Back to Journals » OncoTargets and Therapy » Volume 12
Ponatinib: a novel multi-tyrosine kinase inhibitor against human malignancies
Authors Tan FH, Putoczki TL, Stylli SS ![]() , Luwor RB
, Luwor RB
Received 1 October 2018
Accepted for publication 14 December 2018
Published 18 January 2019 Volume 2019:12 Pages 635—645
DOI https://doi.org/10.2147/OTT.S189391
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Tohru Yamada
Fiona H Tan,1,2 Tracy L Putoczki,1,3,4 Stanley S Stylli,1,5 Rodney B Luwor1
1Department of Surgery, The University of Melbourne, The Royal Melbourne Hospital, Parkville, VIC 3050, Australia; 2Division of Cancer Research, Peter MacCallum Cancer Centre, Melbourne, VIC 3000, Australia; 3Inflammation Division, Walter and Eliza Hall Institute of Medical Research, Parkville, VIC 3052, Australia; 4Department of Medical Biology, The University of Melbourne, Parkville, VIC 3050, Australia; 5Department of Neurosurgery, The Royal Melbourne Hospital, Parkville, VIC 3050, Australia
Abstract: Human malignancies are often the result of overexpressed and constitutively active receptor and non-receptor tyrosine kinases, which ultimately lead to the mediation of key tumor-driven pathways. Several tyrosine kinases (ie, EGFR, FGFR, PDGFR, VEGFR), are aberrantly activated in most common tumors, including leukemia, glioblastoma, gastrointestinal stromal tumors, non-small-cell lung cancer, and head and neck cancers. Iclusig™ (ponatinib, previously known as AP24534) is an orally active multi-tyrosine kinase inhibitor and is currently approved by the US Food and Drug Administration for patients with chronic myeloid leukemia and Philadelphia chromosome-positive acute lymphoblastic leukemia, specifically targeting the BCR-ABL gene mutation, T315I. Due to ponatinib’s unique multi-targeted characteristics, further studies have demonstrated its ability to target other important tyrosine kinases (FGFR, PDGFR, SRC, RET, KIT, and FLT1) in other human malignancies. This review focuses on the available data of ponatinib and its molecular targets for treatment in various cancers, with a discussion on the broader potential of this agent in other cancer indications.
Keywords: ponatinib, cancer treatment, multi-kinase inhibitor, repurposing
Introduction
Receptor tyrosine kinases (RTKs) mediate the interaction between growth factors and their corresponding receptors, resulting in cytoplasmic tyrosine kinase activation. The activation of RTKs regulates several critical cellular mechanisms including cell growth, tissue regeneration, and repair. These processes are tightly regulated in normal cells; however, overexpression and constitutive activation of RTKs have been known to promote tumor survival in several cancer types. There are several known families of RTKs, including the commonly overexpressed EGFR, FGFR, RET, KIT, SRC, BCR-ABL, and PDGFR. Continual activation of these RTKs leads to a cascade of events associated with key pathways known to drive cancers, including STAT3, PI3K/AKT, and RAS/RAF/ERK pathways.2,3 Due to the importance of RTKs and their association with tumor development, they are emerging as promising therapeutic targets.
Tyrosine kinase inhibitors (TKIs) and monoclonal antibodies have been designed to specifically target overexpressed RTKs and subsequent downstream molecules in several cancer types. For instance, successful anti-EGFR (cetuximab, panitumumab, erlotinib, and gefitinib) and anti-HER2 (trastuzumab and pertuzumab) mono-targeted agents have been developed and are currently approved by the US Food and Drug Administration (FDA) for treating patients with colorectal cancer, non-small-cell lung cancer (NSCLC), and breast cancer.4–9 Moreover, SRC proteins are commonly activated as a result of enhanced RTK expression, and the development of SRC inhibitors has also been successful with FDA-approved bosutinib and dasatinib currently used in the treatment of patients with Philadelphia chromosome-positive (Ph+) chronic myeloid leukemia (CML) and acute lymphoblastic leukemia (ALL).10,11 In addition to mono-targeted inhibitors, multi-TKIs have also been developed. Multi-TKIs are agents that target several kinases or pathways simultaneously, and have been widely developed for several diseases, including cancer. For instance, Iclusig™ (ponatinib; ARIAD Pharmaceuticals, Cambridge, MA, USA) a multi-TKI, which targets SRC, ABL, FGFR, PDGFR, and VEGFR, is currently approved by the FDA as a second-line treatment option for patients with Ph+ CML and Ph+ ALL and is the only approved inhibitor to successfully target the ABL-T135I mutation.12 Notably, in preclinical studies, ponatinib has also demonstrated potent anti-tumorigenic properties in other cancers, including lung cancers and glioblastoma (GBM), and Phase II trials are in progress in patients harboring these tumors (NCT02478164, NCT01935336).13,14 This review summarizes ponatinib’s multi-targeted properties and discusses its potential as a prospective anticancer agent for other malignancies.
Ponatinib: preclinical and early-clinical data
Ponatinib (also known as AP24534) is a potent, orally active TKI. The molecular formula of ponatinib is C29H28ClF3N6O, and the chemical name is 3-(imidazo[1,2-b]pyridazin-3-ylethynyl)-4-methyl-N-19benzamide hydrochloride.15 Ponatinib was structurally designed with a carbon–carbon triple bond to target the T315I point mutation within the kinase domain (KD) of BCR-ABL (Figure 1).
 | Figure 1 Chemical structure of ponatinib. |
ARIAD Pharmaceuticals acquired a computational and structure-based drug design platform to develop ponatinib as a potent inhibitor of both native BCR-ABL tyrosine kinase and isoforms that carry mutations responsible for conferring resistance to existing targeted therapies. Furthermore, this inhibitor was structurally designed to involve a carbon–carbon triple bond which protrudes from the purine scaffold, allowing for minimal steric hindrance which is caused by the presence of a large isoleucine residue of the BCR-ABL mutant at position 315.15,16 Such a structural design was primarily based on one of ARIAD’s first ATP-competitive dual SRC/ABL inhibitors known as AP24364, alongside earlier developed inhibitors including nilotinib, dasatinib, and imatinib.15
Preclinical evaluation of ponatinib treatment of CML and Ph+ ALL
Preclinical testing of ponatinib was first reported in 2006 when O’Hare et al demonstrated the potent anti-BCR-ABL effects of ponatinib.12 These in vitro studies utilized Ba/F3 cell lines expressing either native BCR-ABL or BCR-ABL with mutations within the tyrosine KD. The kinase selectivity studies discovered that ponatinib effectively blocked both wild-type and several mutant forms of BCR-ABL kinases, including BCR-ABLT315I, with IC50 values ranging between 0.37 and 2 nM. Furthermore, this study compared the efficacy of ponatinib at several concentrations (0–1,000 nM) against mononuclear cells from primary leukemic blast crisis patient cells specifically driven by wild-type BCR-ABL or BCR-ABLT315I and healthy individuals. The data generated with the cellular proliferation assay showed that ponatinib had induced a specific reduction in viable cells against primary CML cells, showing IC50 values of ~500-fold lower than normal cells, confirming its potent pan-BCR-ABL inhibition.
