")
Back to Journals » International Journal of Nanomedicine » Volume 10 » Issue 1
Poly(n-butylcyanoacrylate) nanoparticles for oral delivery of quercetin: preparation, characterization, and pharmacokinetics and biodistribution studies in Wistar rats
Received 10 January 2015
Accepted for publication 31 March 2015
Published 10 June 2015 Volume 2015:10(1) Pages 3921—3935
DOI https://doi.org/10.2147/IJN.S80706
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Thomas Webster
Mayur Bagad, Zaved Ahmed Khan
Medical Biotechnology Division, School of Biosciences and Technology, VIT University, Vellore Tamil Nadu, India
Background: Quercetin (QT) is a potential bioflavonol and antioxidant with poor bioavailability and very low distribution in the brain. A new oral delivery system comprising of poly(n-butylcyanoacrylate) nanoparticles (PBCA NPs) was introduced to improve the oral bioavailability of QT and to increase its distribution in the brain. Physicochemical characteristics, in vitro release, stability in simulated gastric fluid and intestinal fluids, and pharmacokinetics and biodistribution studies of QT-PBCA NPs coated with polysorbate-80 (P-80) were investigated.
Objective: This study aimed to investigate the physicochemical characteristics, in vitro release, stability in simulated gastric fluid and intestinal fluids, and pharmacokinetics and biodistribution studies of QT-PBCA NPs coated with polysorbate-80 (P-80).
Results: The results showed that QT-PBCA NPs and QT-PBCA NPs coated with P-80 (QT-PBCA+P-80) had mean particle sizes of 161.1±0.44 nm and 166.6±0.33 nm respectively, and appeared spherical in shape under transmission electron microscopy. The mean entrapment efficiency was 79.86%±0.45% for QT-PBCA NPs and 74.58%±1.44% for QT-PBCA+P-80. The in vitro release of QT-PBCA NPs and QT-PBCA+P-80 showed an initial burst release followed by a sustained release when compared to free QT. The relative bioavailability of QT-PBCA NPs and QT-PBCA+P-80 enhanced QT bioavailability by 2.38- and 4.93-fold respectively, when compared to free QT. The biodistribution study in rats showed that a higher concentration of QT was detected in the brain after the NPs were coated with P-80.
Conclusion: This study indicates that PBCA NPs coated with P-80 can be potential drug carriers for poorly water-soluble drugs. These NPs were observed to improve the drugs’ oral bioavailability and enhance their transport to the brain.
Keywords: bioavailability, biodistribution, nanoparticles, pharmacokinetics, poly (n-butylcyanoacrylate), quercetin
Introduction
Quercetin (QT) (3,3′,4′,5,7-pentahydroxyflavone) is a potential flavonoid and is generally found in fruits, vegetables, leaves, and grains.1,2 QT, due to its high antioxidant activity, acts as a free radical scavenger and protects the cells from oxidative stress generated as an outcome of reactive oxygen species.3 It delivers a low intrinsic toxicity and has a surprisingly wide range of beneficial actions, such as antiplatelet, antiradical, anti-inflammatory, antimutagenic, anticarcinogenic, anti-angiogenic, antibacterial, antitumor, antiviral, antianxiety, and cognitive-enhancing effects on the central nervous system.4–6 However, despite the wide spectrum of pharmacological properties, the use of QT in the pharmaceutical field is obstructed due to its hydrophobic nature, physiological medium instability,7 and limited consumption in the brain.5,8 These properties of QT result in low oral bioavailability and restrict its permeability through the blood–brain barrier (BBB). Addressing this problem continues to be spotlighted as a major task while developing formulations for clinical efficacy.
In the past few decades, many pharmaceutical companies have shifted their focus to the formulation of polymeric nanoparticles (NPs) as controlled drug delivery systems in order to improve the oral bioavailability of lipophilic and hydrophobic drugs. This has been achieved through specialized uptake mechanisms, increased half-life (t1/2), minimized toxicity, and increased ability of NPs to cross the physiological barriers such as the BBB.9,10 To improve the oral bioavailability of QT, various attempts have been made by many researchers to formulate various drug delivery systems of current drug therapy, such as poly(D,L-lactide) (PLA) NPs,7 solid lipid NPs,8 lecithin-based novel cationic nanocarriers (LeciPlex),11 chitosan NPs,12 liposomes,13 and poly(D,L-lactide-co-glycolide) (PLGA) NPs,14 but all with limited effects. However, no detailed study has been reported on improving the brain permeability of the drug.
In contrast, poly(n-butylcyanoacrylate) (PBCA) has received considerable attention in recent years due to its ability to overcome the limitations of previous colloidal carriers.15 PBCA is a biodegradable, biocompatible, minimally toxic, bioabsorbable, and bioadhesive polymer that has been extensively investigated in the past few years for controlled and targeted drug delivery.16 A recent study has shown that PBCA NPs have been widely investigated to enhance the oral absorption of poorly soluble polyphenols and isoflavones such as curcumin and puerarin.17,18 Most interestingly, PBCA NPs have been employed as effective delivery systems in the brain because the particles entrap the compounds and prevent rapid elimination or degradation as well as promote penetration through the BBB and distribution in the brain tissue when coated with polysorbate-80 (P-80) on their surfaces.19 There have been several reports of PBCA NPs coated with P-80 that could significantly improve the delivery to the brain of anticancer agents,20 antifungal drugs,21 peptides,22 MRZ 2/576 (N-methyl-d-aspartate receptor antagonists),23 antibiotics,24 nonsteroidal anti-inflammatory drugs,25 and antiviral drugs.26 However, till date QT-PBCA NPs coated with 1% P-80 have not been reported to have improved their bioavailability and enhanced the distribution of QT in the brain via oral administration. In this study, QT-PBCA NPs were prepared by the anionic emulsion polymerization method and the physicochemical characteristics of NPs, in vitro release, and stability of NPs were investigated. Additionally, pharmacokinetics and biodistribution studies of QT-PBCA NPs and QT-PBCA+P-80 (coated with P-80) after oral delivery to male Wistar rats were evaluated with free QT solution.
Materials and methods
Materials
QT was obtained from Sigma-Aldrich (St. Louis, MO, USA), and Pluronic® F-68 was provided by the HiMedia Labs (India). The monomer, n-butylcyanoacrylate (n-BCA) was a generous gift sample from Tong Shen Enterprise Co., Ltd (Taiwan) and was utilized without any further purification. P-80, acetonitrile high-performance liquid chromatography (HPLC) grade, and methanol HPLC grade were purchased from S.D. Fine-Chem Ltd., (Mumbai, India). All other reagents used in the study were of analytical regent grade.
