers using a new mechanism-based PK–PD modelling approach. J Pharmaceut Sci. 2009;98(10):3816–3828. doi:10.1002/jps.21658
28. Chen CI, Cao Y, Trudel S, et al. An open-label, pharmacokinetic study of lenalidomide and dexamethasone therapy in previously untreated multiple myeloma (MM) patients with various degrees of renal impairment – validation of official dosing guidelines. Leukemia Lymphoma. 2020;61(8):1860–1868. doi:10.1080/10428194.2020.1747064
29. Bridoux F, Chen N, Moreau S, et al. Pharmacokinetics, safety, and efficacy of lenalidomide plus dexamethasone in patients with multiple myeloma and renal impairment. Cancer Chemother Pharmacol. 2016;78(1):173–182. doi:10.1007/s00280-016-3068-9
30. Lee S, Yang S, Shim W-S, et al. Development and validation of an improved HPLC-MS/MS method for quantifying total and unbound lenalidomide in human plasma. Pharmaceutics. 2024;16(10):1340. doi:10.3390/pharmaceutics16101340
31. U.S. Department of Health and Human Services FaDA. Guidance for industry. Population Pharmacokinetics. Available from: https://www.fda.gov/media/128793/download.
32. Bauer RJ. NONMEM tutorial part ii: estimation methods and advanced examples. CPT Pharmacometrics Syst Pharmacol. 2019;8(8):538–556. doi:10.1002/psp4.12422
33. Du Bois D, Du Bois EF.A formula to estimate the approximate surface area if height and weight be known. 1916. Nutrition. 1989;5(5):303–311.
34. Cockcroft DW, Gault H. Prediction of creatinine clearance from serum creatinine. Nephron. 1976;16(1):31–41. doi:10.1159/000180580
35. Inker LA, Eneanya ND, Coresh J, et al. New creatinine- and cystatin C–Based equations to estimate GFR without race. N Engl J Med. 2021;385(19):1737–1749. doi:10.1056/nejmoa2102953
36. Chen N, Lau H, Kong L, et al. Pharmacokinetics of lenalidomide in subjects with various degrees of renal impairment and in subjects on hemodialysis. J Clin Pharmacol. 2007;47(12):1466–1475. doi:10.1177/0091270007309563
37. Holford N. Absorption and Half-Life. Transl Clin Pharmacol. 2016;24(4):157. doi:10.12793/tcp.2016.24.4.157
38. Chen N, Zhou D-B, Yu L, et al. Pharmacokinetics (PK) of lenalidomide when administered alone or in combination with dexamethasone in chinese patients (Pts) with relapsed/refractory multiple myeloma (RRMM): the MM-021 trial. Blood. 2012;120(21):5046. doi:10.1182/blood.v120.21.5046.5046
39. Agency EM. Summary of the European Public Assessment Report (EPAR) for Revlimid. Center for Drug Evaluation and Research. 2025. Available from: https://www.ema.europa.eu/en/documents/product-information/revlimid-epar-product-information_en.pdf.
40. Stevens PE, Ahmed SB, Carrero JJ, et al. KDIGO 2024 clinical practice guideline for the evaluation and management of chronic kidney disease. Kidney Int. 2024;105(4):S117–S314. doi:10.1016/j.kint.2023.10.018
41. Mikhael J, Singh E, Rice MS. Real-world renal function among patients with multiple myeloma in the United States. Blood Cancer J. 2021;11(5). doi:10.1038/s41408-021-00492-6
42. Ku E, Kopple JD, Johansen KL, et al. Longitudinal weight change during CKD progression and its association with subsequent mortality. Am J Kidney Dis. 2018;71(5):657–665. doi:10.1053/j.ajkd.2017.09.015
43. Pharmacokinetics in patients with impaired renal function – study design, data analysis, and impact on dosing guidance for industry. 2024.
44. Guideline on the evaluation of the pharmacokinetics of medicinal products in patients with decreased renal function. 2016.
45. Korbet SM, Schwartz MM. Multiple Myeloma. J Am Soc Nephrol. 2006;17(9):2533–2545. doi:10.1681/asn.2006020139
46. Strougo A, Yassen A, Krauwinkel W, Danhof M, Freijer J. A semiphysiological population model for prediction of the pharmacokinetics of drugs under liver and renal disease conditions. Drug Metab Dispos. 2011;39(7):1278–1287. doi:10.1124/dmd.110.037838
47. (NCBI) NCfBI. dbSNP RefSNP Report: rs1045642. 2024. dbSNP (Short Genetic Variations). Available from: https://www.ncbi.nlm.nih.gov/snp/rs1045642.
48. (NCBI) NCfBI. dbSNP RefSNP Report: rs2032582. 2024. dbSNP (Short genetic variations). Available from: https://www.ncbi.nlm.nih.gov/snp/rs2032582.
49. (NCBI) NCfBI. dbSNP RefSNP Report: rs1128503. 2024. dbSNP (Short Genetic Variations). Available from: https://www.ncbi.nlm.nih.gov/snp/rs1128503.
50. Delanaye P, Guerber F, Scheen A, et al. Discrepancies between the Cockcroft–Gault and Chronic Kidney Disease Epidemiology (CKD-EPI) equations: implications for refining drug dosage adjustment strategies. Clin Pharmacokinet. 2017;56(2):193–205. doi:10.1007/s40262-016-0434-z
51. Dimopoulos M, Alegre A, Stadtmauer EA, et al. The efficacy and safety of lenalidomide plus dexamethasone in relapsed and/or refractory multiple myeloma patients with impaired renal function. Cancer. 2010;116(16):3807–3814. doi:10.1002/cncr.25139
52. Ludwig H, Rauch E, Kuehr T, et al. Lenalidomide and dexamethasone for acute light chain-induced renal failure: a phase II study. Haematologica. 2015;100(3):385–391. doi:10.3324/haematol.2014.115204
53. Barlogie B. Thalidomide and CC-5013 in multiple myeloma: the University of Arkansas experience. Semin Hematol. 2003;40:33–38. doi:10.1053/j.seminhematol.2003.09.005
54. Verbraecken J, Van De Heyning P, De Backer W, Van Gaal L. Body surface area in normal-weight, overweight, and obese adults. A comparison study. Metabolism. 2006;55(4):515–524. doi:10.1016/j.metabol.2005.11.004
55. Song Z, Ma L, Bao L, et al. Toward therapeutic drug monitoring of lenalidomide in hematological malignancy? Results of an observational study of the exposure-safety relationship. Front Pharmacol. 2022;13. doi:10.3389/fphar.2022.931495.
56. Richardson PG, Blood E, Mitsiades CS, et al. A randomized phase 2 study of lenalidomide therapy for patients with relapsed or relapsed and refractory multiple myeloma. Blood. 2006;108(10):3458–3464. doi:10.1182/blood-2006-04-015909
 © 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.