The efficacy of ponatinib was also examined in several in vivo CML mouse models expressing wild-type BCR-ABL or BCR-ABLT315I. The potent effect of ponatinib was illustrated using a Ba/F3 BCR-ABL-expressing mouse model where ponatinib was orally administered daily for 19 days. The median survival rate of mice treated with either 2.5 or 5 mg/kg ponatinib was prolonged to 27.5 and 30 days, respectively (P<0.01 for both doses), compared to vehicle-treated mice, which survived only 19 days following the intravenous injection of Ba/F3 cells.12 Ponatinib treatment was also observed to increase the survival rates of mice challenged with intravenously injected BAF/3 cells expressing BCR-ABLT315I. Ponatinib treatment (5, 15, and 25 mg/kg) prolonged the median survival in a dose-dependent manner (19.5, 26, and 30 days, respectively; P<0.01 for all doses) compared to vehicle-treated mice which survived only 16 days.12 However, in comparison, dasatinib administered at doses as high as 300 mg/kg did not enhance the survival of mice injected with BAF/3 cells expressing BCR-ABLT315I, highlighting the limited efficacy of dasatinib as a TKI against this BCR-ABL mutation. Ponatinib doses as low as 30 and 50 mg/kg have been shown to induce tumor stasis or regression in a subcutaneous BCR-ABLT315I mouse model.12 Overall, these initial preclinical findings demonstrated the potential inhibitory effect of ponatinib on native and mutant BCR-ABL cells.
Following further clinical evaluation (summarized in Table 1), the FDA approved the use of ponatinib hydrochloride as a third-line treatment option for adult patients with T315I-postive CML (including accelerated phase, chronic phase, or blast phase) or those with T315I-positive Ph+ ALL on the 14th of December 2012.17 Ponatinib treatment is provided to patients who are intolerant or resistant to two or more prior TKI therapies, including the use of dasatinib, nilotinib, and/or imatinib. The FDA approval was based on data obtained from the Phase II PACE trial of ponatinib.18 The PACE trial evaluated the efficacy and safety of ponatinib in CML and Ph+ ALL patients who were resistant or did not respond to dasatinib or nilotinib treatment and included those who harbored the T315I mutation. Overall, within a minimum follow-up of 48 months, this study demonstrated ponatinib’s continual efficacy and durability in this heavily pretreated population to retain long-term molecular and cytogenic responses.19
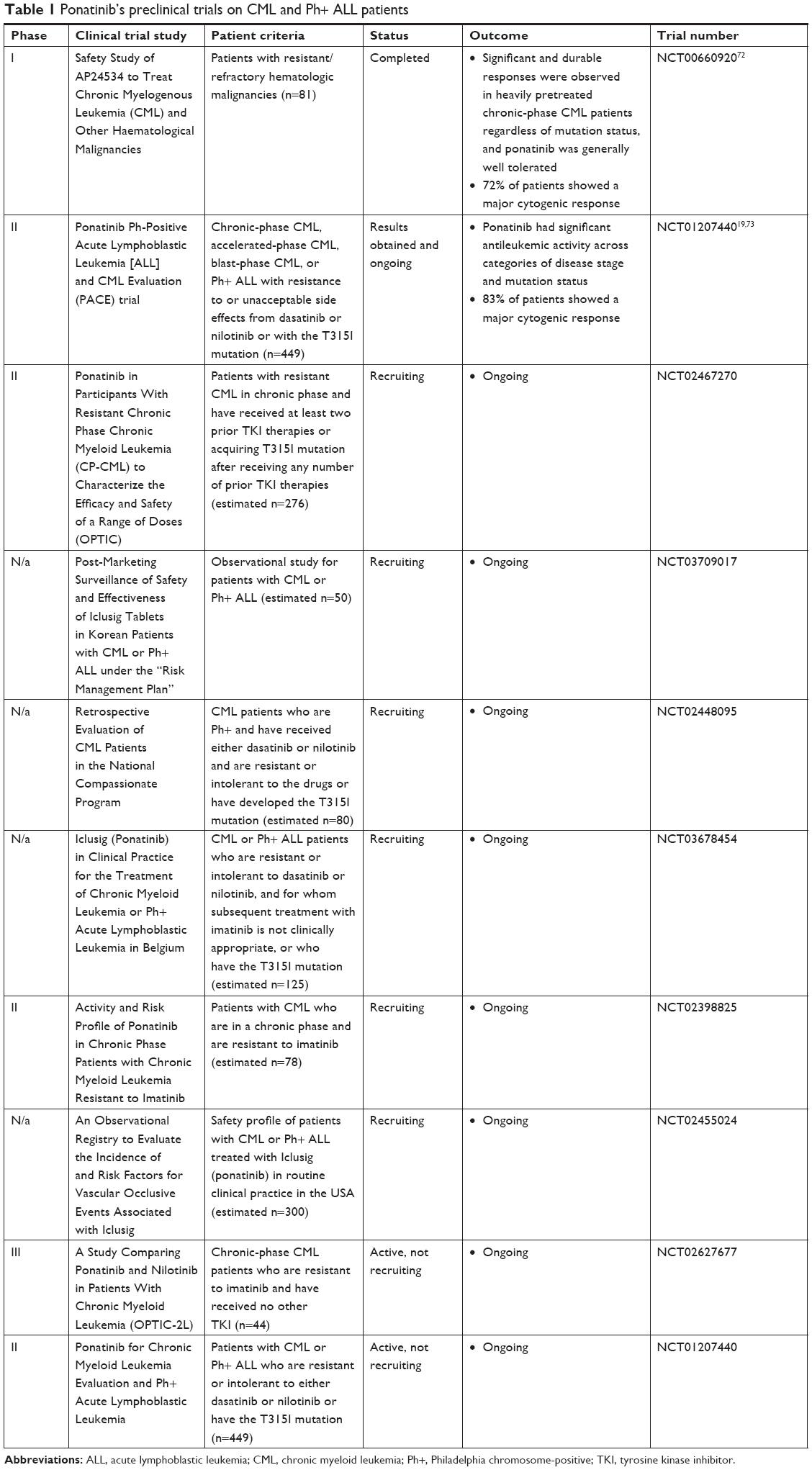 | Table 1 Ponatinib’s preclinical trials on CML and Ph+ ALL patients |
Despite the successful development and FDA approval of ponatinib and other TKIs (such as dasatinib, nilotinib, and imatinib), one of the main challenges within the research and management of TKIs is understanding and controlling the TKI-associated vascular adverse events (VAEs). In particular, the emergence of these TKIs for the treatment of hematologic malignancies has evidently shown that their use in patient treatment is linked with vascular toxicity.20,21 For instance, ponatinib-related VAEs have been reported in as many as 20%–40% of patients receiving the standard 45 mg dose, and ponatinib has also been associated with an increased risk of cardiovascular events and hypertension. However, the cause of these events is unclear.22,23 In an attempt to identify the cause, studies have demonstrated ponatinib’s inhibitory effect on human vascular endothelial cells (HUVECs) and human microvascular endothelial cell line HMEC-1 with IC50 values ranging between 100 and 200 nM.24,25 Moreover, these studies further analyzed the inhibitory effect of ponatinib on HUVECs overexpressing VEGFR2, and this effect was observed to be reduced compared to the control cells.24 VEGFR2 is one of ponatinib’s targets but also serves as a key driver of angiogenesis and as a survival factor of endothelial cells.12,26 Although these studies are not conclusive, they provide an insight into ponatinib’s possible antiangiogenic effect that is possibly mediated by VEGFR2 inhibition, providing an explanation for TKI-associated VAEs.
Mechanisms of resistance to ponatinib