Preparation of QT-PBCA NPs
QT-PBCA NPs were prepared according to the anionic emulsion polymerization method with some modifications.27,28 Briefly, 1% (w/v) of Pluronic F-68 was dissolved in the acidic medium containing 0.01 N HCl (pH 2–3) at room temperature. QT (0.1% w/v) powder was then added to the above system after dissolving the powder separately in a minimum quantity of ethanol. Finally, the monomer n-BCA (1% w/v) was injected drop by drop into the stirred medium under continuous magnetic stirring. Polymerization reaction was carried for 4 hours with constant stirring at 400 rpm, under room temperature. 0.1 N NaOH was used to adjust the pH of the resulting suspension to 6.8±0.1. The polymerization reaction mixture was stirred for an additional 2 hours to ensure complete removal of ethanol in the mixture under constant stirring, and the reaction was gradually completed. The QT-PBCA suspension was obtained after the above system was filtered through a 0.2 μm syringe filter membrane (HiMedia Labs) to remove the aggregates. The NPs were separated by ultracentrifugation at 16,000 rpm for 60 minutes and rinsed with distilled water at least three times. The formed pellets were resuspended in distilled water and lyophilized with trehalose dihydrate (1%) acting as cryoprotectants,29 using a lyophilizer (Lark, India). Drug-free NPs (blank PBCA NPs) were made in the same method as that for preparing QT-containing NPs except that in the former the drug was absent.
Later, for coating the lyophilized powder of QT-PBCA NPs were resuspended in phosphate buffered saline (PBS), containing 1% P-80 solution at a final concentration of 20 mg/mL. Subsequently, the NPs were incubated for 30 minutes at 20°C under constant stirring, and were finally lyophilized.30
Analysis of particle size, zeta potential, and morphology
Particle size and surface charges (zeta potential) of the QT-PBCA NPs and QT-PBCA+P-80 were determined by light scattering based on laser diffraction using the Malvern Zetasizer (Nano Sizer ZS90; Malvern Instruments, Inc., Malvern, UK) after suitable dilution. Analysis (n=3) was carried out for 120 seconds at room temperature at a 90° angle of detection. The apparatus consisted of a He–Ne laser (5 mW) and a sample-holding cell of 5 mL capacity. The values of the particle sizes and zeta potential were presented as mean ± standard deviations (SDs) from three replicate samples.
The morphologies of QT-PBCA NPs and QT-PBCA+P-80 were observed under a transmission electron microscope (TEM) (JEOL JEM-1200EX; Tokyo, Japan) using the negative staining method. The sample was made by dispersing the NPs in distilled water, and one drop was added to a copper grid to make a thin liquid film. Later, the film was negatively stained by adding 2% (w/v) phosphotungstic acid (pH 7.0). The grid was dried at room temperature and the morphology of NPs was examined under TEM. Subsequently, photographs were taken at 30,000× magnification and 80 kV voltage.
Determination of drug entrapment efficiency of QT-PBCA NPs
The amount of QT entrapped in the PBCA NPs and coated with 1% P-80 was measured by the ultracentrifugation method (C-24 BL; Remi Laboratory Instruments, India), in accordance to the previous reports.31 The freshly prepared NP suspension was centrifuged at 20,000 rpm for 60 minutes at 4°C to obtain pellets of NPs and the supernatant was placed in a separate tube. The obtained pellet was dissolved in methanol to extract QT that was diluted appropriately. The concentration of QT was measured by UV–VIS absorbance spectroscopy at 374 nm by using a double-beam UV–VIS spectrophotometer (AU-2700; Systronics, India). Drug entrapment efficiency (DEE) was calculated as DEE (%) = (amount of QT in NPs/initial amount of QT) ×100.
Additionally, for the spectroscopic scan analysis from 200 to 700 nm, 1 mL each of free QT solution and QT-PBCA NP suspension were used.
Fourier transform infrared spectral study
Fourier transform infrared (FTIR) spectra (Spectrum RX-1; PerkinElmer, Waltham, MA, USA) were taken to investigate the possible chemical interactions between QT after conjugating with PBCA NPs. Free QT, QT-PBCA NPs, and blank PBCA NPs were crushed with KBr to obtain the pellets by applying a pressure of 300 kg/cm2. FTIR spectra of the above sample were obtained by averaging 32 interferograms with a resolution of 2 cm−1 in the range of 1,000–4,000 cm−1.
Differential scanning calorimetry
The physical state of QT entrapped in PBCA NPs was characterized using differential scanning calorimetry (DSC) thermogram analysis (Mettler Toledo DSC 822e; India). Each sample (10–20 mg of free QT, QT-PBCA NPs, and blank PBCA NPs and n-BCA) was sealed separately in standard aluminum pans. The samples were purged in the DSC with pure dry nitrogen gas set at a flow rate of 40 mL/min. The change in temperature was set at 20°C/min, and the heat flow was recorded from 0°C to 350°C.
X-ray diffraction
X-ray diffraction (XRD) patterns of free QT, QT-PBCA NPs, and blank PBCA NPs were obtained using an XRD instrument (D8 ADVANCE; Bruker, India). Measurements were carried out at a voltage of 40 kV and current of 20 mA. The diffractograms were performed with a scanning rate of 2°/min over a range of 4°−40° (2θ).
Drug stability study of simulated gastric fluid and simulated intestinal fluid
The stabilities of QT-PBCA NPs and QT-PBCA+P-80 were evaluated in simulated gastric fluid (SGF) and simulated intestinal fluid (SIF).32 Briefly, 1 mL of QT-PBCA and QT-PBCA+P-80 (equivalent to 20 mg) was suspended in 10 mL of both SGF and SIF. This mixture was incubated for 3 hours in the case of SGF (pH 1.2), while for SIF (pH 6.8) the time interval was 6 hours. The tubes were placed in an orbital shaker and were maintained at 37°C and at 100 rpm. At different time intervals, a known volume of suspension was taken out and centrifuged at 5,000 rpm for 15 minutes. The supernatant was separated to quantify the amount of the drug by UV–VIS spectroscopy at 374 nm. Later, the amount of the remaining drug after the incubation for 3 hours in SGF and 6 hours in SIF from each formulation was determined.