As with many targeted therapies, tumor resistance is expected to arise to ponatinib and other malignancies. Several studies have explored this notion, and here we highlight these studies and discuss implications of the mechanisms of resistance. A recent study by Lu et al evaluated resistance mechanisms by exposing TKI-naïve and dasatinib-resistant BCR-ABL1-positive K562 cells to ponatinib with continuous, long-term co-culture. Enhanced Axl overexpression was detected in the ponatinib-resistant TKI-naïve cells, while mutations in the BCR-ABL1 KD including T315I and compound mutation G250E/E255K were observed in the dasatinib-resistant cells.27 Zabriskie et al thoroughly investigated existing mutations in the BCR-ABL1 kinase, particularly focusing on additional mutations paired with the T315I mutation commonly seen in CML.28 They observed that many compound mutations conferred resistance to ponatinib with the E255V/T315I compound mutation conferring the greatest resistance to ponatinib in in vitro proliferation assays. Further evidence that this mutation confers resistance to ponatinib was demonstrated when E255V/T315I was observed in a specimen of a Ph+ ALL patient who showed transient response to ponatinib but underwent rapid hematologic relapse and was discontinued from ponatinib treatment.28 The I315M mutation has also been shown to confer resistance to ponatinib in cell-based assays and in another Ph+ ALL patient.1,28,29
However, the presence of these mutations in patient samples is low. Parker et al examined low-level mutations in a large set of CML and Ph+ ALL patient specimens, and although low-level mutations were observed in 15% of samples, they did not undergo clonal expansion during ponatinib treatment and did not correlate with overall outcome.30 Similarly, single or compound mutation identified from 267 chronic-phase CML patients did not confer primary and/or secondary resistance to ponatinib.31 Therefore, clinical decisions and policy changes regarding the use of ponatinib cannot currently be made based on the current knowledge of resistance mechanisms to ponatinib. An analysis with larger sample sizes and longer treatment times may be required to increase our understanding of the key drivers of resistance to ponatinib in the future.
The multi-kinase inhibitory properties of ponatinib
ABL and solid tumors
The ABL family of kinases (also known as Arg), ABL1 and ABL2, has been recently shown to contribute to the progression of solid tumors (including colorectal cancer, chondrosarcoma, gastric cancer, ovarian cancer, and melanoma cancer) through activation pathways which are distinct from those observed in ABL-induced leukemias.32–36 In fact, studies have shown that solid tumors, which are induced by ABL kinases, are not linked to the event of chromosome translocation as seen during the progression of leukemia and are rather driven by elevated ABL1 or ABL2 expression, amplification, protein expression, metabolic stress, and/or inactivation of negative regulatory proteins.32,37 Several reports have identified that multiple RTKs can contribute to enhanced ABL activation, including EGFR, PDGFR, and the hepatocyte growth factor receptor (MET), all of which are known to facilitate changes in cell proliferation, migration, invasion, and survival.38–41
FLT3, c-KIT, VEGFR, PDGFR, and SRC
As mentioned previously, ponatinib was initially designed to inhibit BCR-ABL; however, studies have also identified ponatinib to target several other kinases, and ponatinib is therefore classified as a multi-TKI. O’Hare et al utilized the Kinase Hotspot assay (a direct measurement of the substrate phosphorylation) to profile ponatinib against >100 kinases in primary CML cell lines and identified several underlying targets, including FLT3 (IC50 0.3–2 nmol/L) and c-KIT (IC50 8–20 nmol/L).12 Ponatinib was also effective against the members of the tyrosine kinase families including FGFR (IC50 2.2 nmol/L), VEGFR (IC50 1.5 nmol/L), PDGFR (IC50 1.1 nmol/L), and c-SRC (IC50 5.4 nmol/L).12,42–44 Ponatinib’s molecular targets have thus expanded to other cancer types as summarized in Table 2.