In vitro release studies
The in vitro release of QT from QT-PBCA NPs and QT-PBCA+P-80 was carried out in PBS (0.01 M PBS, pH 7.4) medium using a dialysis bag method.33,34 Briefly, lyophilized NPs equivalent to 2 mg of QT (QT-PBCA NPs and QT-PBCA+P-80) or free QT were suspended in 2 mL of PBS (pH 7.4) using cellulose dialysis tube bags (with molecular weight cut-off size 12,000 Da). The bags were dipped into 200 mL PBS (pH 7.4) containing 0.01% (v/v) Polysorbate-80 to maintain the sink condition. Dialysis was carried out at 37°C±1°C with magnetic stirring at 300 rpm. At regular time intervals, 1 mL of the sample was withdrawn and the same volume of fresh PBS was added to maintain a constant volume. The amount of QT in the medium was analyzed by using the HPLC-based method mentioned below. The release from each formulation was estimated at triplicate and the results were expressed as mean ± SD.
Formulation stability studies
Formulation stability upon storage at room temperatures was studied to evaluate the stability of the NP formulation containing QT. After the freeze drying process, the QT-PBCA NPs were stored in desiccators under vacuum at room temperature over a period of 180 days. The sampling time points were 0, 30, 60, 120, and 180 days. The formulations were studied for their particle size, polydispersity index, zeta potential, entrapment efficiency, as well as for any changes in the physical appearance of the NPs.
In vivo oral pharmacokinetics and biodistribution studies in rats
Animals
The study was performed in male Wistar albino rats, having a mean weight of 180–220 g, procured from the animal house facility at VIT University, Vellore, Tamil Nadu, India. The animal experiments were duly approved by the Institutional Animal Ethics Committee (IAEC) of VIT University, Vellore, Tamil Nadu, India (VIT/IAEC/VIIth/32). The rats were housed four per cage under standard conditions at a temperature of 25°C±2°C and relative humidity of 50%–60% under natural light. They had free access to standard rodent diet of commercial pelleted feed from Hindustan Lever Ltd (Mumbai, India) and water ad libitum prior to each experiment. The animals were fasted overnight before the start of the experiment and allowed free access of water ad libitum.
Dosing and sampling
QT-PBCA NPs and QT-PBCA+P-80 were freshly prepared for oral administration. The rats were allocated randomly in four groups (n=5). Group I was administered with a free QT solution (50 mg/kg). Group II was treated with QT-PBCA NPs, and group III received QT-PBCA+P-80, whereas group IV served as control. All formulations were given orally at a dose of 50 mg/kg body weight via oral feeding cannula (16 no).35 Blood samples (approximately 0.3 mL) were taken from the tail vein under mild anesthesia and withdrawn into heparinized centrifuge tubes at predetermined time intervals. Plasma was immediately separated by centrifuging the blood samples at 3,000 rpm for 10 minutes at 4°C and stored at −80°C until analysis. To determine the organ distribution of the drug at distinct time points, the animals were sacrificed (n=5) by cervical dislocation. Tissue samples from the brain, liver, heart, spleen, and kidney were quickly removed and stored at −80°C before analysis.36
Plasma and tissue sample preparation
The plasma sample of 100 μL was extracted with 1 mL of methanol on a vortex mixer for 5 minutes. The tubes were then centrifuged at 10,000 rpm for 10 minutes at 4°C. The clear supernatant was collected and filtered (0.20 μm syringe filters). Twenty microliter of the sample was injected into the HPLC system for determination of QT in plasma. Tissue samples (not more than 100 mg) were homogenized with 500 μL in 1× PBS (pH 7.4). The tissue homogenates were centrifuged at 10,000 rpm for 10 minutes at 4°C and the clear supernatant thus obtained was used further. Then, 200 μL aliquots of the clear tissue homogenates with 300 μL of methanol were added and the dispersion was vortexed for 2 minutes. The samples were later centrifuged at 10,000 rpm for 10 minutes at 4°C. The clear supernatant was filtered (0.20 μm syringe filters) and was injected into the HPLC system for determining QT in tissue samples.
Pharmacokinetics analysis
The pharmacokinetics analysis of plasma concentration–time profile was carried out by a noncompartmental model using Kinetica software, version 5.2 (Thermo Fisher Scientific Inc., Waltham, MA, USA). All data were expressed as mean ± SD and the level of significance was taken as P<0.05.
HPLC analysis of QT
QT content was analyzed by reverse phase-HPLC with a UV detector and a vacuum degasser run by Autochro-3000 software (Acme 9000 series; Young Lin, USA). Chromatographic separation was achieved by using a reverse-phase C18 column (150 mm ×4.6 mm, pore size 5 μm, Kromasil). The mobile phase was made up of methanol and water (70:30 v/v) (pH adjusted to 3.64 with glacial acetic acid). The separation was performed under an isocratic condition with a constant flow rate 1.0 mL/min, injection volume 20 μL, column temperature 25°C, and a detection wavelength of 374 nm.33 The calibration curve for QT was linear within the range of 0.2–100 μg/mL concentrations in the mobile phase with correlation coefficients of R2=0.9996. The detection limit (signal-to-noise ratio of 3:1) and the quantification limit (signal-to-noise ratio of 10:1) were found to be 0.05 μg/mL and 0.1 μg/mL, respectively. The mean recovery of QT in plasma and tissue homogenates were within the range of 94.72%–104.54% at the present assay condition. The precision of QT calculated as relative SD was lower than 10% for intra- and inter-day assays.
Results and discussion
Preparation and physicochemical characterization of QT-PBCA NPs
The schematic illustration of the fabrication of QT-PBCA NPs is shown in Figure 1. Based on the anionic emulsion polymerization process, the swollen structures formed by the micelles were taken up by the BCA monomer.37 Throughout the polymerization process, hydroxyl ions in the medium initiated the reaction of monomeric units to form oligomers that were stabilized by the surfactant.37,38 The freshly formed particles further absorbed monomer molecules until the monomer completely disappeared and gradually polymerized into nanosized particles. The hydroxyl ions present in the aqueous solution promoted the polymerization of the remaining monomers.38 Concurrently, QT is a hydrophobic drug, which might have gradually dispersed in the hydrophobic part of the swollen micelles. It might have further been incorporated in polymer NPs by emulsion during the polymerization.18 This rapid polymerization reaction occurred gradually when the pH was adjusted stepwise (pH >5.0).39
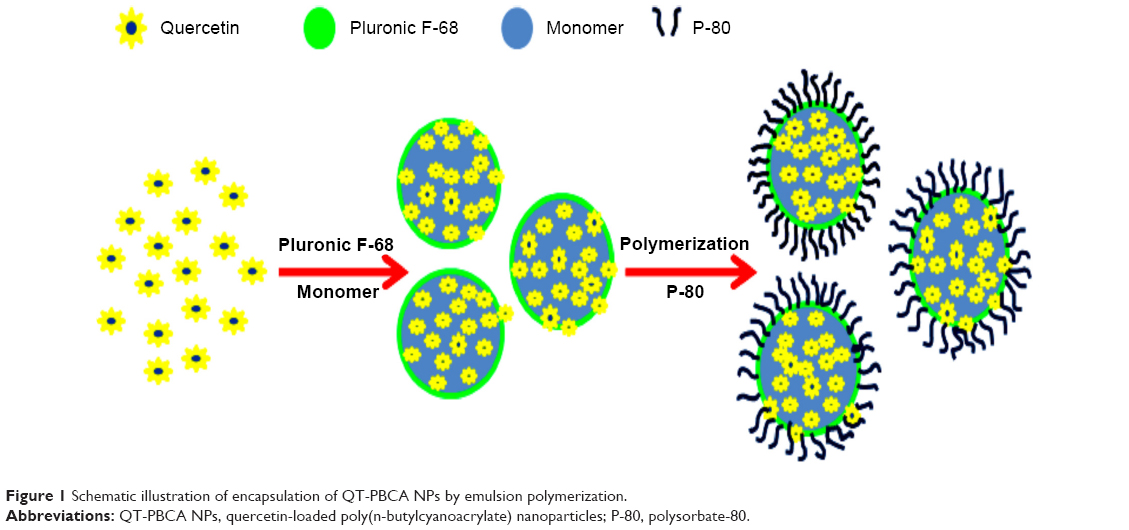 | Figure 1 Schematic illustration of encapsulation of QT-PBCA NPs by emulsion polymerization. |
Based on previous studies, Pluronic F-68 (triblock copolymer poloxamer 188) is biocompatible, has low toxicity effect, and is approved for pharmaceutical applications.40 Recent studies have suggested that drug formulations of Pluronic F-68 have a potential for improving the pharmacokinetics of orally administered P-glycoprotein (P-gp) and/or CYP3A4 substrates in vivo.41 The findings show that Pluronic F-68 produces a protective hydrophilic coating that preserves the structural integrity of the PBCA NPs by protecting them from chemicals and enzymatic degradation until absorption. It also increases the encapsulation of QT-PBCA NPs, which in turn increases the bioavailability of QT and gives longer persistence due to sustained release. Pluronic F-68 was also used as a steric stabilizer for covering the surface of particles after the polymerization reaction was complete. In addition, it was used for obtaining a stable NP form that would increase the size of the NPs when its percentage was more than 3%.34
Figure 2A and B show the particle size distribution pattern of QT-PBCA NPs and QT-PBCA+P-80 with an average size of 161.1±0.44 nm and 166.6±0.33 nm, respectively. Particle size distribution is an important parameter for a drug delivery system and the particles in the nanometer range can go through capillary distribution.42 This also leads to uniform perfusion at the preferred targeted site.42,43 It could be concluded that the larger particle size is due to the higher dehydration of propylene oxide and ethylene oxide.37 Since the NP diameter did not change significantly after coating, the exact nature of orientation of P-80 upon naked PBCA NPs needs to be further investigated. However, it can be assumed that P-80 did not interact with porous, polymeric PBCA NPs, which could have brought about a deviation from the constant size ranges of all PBCA NPs.32
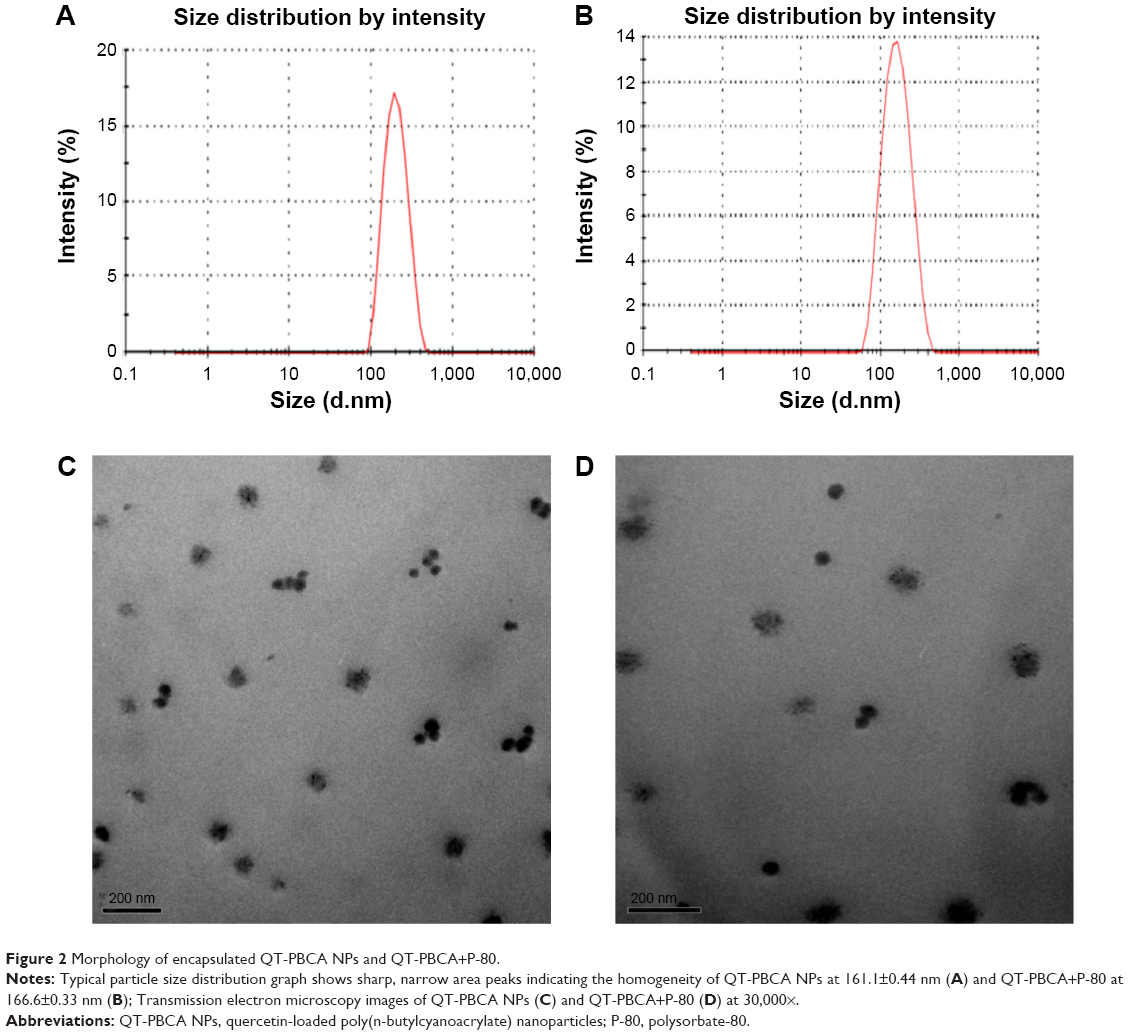 | Figure 2 Morphology of encapsulated QT-PBCA NPs and QT-PBCA+P-80. |
The zeta potential of optimized QT-PBCA NPs and QT-PBCA+P-80 was approximately −7.28±0.60 mV and −25.57±0.81 mV (n=3), respectively. A higher magnitude of zeta potential in PBCA NPs indicates that the strong electrostatic repulsion prevents particle aggregation and gives good stability to the NPs’ suspension.44 The P-80 coating reduced the zeta potential significantly, suggesting a possible masking effect.