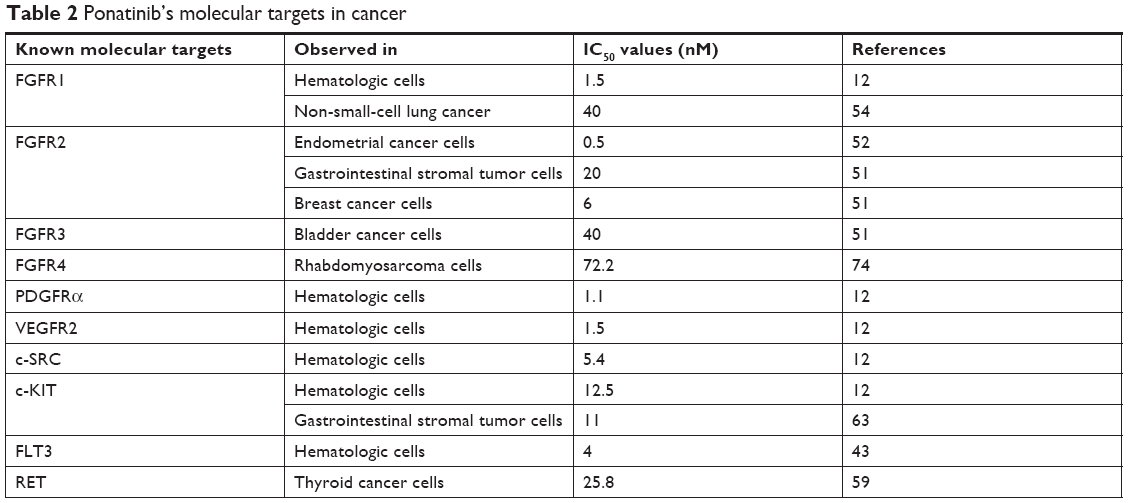 | Table 2 Ponatinib’s molecular targets in cancer |
FGFR family
The FGFR family of tyrosine kinase receptors consists of four highly conserved family members (FGFR1–4) and has been identified to play a role in the progression of multiple tumor types including squamous cell lung cancers, breast cancers, gastric cancers, and endometrial cancers. Next-generation sequencing of 4,853 patient samples with varying cancers identified that 343 (7.1%) samples contained FGFR aberrations. Of these 343 cases, most of the aberrations were FGFR gene amplifications (66%), while 26% were FGFR mutations and 8% were FGFR rearrangements.45 FGFR gene fusions have also been reported in several cancer types.46,47 Lymphomas and leukemias have also been identified with FGFR rearrangements and FGFR fusions.48–50
Thus, FGFR represents a viable target in cancer therapy.13 In fact, studies highlight the potential of ponatinib as an FGFR inhibitor in several cancer types. For example, Gozgit et al engineered Ba/F3 cells to express activated FGFR1–4 and demonstrated a potent inhibition with ponatinib treatment against the FGFR-mediated signaling pathway with IC50 values of <40 nmol/L when compared to parental Ba/F3 cells.51 This study further examined the effect on ponatinib amongst a panel of 14 FGFR-amplified or FGFR-mutated cancer cell lines (endometrial, gastric, breast, lung, bladder, and colon), and similar IC50 values <40 nmol/L were observed. Ponatinib’s potency to reduce tumor growth was also observed in endometrial, bladder, and gastric cancer mouse models with FGFR mutations. Kim et al also investigated ponatinib’s inhibitory properties against FGFR kinases, in particular in endometrial cancer cells (MFE-296 and AN3CA) harboring activating FGFR2 mutations. Ponatinib treatment resulted in significant inhibition of cell proliferation, migration, and invasion compared to the control-treated cells.52 Additionally, this study further explored ponatinib’s effect on the downstream pathways associated with FGFR2 activation and highlighted the inhibitory effect on cell proliferation in endometrial cancer cells through ERK, AKT, STAT5, and phospholipase Cγ. Another member of the FGFR family of kinases, FGFR4, has been reported to be commonly amplified and activated in patients with rhabdomyosarcoma (RMS).53 Li et al identified ponatinib as a potent inhibitor against the growth of RMS cells (RMS772; IC50 <70 nmol/L) which were engineered to harbor mutated or wild-type FGFR4.74 This observation was further supported with the use of a mutated FGFR4 RMS mouse model, where ponatinib significantly reduced tumor growth compared to control tumors. Moreover, ponatinib has also been demonstrated to inhibit the growth and suppression of clonogenicity in NSCLC cell lines with elevated FGFR1 expression and the growth of primary lung cancer culture cells (IC50 <50 nmol/L).54,55 Taken together, these studies highlight ponatinib as a multi-targeted inhibitor which potentially acts as a pan-FGFR inhibitor in a range of cancer types, further broadening its clinical application.
Ponatinib and other targets
As mentioned previously, O’Hare et al not only identified ponatinib as a potent BCR-ABL inhibitor but also demonstrated its ability to target FLT3 kinases. In relation to hematologic malignancies, activating mutations of FLT3 have been observed in up to 30% of newly diagnosed patients with AML who were associated with poor prognosis in terms of relapse and overall survival when treated with standard therapies, and ponatinib was later found to be an attractive therapeutic agent.56–58 To explore ponatinib’s potential applications in hematologic malignancies beyond BCR-ABL-driven CML, Gozgit et al evaluated the cellular activity of ponatinib against FLT3 in a panel of leukemic cell lines (MV4-11, Kasumi-1, KG1, and EOL1) with dysregulated FLT3 expression and identified ponatinib to inhibit phosphorylation of activated FLT3, reduce cell viability, and induce apoptosis at concentrations <10 nmol/L.43 This observation was further supported in an MV4-11 mouse xenograft model where daily oral dose of ponatinib for 4 weeks resulted in a dose-dependent inhibition of p-FLT3 signaling and tumor regression.43 In addition, phosphorylated KIT, FGFR1, and PDGFR protein expression was also observed to be reduced at concentrations <100, <10, and <3 nmol/L, respectively. Due to these findings, this study suggests that further investigation of ponatinib in patients with FLT3-driven AML malignancies and other hematologic tumors driven by KIT, FGFR1, or PDGFR is warranted.
Ponatinib has also been demonstrated in other studies to target tumor-driving molecules in various cancers. De Falco et al showed that ponatinib can act as a potent inhibitor of the RET transmembrane RTK in thyroid cancers.59 Activated RET mutations are commonly seen in ~98% of medullary thyroid carcinomas and an estimated 50% of patients with sporadic thyroid tumors harboring somatic RET mutations.60–62 In this study, ponatinib was observed to inhibit immunopurified RET kinase at an IC50 value of 25.8 nM in NIH3T3 and RAT1 fibroblasts (stably expressing oncogenic RET mutations) and TT thyroid cancer cell lines. In addition, ponatinib also demonstrated a reduction in kinase activity of RET/V804M, an RET mutant known to confer resistance to other TKIs. Daily ponatinib treatment (30 mg/kg) by oral gavage for 3 weeks significantly reduced tumor growth compared to the vehicle-treated mice in a subcutaneous TT xenograft mouse model. Moreover, RET phosphorylation was also significantly decreased, as early as 5 hours after the commencement of ponatinib treatment. This study suggests that further studies evaluating ponatinib’s potential as an anti-RET inhibitor in patients affected by thyroid cancers and possibly other RET-related malignancies are warranted.
Another target of ponatinib is KIT.12,63 Garner et al evaluated the efficacy of ponatinib in gastrointestinal stromal tumor (GIST)-derived cell lines and demonstrated its potent effect (IC50 ≤11 nmol/L) on wild-type KIT and KIT with mutations within exon 11 (V559D and V560G), a primary “hotspot” which harbors activating mutations.63 This study also evaluated ponatinib’s potent inhibitory effect on the growth of a GIST-1 patient-derived xenograft (PDX) mouse model. Ponatinib treatment (30 mg/kg) resulted in complete tumor regression which was maintained 6 weeks following the withdrawal of ponatinib. In comparison, imatinib (300 mg/kg), sunitinib (80 mg/kg), and regorafenib (100 mg/kg) treatment demonstrated tumor inhibition to some extent but were all inferior to ponatinib in this model.
Furthermore, Sun et al demonstrated ponatinib’s inhibitory action against the multidrug-resistant (MDR) phenotype which is a result of the overexpression of ABC transporter family seen in various cancer cells.64 In fact, there are several ABC proteins that are known to be responsible for developing the MDR phenotype in cancer cells which include multidrug resistance proteins (MRPs).65 In this study, the overexpression of MRP7 was demonstrated in HEK/MRP7-transfected human embryonic kidney cells, and in the presence of 0.5 μM ponatinib, MRP7 gene expression was downregulated and MRP7 protein expression was reduced in a dose-dependent manner (0, 0.1, 0.25, and 0.5 μM). Although this study did not perform in vivo assays to further evaluate the findings, it highlighted the great potential of ponatinib in MDR cancer patients.
Based on preclinical studies demonstrating ponatinib’s potent effect against models involving FGFR2, RET, and KIT mutations, clinical trials on the treatment of patients with biliary cancer, solid cancers, and advanced NSCLC have commenced (NCT02265341, NCT02272998, and NCT01813734; Table 3).54,59,66
 | Table 3 Ponatinib’s clinical trials in other cancer types |
Preclinical evaluation of ponatinib in other tumor types
Ponatinib’s inhibitory effect against tumorigenic properties has also been evaluated in other cancers. Most notably, Wylie et al recently demonstrated that ponatinib exhibited antiproliferative effects against a large series of cell lines derived from different cancers.67 A study conducted by Zhang et al assessed ponatinib’s inhibitory effect against a commonly used GBM cell line U87MG in both in vitro and in vivo models.66 Ponatinib was observed to inhibit cell viability and induce apoptotic events with elevated sub-G1 DNA content, a commonly used apoptotic marker, in a dose-dependent manner (0.78–200 nM). Furthermore, this study observed inhibition of cell migration and cell invasion with ponatinib treatment at concentrations ranging between 1.25 and 20 nM. In a GBM subcutaneous xenograft mouse model, ponatinib markedly reduced tumor growth in a dose-dependent manner (5 and 10 mg/kg) after daily injection and also induced cell apoptosis in the tumor. In addition, Laramy et al investigated the variations in drug distribution and free fraction at various tumor implantation sites and how this influenced ponatinib’s efficacy in a PDX model of GBM.68 Comparing a heterotopic model (flank) and an orthotopic (intracranial) model, a daily oral dose of ponatinib (30 mg/kg) was observed to be more effective in reducing tumor growth of the flank (days of survival 32 vs 40) but not in the intracranial model (days of survival 40 vs 40). This study demonstrated ponatinib’s limited drug distribution or potential inability to cross the blood–brain barrier. Nonetheless, ponatinib is currently being evaluated in GBM patients.
The antitumor effects of ponatinib have also been demonstrated to reduce neuroblastoma cells in vitro and in vivo. Whittle et al exposed ponatinib to a range of neuroblastoma cell lines (CHP-134, CHP-212, NGP, LAN-5, SH-EP, SK-N-AS, SK-N-BE(2), and SK-N-SH) and observed that ponatinib reduced cell viability (<20 μM) and cell migration (1 μM).69 Daily ponatinib treatment (30 mg/kg) by oral gavage for 5 days per week for 3 weeks reduced tumor size and vascularity in SK-N-AS and SK-N-SH xenograft models. This was supported by the observation of reduced CD31 staining in the ponatinib-treated cohort compared to the untreated cohort, suggesting the possible antiangiogenic effects of ponatinib. Though these studies did not specifically asses which RTKs are inhibited by ponatinib, they demonstrate the potential for further investigations into the use of ponatinib.
Several mutated or overexpressed receptors along with several tumor-driving molecules are known to play a significant role during carcinogenesis, and therefore, combinational targeted therapies have been investigated to alleviate tumor burden driven by several cancer-promoting pathways. In fact, a number of in vitro and in vivo studies have highlighted the significant potential of ponatinib alone and in combination with other targeted inhibitors to develop a new therapeutic strategy to treat advanced cancers. Gozgit et al investigated various combinational treatments to target known activating mutations in FGFR2, which commonly occur along with genetic alterations in the mammalian target of rapamycin (mTOR) pathway.51 Their study examined the combinatorial treatment of ponatinib (anti-FGFR2 inhibitor) and ridaforolimus (anti-mTOR agent) and demonstrated the dual inhibition of both molecules in vitro and also induced tumor regression in an endometrial cancer model. Furthermore, Sokolov discovered that the combination of blinatumomab (a monoclonal antibody) and ponatinib resulted in improved long-term results in the treatment of relapsed Ph+ ALL.70 Based on these results, a Phase II clinical trial (NCT03263572) on this dual combination is currently recruiting Ph+ ALL patients.71
Conclusion
In summary, human malignancies are complex in nature and often involve multiple intrinsic cancer-driven pathways, and thus, the inhibition of single targets alone is likely to be therapeutically ineffective. Therefore, the use of a multiple targeted inhibitor like ponatinib alone or in combination with other agents that target other RTKs or cancer-related pathways provides an opportunity to inhibit cancer progression.