The QT-PBCA NPs and QT-PBCA+P-80 NPs were spherical or ellipsoidal in shape with a smooth surface and was slightly aggregated under TEM as shown in Figure 2C and D. The aggregation could be due to the inherent adhesive property of PBCA.44
The mean entrapment efficiency was 79.86%±0.45% for QT-PBCA NPs and 74.58%±1.44% for QT-PBCA NPs coated with P-80. Our finding suggests that the higher the entrapment efficiency, the better the interaction of QT with PBCA NPs. An increase in the content of monomer (with a constant amount of initial drug) enhanced the drug encapsulation efficiency.45 In contrast, coating QT-PBCA NPs with 1% P-80 slightly reduced the DEE of the NPs. This may be due to the drug escaping into the medium during the process of coating.
Additionally, the confirmation of the encapsulation as well as binding of QT-PBCA NPs formulation was carried out by using a UV–Vis spectrophotometer. The free QT powder in ethanolic solution showed two absorption peaks at 254 nm and 374 nm related to the conjugation in the B-ring and A-ring respectively.46 The absorbance of QT-PBCA NPs showed the characteristic peak of turbidity, which is indicative of NPs’ formation. These results support the successful entrapment of QT into the PBCA NPs as presented in Figure 3A. The prepared QT-PBCA NPs were able to be homogenously dispersed into aqueous media without apparent drug aggregates. In comparison, free QT dispersed in water, presented in bulk, and attached on the surface of the container showed poor water solubility as shown in Figure 3B.
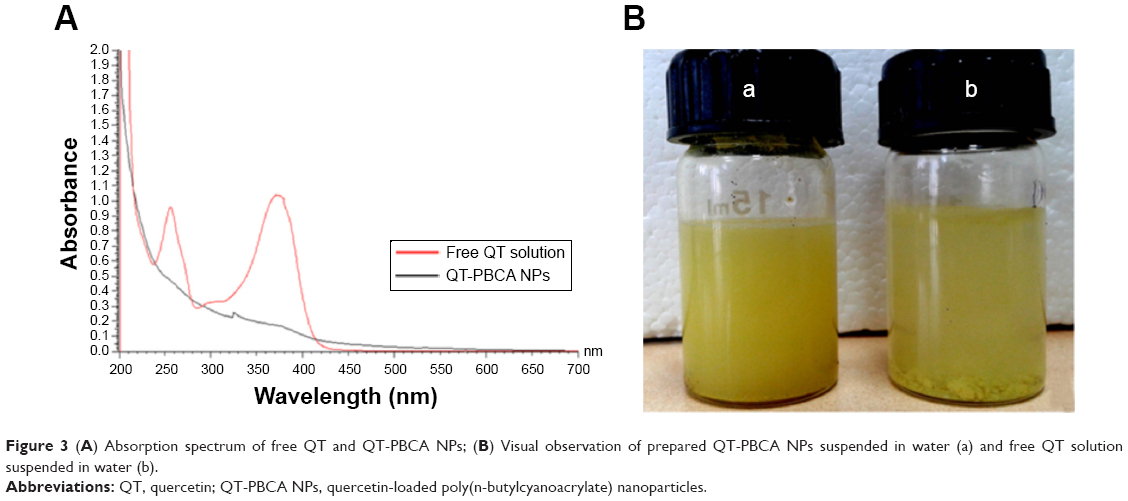 | Figure 3 (A) Absorption spectrum of free QT and QT-PBCA NPs; (B) Visual observation of prepared QT-PBCA NPs suspended in water (a) and free QT solution suspended in water (b). |
Figure 4 shows the FTIR spectrum of n-BCA monomer, blank PBCA NPs, QT-PBCA NPs, and free QT. It confirms drug encapsulation in the NPs and a possible reaction between QT and the polymer during the preparation of NPs. For free QT, major characteristic broader bands corresponding to the OH phenolic stretch at 3,385.07 cm−1 (shown by arrows marked in Figure 4D), to C=O absorption band at 1,666.5 cm−1, bands of the C–C stretches at 1,612.49 cm−1, C–H bending at 1,433.11 cm−1, and the region for C–O stretches at 1,244.09 cm−1. These results are similar to the previous report.14 The absorbance bands of the n-BCA monomer are C–H stretching at 2,962.66 cm−1, C=N stretch at 2,249.83 cm−1, C=O absorbance at 1,751.36 cm−1, and C–O stretch was observed at 1,255 cm−1. The QT-PBCA NPs and blank PBCA NPs of spectrum exhibit the broader peak at 3,410.15 cm−1 (OH stretching) corresponding to hydroxyl radicals and is slightly shifted or enlarged during the term of energy absorption. This shift in the peak of QT-PBCA NPs has been stronger than that of free QT. The C=N peak at 2,249.83 cm−1 on n-BCA is also present in the spectra of blank PBCA NPs and QT-PBCA, indicating that C=N did not take part in the polymerization reaction39 (arrows marked in Figure 4A–C). In short, the characteristic bands of QT (1,600–1,400 cm−1) were observed in loaded NPs and did not participate in the polymerization reaction. This indicated the presence and encapsulation of QT into PBCA NPs by possible intermolecular forces such as the hydrogen bond.
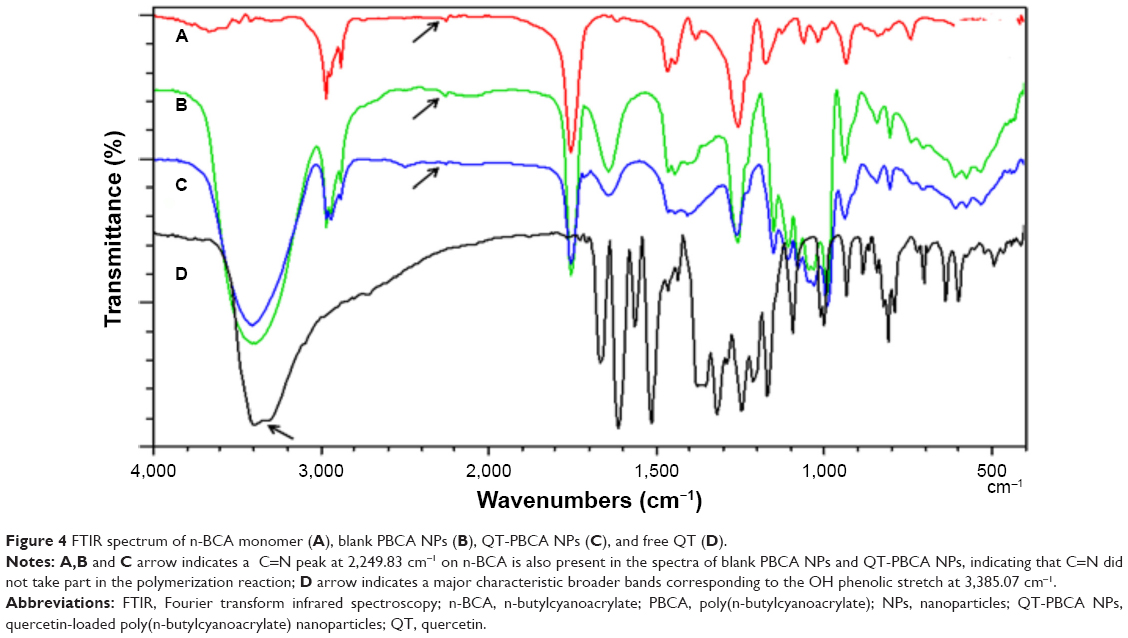 | Figure 4 FTIR spectrum of n-BCA monomer (A), blank PBCA NPs (B), QT-PBCA NPs (C), and free QT (D). |
The DSC curves of the QT-PBCA, blank PBCA NPs, and free QT are presented in Figure 5. The free QT showed two endothermic peaks at 112.80°C and at 323.63°C respectively. These results are in agreement with previously published data.14 The DSC thermogram of QT-PBCA NPs shows a sharp endothermic peak at 168.08°C. However, blank PBCA NPs exhibit two endothermic peaks at 94.47°C and 192.6°C, respectively. It was suggested that the QT peak at 323.63°C was not visible in QT-PBCA NPs, possibly due to the entrapment of drug molecules into PBCA NPs via molecular dispersion form.47 It is also summarized that QT-PBCA NPs were in the noncrystalline state, but in high-energy amorphous form that supports the entrapment of QT in PBCA NPs. These results indicate that the hydrophobic drug strongly interacted with the PBCA NPs and may be potentially released slowly in vivo.
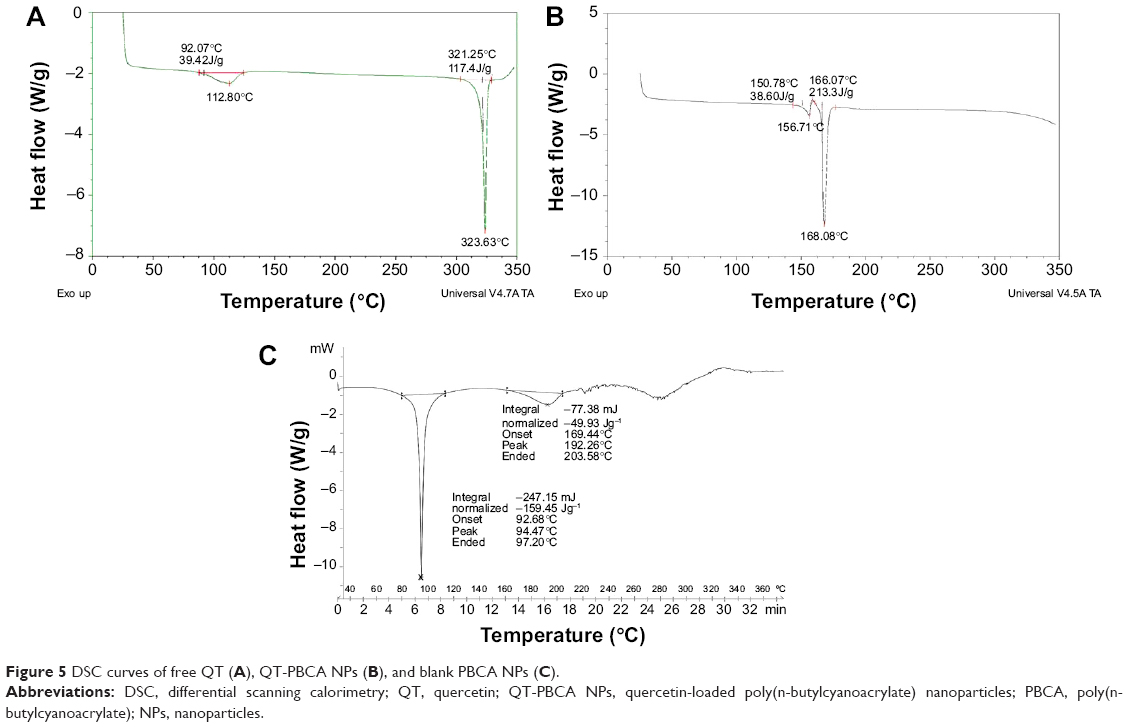 | Figure 5 DSC curves of free QT (A), QT-PBCA NPs (B), and blank PBCA NPs (C). |
X-ray powder diffraction data of free QT, QT-PBCA NPs, and blank PBCA NPs are shown in Figure 6. The XRD pattern of free QT showed a number of distinct peaks characteristic of high crystalline nature.14 In short, no diffraction peaks were observed in QT-PBCA NPs compared to free QT. This finding is consistent with the result given above from the DSC analysis, providing evidence that free QT in the QT-PBCA NPs was indeed converted from a crystal to an amorphous state.