Acknowledgments
RBL is a recipient of the Victorian Cancer Agency Mid-Career Research Fellowship (MCRF15017). TLP is a recipient of the National Health and Medical Research Council Project Grants (1080498).
Disclosure
The authors report no conflicts of interest in this work.
References
O’Hare T, Shakespeare WC, Zhu X, et al. AP24534, a pan-BCR-ABL inhibitor for chronic myeloid leukemia, potently inhibits the T315I mutant and overcomes mutation-based resistance. Cancer Cell. 2009;16(5):401–412. | ||
Jorissen RN, Walker F, Pouliot N, Garrett TP, Ward CW, Burgess AW. Epidermal growth factor receptor: mechanisms of activation and signalling. Exp Cell Res. 2003;284(1):31–53. | ||
Porta R, Borea R, Coelho A, et al. FGFR a promising druggable target in cancer: molecular biology and new drugs. Crit Rev Oncol Hematol. 2017;113:256–267. | ||
Cohen MH, Johnson JR, Chen YF, Sridhara R, Pazdur R. FDA drug approval summary: erlotinib (Tarceva) tablets. Oncologist. 2005;10(7):461–466. | ||
Fukuoka M, Yano S, Giaccone G, et al. Multi-institutional randomized phase II trial of gefitinib for previously treated patients with advanced non-small-cell lung cancer (The IDEAL 1 Trial) [corrected]. J Clin Oncol. 2003;21(12):2237–2246. | ||
van Hellemond IE, Creemers GJ, van Warmerdam LJ, de Jong FA, Koornstra RH. Panitumumab monotherapy as a second-line treatment in metastasised colorectal cancer: a single centre experience. Clin Oncol (R Coll Radiol). 2014;26(3):135–141. | ||
Giusti RM, Shastri KA, Cohen MH, Keegan P, Pazdur R. FDA drug approval summary: panitumumab (Vectibix). Oncologist. 2007;12(5):577–583. | ||
Agus DB, Gordon MS, Taylor C, et al. Phase I clinical study of pertuzumab, a novel HER dimerization inhibitor, in patients with advanced cancer. J Clin Oncol. 2005;23(11):2534–2543. | ||
Baselga J, Norton L, Albanell J, Kim YM, Mendelsohn J. Recombinant humanized anti-HER2 antibody (Herceptin) enhances the antitumor activity of paclitaxel and doxorubicin against HER2/neu overexpressing human breast cancer xenografts. Cancer Res. 1998;58(13):2825–2831. | ||
Cortes JE, Khoury HJ, Kantarjian HM, et al. Long-term bosutinib for chronic phase chronic myeloid leukemia after failure of imatinib plus dasatinib and/or nilotinib. Am J Hematol. 2016;91(12):1206–1214. | ||
Shah NP, Rousselot P, Schiffer C, et al. Dasatinib in imatinib-resistant or -intolerant chronic-phase, chronic myeloid leukemia patients: 7-year follow-up of study CA180-034. Am J Hematol. 2016;91(9):869–874. | ||
O’Hare T, Shakespeare WC, Zhu X, et al. AP24534, a pan-BCR-ABL inhibitor for chronic myeloid leukemia, potently inhibits the T315I mutant and overcomes mutation-based resistance. Cancer Cell. 2009;16(5):401–412. | ||
Ren M, Hong M, Liu G, et al. Novel FGFR inhibitor ponatinib suppresses the growth of non-small cell lung cancer cells overexpressing FGFR1. Oncol Rep. 2013;29(6):2181–2190. | ||
Zhang J, Zhou Q, Gao G, et al. The effects of ponatinib, a multi-targeted tyrosine kinase inhibitor, against human U87 malignant glioblastoma cells. Onco Targets Ther. 2014;7:2013–2019. | ||
Huang WS, Metcalf CA, Sundaramoorthi R, et al. Discovery of 3-[2-(imidazo[1,2-b]pyridazin-3-yl)ethynyl]-4-methyl-N-{4-[(4-methylpiperazin-1-yl)methyl]-3-(trifluoromethyl)phenyl}benzamide (AP24534), a potent, orally active pan-inhibitor of breakpoint cluster region-abelson (BCR-ABL) kinase including the T315I gatekeeper mutant. J Med Chem. 2010;53(12):4701–4719. | ||
Cortes JE, Kantarjian H, Shah NP, et al. Ponatinib in refractory Philadelphia chromosome-positive leukemias. N Engl J Med. 2012;367(22):2075–2088. | ||
FDA approval for ponatinib hydrochloride. 2014 [cited June 3, 2016]. Available from: http://www.cancer.gov/about-cancer/treatment/drugs/fda-ponatinibhydrochloride. Accessed January 10, 2019. | ||
Sanford D, Kantarjian H, Skinner J, Jabbour E, Cortes J. Phase II trial of ponatinib in patients with chronic myeloid leukemia resistant to one previous tyrosine kinase inhibitor. Haematologica. 2015;100(12):e494–e495. | ||
Cortes JE, Kim D-W, Pinilla J, et al. PACE: a pivotal phase 2 trial of ponatinib in patients with CML and Ph+ ALL resistant or intolerant to dasatinib or nilotinib, or with the T315I mutation. Clin Lymphoma Myeloma Leuk. 2013;13:S384. | ||
Valent P, Hadzijusufovic E, Schernthaner GH, Wolf D, Rea D, Le Coutre P. Vascular safety issues in CML patients treated with BCR/ABL1 kinase inhibitors. Blood. 2015;125(6):901–906. | ||
Dahlén T, Edgren G, Lambe M, et al. Cardiovascular events associated with use of Tyrosine kinase inhibitors in Chronic Myeloid Leukemia: A population-based cohort study. Ann Intern Med. 2016;165(3):161–166. | ||
Cortes JE, Kim DW, Pinilla-Ibarz J, et al; PACE Investigators. A phase 2 trial of ponatinib in Philadelphia chromosome-positive leukemias. N Engl J Med. 2013;369(19):1783–1796. | ||
Gagnieu M-C. Cardio-vascular events occurring on ponatinib in chronic phase chronic myeloid leukemia patients, preliminary analysis of a multicenter cohort. Blood. 2013;122(21). | ||