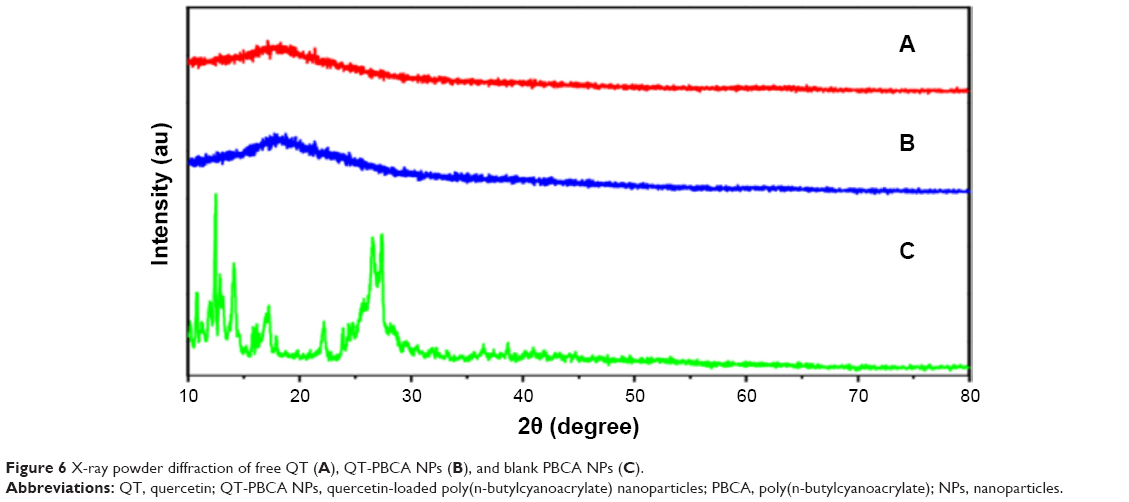 | Figure 6 X-ray powder diffraction of free QT (A), QT-PBCA NPs (B), and blank PBCA NPs (C). |
The stability in SGF and SIF of QT-PBCA NPs was developed for oral administration. It was important to estimate the protective effect of P-80 coating on the poorly water soluble QT drug. The free QT was evaluated after 3 hours of incubation in SGF (pH 1.2). The percentages of drug remaining for pure QT, QT-PBCA NPs, and QT-PBCA+P-80 in SGF were 45.79%±4.41%, 68.33%±2.51%, and 81.38%±3.55% respectively as shown in Figure 7A. This result suggested that QT protection had been increased by 35.59% due to NPs’ coating with P-80. Likewise, free QT was evaluated after 6 hours of incubation in SIF. The percentages of the drug remaining for pure QT, QT-PBCA NPs, and QT-PBCA+P-80 in SIF were 39.66%±1.52%, 64.38%±4.16%, and 71.33%±5.13%, respectively, as shown in Figure 7B. Hence, it can be hypothesized that the long chains of P-80 could have also organized such a protective brush, preventing the degradation of the drug and exerting a protective effect upon the QT-PBCA NPs from gastrointestinal (GI) fluids. Therefore, in both simulated gastric and intestinal fluids, there was an increased protection of QT by 1.8-fold with the use of QT-PBCA+P-80 NPs. These results are in agreement with the previously reported studies.32
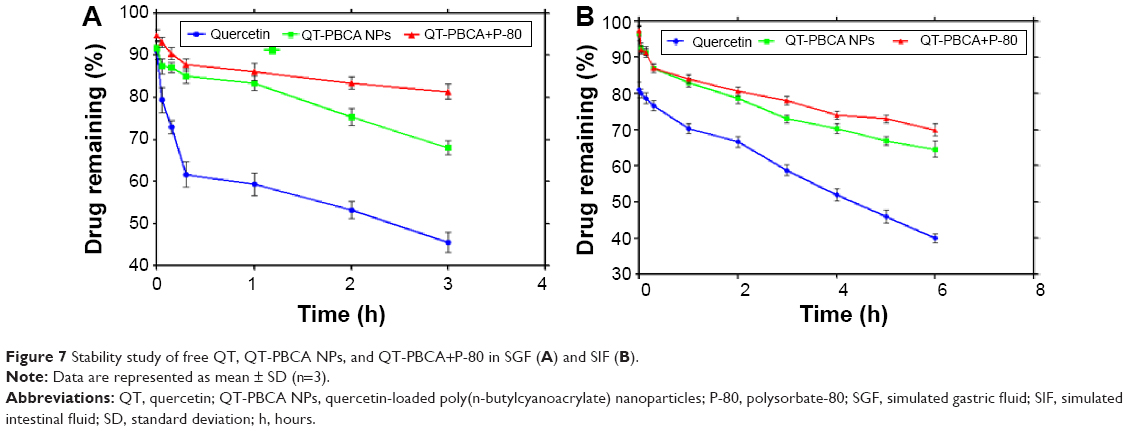 | Figure 7 Stability study of free QT, QT-PBCA NPs, and QT-PBCA+P-80 in SGF (A) and SIF (B). |
In vitro drug release study
The results of drug release profiles of free QT, QT-PBCA NPs, and QT-PBCA+P-80 NPs into PBS (pH 7.4), are shown in Figure 8. The release of QT from the solution was found to be an initial burst release of nearly 83.66%±1.51% within 8 hours. In contrast, the release profile of QT from QT-PBCA NPs exhibited an initial burst release during the first 6 hours of the experiment. It was further revealed that some of the drug molecules entrapped near the surface of PBCA NPs were released first. This was accompanied by a slower and more sustained release of QT from NPs at the rate of 66.38%±4.58% over the period of 48 hours. Therefore, the release profile of QT-PBCA+P-80 was 57.33%±4.11% within 48 hours. Both the formulations revealed that the entrapment of QT into PBCA NPs exhibited a biphasic release pattern with an initial burst effect followed by a sustained and controlled release.19 The results showed that coating PBCA NPs with P-80 slightly decreased the release of QT from the NPs (9.05%) compared to that from QT-PBCA NPs (P<0.05). This result was in agreement with earlier reports that stated that drug-loaded PBCA NPs provided a controlled and sustained release pattern.37
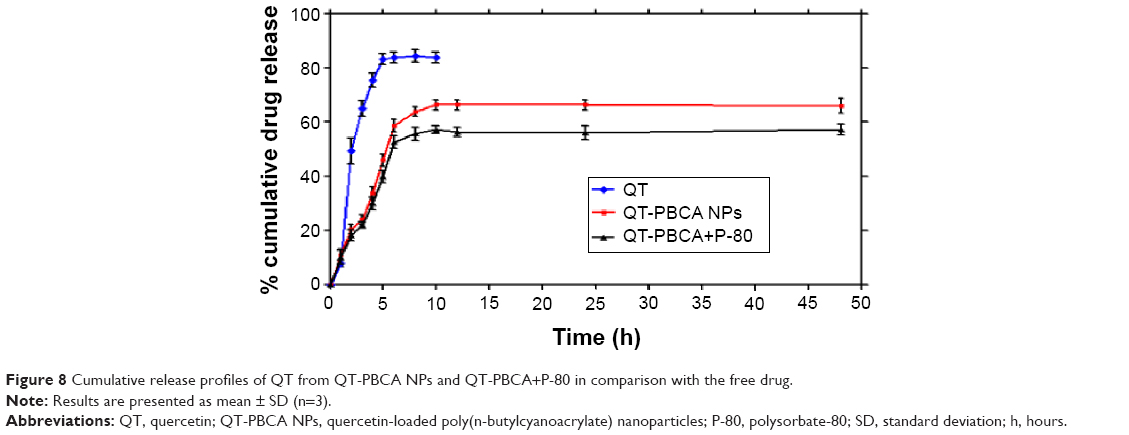 | Figure 8 Cumulative release profiles of QT from QT-PBCA NPs and QT-PBCA+P-80 in comparison with the free drug. |
Stability studies
Table 1 shows a slight variance in the particle size, polydispersity index, zeta potential, and entrapment efficiency of QT-PBCA NPs after a storage period of 180 days at room temperature. The results related to the particle size, polydispersity index, zeta potential, and entrapment efficiency showed that at storage conditions in amber vials at room temperature, QT-PBCA NPs were stable as there was no significant change (P<0.05) after 30, 60, 120, and 180 days compared to initial results. There was no change in the physical appearance of QT-PBCA NPs after 30, 60, 120, and 180 days.