Gover-Proaktor A, Granot G, Shapira S, et al. Ponatinib reduces viability, migration, and functionality of human endothelial cells. Leuk Lymphoma. 2017;58(6):1455–1467. | ||
Valent P, Hadzijusufovic E, Hoermann G, et al. Risk factors and mechanisms contributing to TKI-induced vascular events in patients with CML. Leuk Res. 2017;59:47–54. | ||
Basagiannis D, Zografou S, Murphy C, et al. VEGF induces signalling and angiogenesis by directing VEGFR2 internalisation through macropinocytosis. J Cell Sci. 2016;129(21):4091–4104. | ||
Lu L, Kok CH, Saunders VA, et al. Modelling ponatinib resistance in tyrosine kinase inhibitor-naïve and dasatinib resistant BCR-ABL1+ cell lines. Oncotarget. 2018;9(78):34735–34747. | ||
Zabriskie MS, Eide CA, Tantravahi SK, et al. BCR-ABL1 compound mutations combining key kinase domain positions confer clinical resistance to ponatinib in Ph chromosome-positive leukemia. Cancer Cell. 2014;26(3):428–442. | ||
Eide CA, Adrian LT, Tyner JW, et al. The ABL switch control inhibitor DCC-2036 is active against the chronic myeloid leukemia mutant BCR-ABLT315I and exhibits a narrow resistance profile. Cancer Res. 2011;71(9):3189–3195. | ||
Parker WT, Yeung DT, Yeoman AL, et al. The impact of multiple low-level BCR-ABL1 mutations on response to ponatinib. Blood. 2016;127(15):1870–1880. | ||
Deininger MW, Hodgson JG, Shah NP, et al. Compound mutations in BCR-ABL1 are not major drivers of primary or secondary resistance to ponatinib in CP-CML patients. Blood. 2016;127(6):703–712. | ||
Ganguly SS, Fiore LS, Sims JT, et al. c-Abl and Arg are activated in human primary melanomas, promote melanoma cell invasion via distinct pathways, and drive metastatic progression. Oncogene. 2012;31(14):1804–1816. | ||
Chen WS, Kung HJ, Yang WK, Lin W. Comparative tyrosine-kinase profiles in colorectal cancers: enhanced arg expression in carcinoma as compared with adenoma and normal mucosa. Int J Cancer. 1999;83(5):579–584. | ||
O’Donovan M, Russell JM, O’Leary JJ, Gillan JA, Lawler MP, Gaffney EF. Abl expression, tumour grade, and apoptosis in chondrosarcoma. Mol Pathol. 1999;52(6):341–344. | ||
Wu CW, Li AF, Chi CW, et al. Arg tyrosine kinase expression in human gastric adenocarcinoma is associated with vessel invasion. Anticancer Res. 2003;23(1A):205–210. | ||
Niyazi M, Ghazizadeh M, Konishi H, Kawanami O, Sugisaki Y, Araki T. Expression of p73 and c-Abl proteins in human ovarian carcinomas. J Nippon Med Sch. 2003;70(3):234–242. | ||
Simpson L, He X, Pins M, et al. Renal medullary carcinoma and ABL gene amplification. J Urol. 2005;173(6):1883–1888. | ||
Li R, Knight JF, Park M, Pendergast AM. Abl kinases regulate HGF/Met signaling required for epithelial cell scattering, tubulogenesis and motility. PLoS One. 2015;10(5):e0124960. | ||
Plattner R, Kadlec L, De Mali KA, Kazlauskas A, Pendergast AM. c-Abl is activated by growth factors and Src family kinases and has a role in the cellular response to PDGF. Genes Dev. 1999;13(18):2400–2411. | ||
Furstoss O, Dorey K, Simon V, Barilà D, Superti-Furga G, Roche S. c-Abl is an effector of Src for growth factor-induced c-myc expression and DNA synthesis. EMBO J. 2002;21(4):514–524. | ||
Dixit A, Verkhivker GM. Hierarchical modeling of activation mechanisms in the ABL and EGFR kinase domains: thermodynamic and mechanistic catalysts of kinase activation by cancer mutations. PLoS Comput Biol. 2009;5(8):e1000487–22. | ||
European Medicines Agency. Iclusig (Ponatinib): summary of product characteristics. 2014 [cited July 13, 2016]. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/002695/WC500145646.pdf. Accessed January 10, 2019. | ||
Gozgit JM, Wong MJ, Wardwell S, et al. Potent activity of ponatinib (AP24534) in models of FLT3-driven acute myeloid leukemia and other hematologic malignancies. Mol Cancer Ther. 2011;10(6):1028–1035. | ||
Rivera VM, et al. Potent antitumor activity of AP24534, an orally active inhibitor of Bcr-Abl variants including T315I, in in vitro and in vivo models of chronic myeloid leukemia (CML). Blood. 2007;110(11):1032. | ||
Helsten T, Elkin S, Arthur E, et al. The FGFR landscape in cancer: analysis of 4,853 tumors by next-generation sequencing. Clin Cancer Res. 2016;22(1):259–267. | ||
Costa R, Carneiro BA, Taxter T, et al. FGFR3-TACC3 fusion in solid tumors: mini review. Oncotarget. 2016;7(34):55924–55938. | ||
Gallo LH, Nelson KN, Meyer AN, Donoghue DJ. Functions of fibroblast growth factor receptors in cancer defined by novel translocations and mutations. Cytokine Growth Factor Rev. 2015;26(4):425–449. | ||
Maeda T, Yagasaki F, Ishikawa M, Takahashi N, Bessho M. Transforming property of TEL-FGFR3 mediated through PI3-K in a T-cell lymphoma that subsequently progressed to AML. Blood. 2005;105(5):2115–2123. | ||
Nakamura Y, Ito Y, Wakimoto N, Kakegawa E, Uchida Y, Bessho M. A novel fusion of SQSTM1 and FGFR1 in a patient with acute myelomonocytic leukemia with t(5;8)(q35;p11) translocation. Blood Cancer J. 2014;4:e265. | ||
Yagasaki F, Wakao D, Yokoyama Y, et al. Fusion of ETV6 to fibroblast growth factor receptor 3 in peripheral T-cell lymphoma with a t(4;12)(p16;p13) chromosomal translocation. Cancer Res. 2001;61(23):8371–8374. | ||
Gozgit JM, Wong MJ, Moran L, et al. Ponatinib (AP24534), a multitargeted pan-FGFR inhibitor with activity in multiple FGFR-amplified or mutated cancer models. Mol Cancer Ther. 2012;11(3):690–699. | ||
Kim DH, Kwak Y, Kim ND, Sim T. Antitumor effects and molecular mechanisms of ponatinib on endometrial cancer cells harboring activating FGFR2 mutations. Cancer Biol Ther. 2016;17(1):65–78. | ||
Taylor JG, Cheuk AT, Tsang PS, et al. Identification of FGFR4-activating mutations in human rhabdomyosarcomas that promote metastasis in xenotransplanted models. J Clin Invest. 2009;119(11):3395–3407. | ||
Ren M, Hong M, Liu G, et al. Novel FGFR inhibitor ponatinib suppresses the growth of non-small cell lung cancer cells overexpressing FGFR1. Oncol Rep. 2013;29(6):2181–2190. | ||
Singleton KR, Hinz TK, Kleczko EK, et al. Kinome RNAi screens reveal synergistic targeting of mTOR and FGFR1 pathways for treatment of lung cancer and HNSCC. Cancer Res. 2015;75(20):4398–4406. | ||
Birg F, Courcoul M, Rosnet O, et al. Expression of the FMS/KIT-like gene FLT3 in human acute leukemias of the myeloid and lymphoid lineages. Blood. 1992;80(10):2584–2593. | ||
Brasel K, Escobar S, Anderberg R, de Vries P, Gruss HJ, Lyman SD. Expression of the flt3 receptor and its ligand on hematopoietic cells. Leukemia. 1995;9(7):1212–1218. | ||
Piacibello W, Fubini L, Sanavio F, et al. Effects of human FLT3 ligand on myeloid leukemia cell growth: heterogeneity in response and synergy with other hematopoietic growth factors. Blood. 1995;86(11):4105–4114. | ||
De Falco V, Buonocore P, Muthu M, et al. Ponatinib (AP24534) is a novel potent inhibitor of oncogenic RET mutants associated with thyroid cancer. J Clin Endocrinol Metab. 2013;98(5):E811–E819. | ||
Santoro M, Rosati R, Grieco M, et al. The ret proto-oncogene is consistently expressed in human pheochromocytomas and thyroid medullary carcinomas. Oncogene. 1990;5(10):1595–1598. | ||
Blaugrund JE, Johns MM, Eby YJ, et al. RET proto-oncogene mutations in inherited and sporadic medullary thyroid cancer. Hum Mol Genet. 1994;3(10):1895–1897. | ||
Eng C, Mulligan LM, Smith DP, et al. Mutation of the RET protooncogene in sporadic medullary thyroid carcinoma. Genes Chromosomes Cancer. 1995;12(3):209–212. | ||
Garner AP, Gozgit JM, Anjum R, et al. Ponatinib inhibits polyclonal drug-resistant KIT oncoproteins and shows therapeutic potential in heavily pretreated gastrointestinal stromal tumor (GIST) patients. Clin Cancer Res. 2014;20(22):5745–5755. | ||
Sun YL, Kumar P, Sodani K, et al. Ponatinib enhances anticancer drug sensitivity in MRP7-overexpressing cells. Oncol Rep. 2014;31(4):1605–1612. | ||
Sun YL, Patel A, Kumar P, Chen ZS. Role of ABC transporters in cancer chemotherapy. Chin J Cancer. 2012;31(2):51–57. | ||
Zhang J, Zhou Q, Gao G, et al. The effects of ponatinib, a multi-targeted tyrosine kinase inhibitor, against human U87 malignant glioblastoma cells. Onco Targets Ther. 2014;7:2013–2019. | ||
Wylie AA, Schoepfer J, Jahnke W, et al. The allosteric inhibitor ABL001 enables dual targeting of BCR-ABL1. Nature. 2017;543(7647):733–737. | ||
Laramy JK, Kim M, Gupta SK, et al. Heterogeneous binding and central nervous system distribution of the multitargeted kinase inhibitor ponatinib restrict orthotopic efficacy in a patient-derived xenograft model of glioblastoma. J Pharmacol Exp Ther. 2017;363(2):136–147. | ||
Whittle SB, Patel K, Zhang L, et al. The novel kinase inhibitor ponatinib is an effective anti-angiogenic agent against neuroblastoma. Invest New Drugs. 2016;34(6):685–692. | ||
Sokolov AN. Blinatumomab + tyrosine kinase inhibitors in the treatment of relapsed philadelphia chromosome positive acute lymphoblastic leukemia patients – clinical efficacy and peripheral blood lymphocytes subpopulations kinetics. Blood. 2016;128(22):4024. | ||
M.D. Anderson Cancer Center, Amgen, Takeda. Blinatumomab and Ponatinib in Patients with Philadelphia Chromosome (Ph)-Positive and/or BCR-ABL Positive Acute Lymphoblastic Leukemia (ALL). Available from: https://clinicaltrials.gov/ct2/show/NCT03263572. NML identifier: NCT03263572. Accessed January 10, 2019. | ||
Mauro MJ, Courtes JE, Kantarjian H. Safety and durability of ponatinib in patients with Philadelphia chromosome-positive (Ph+) leukemia: Long-term follow-up of an ongoing phase I study. J Clin Oncol. 2013;31. | ||
Hochhaus A. Molecular responses with ponatinib in patients with philadelphia chromosome positive (Ph+) leukemia: results from the pace trial. Blood. 2012;120:3763. | ||
Li SQ, Cheuk AT, Shern JF, et al. Targeting wild-type and mutationally activated FGFR4 in rhabdomyosarcoma with the inhibitor ponatinib (AP24534). PLoS One. 2013;8(10):e76551. |
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.