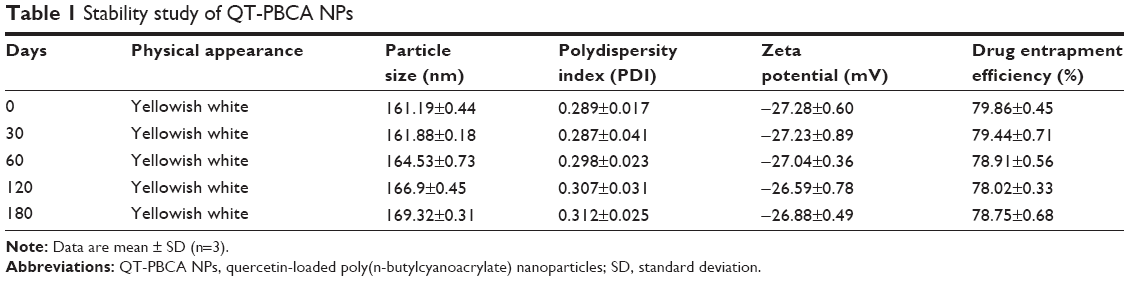 | Table 1 Stability study of QT-PBCA NPs |
In vivo pharmacokinetics and biodistribution studies in Wistar rats
The mean QT concentration–time profiles in the plasma after oral administration of a single dose of 50 mg/kg of QT in different formulations are shown in Figure 9, and the corresponding pharmacokinetics parameters are summarized in Table 2.
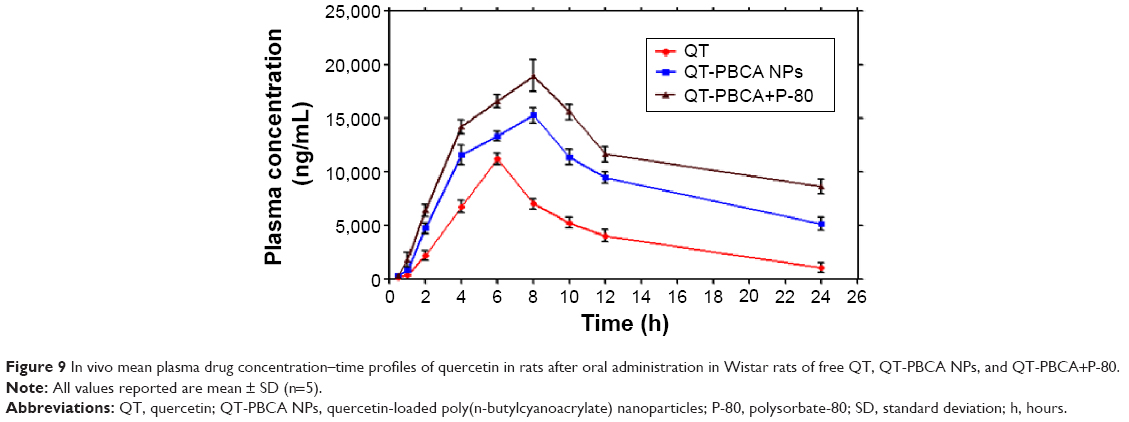 | Figure 9 In vivo mean plasma drug concentration–time profiles of quercetin in rats after oral administration in Wistar rats of free QT, QT-PBCA NPs, and QT-PBCA+P-80. |
 | Table 2 Plasma pharmacokinetics parameters of free QT solution, QT-PBCA NPs, and QT-PBCA+P-80, after single oral administration of 50 mg/kg body weight in rats |
After oral administration of free QT solution in rats, the maximum plasma concentration (Cmax) achieved was 11.26±0.09 μg/mL in 6 hours. Plasma concentration declined rapidly after 6 hours, indicating that the drug was distributed and metabolized rapidly. Elsewhere, it is reported that QT is quickly metabolized by the intestinal enzymes.48 A complete lack of QT and its metabolites implies that the metabolism of QT seems to occur primarily in the enterocyte.48 An earlier hypothesis suggests that the QT metabolites are strongly bound to albumin, which could delay their evacuation, and that they are preferentially excreted in bile.49 The Cmax of QT-PBCA NPs and QT-PBCA+P-80 were 15.30±0.12 and 19.11±0.25 μg/mL respectively. The Cmax of QT-PBCA NPs and QT-PBCA+P-80 were increased ~1.4- and ~1.7-fold respectively and were significantly different (P<0.05) from that of free QT. The small size of QT-PBCA NPs causes a high amount of QT in plasma. The coating of P-80 on the surface QT-PBCA helps in keeping the formulation in the circulation for a prolonged period.17,20 The area under the concentration–time curve (AUCtotal) of QT-PBCA NPs was 248.63±6.82 μg·h/mL; for QT-PBCA+P-80, it was 313.97±7.88 μg·h/mL. The areas were ~2.4- and 4.1-fold higher than the AUCtotal of free QT (104.22±2.05 μg·h/mL) (P<0.05). The mean residence time (MRT) of QT-PBCA NPs and QT-PBCA+P-80 were 23.01±7.52 and 30.26±7.78 hours, respectively and they were higher when compared to the MRT of free QT (11.72±1.75 hours). Therefore, t1/2 of QT from the PBCA NPs increased to ~14 hours, and that from the QT-PBCA+P-80 enhanced to ~19 hours; for free QT, t1/2 was 6 hours. In the present study, the difference in pharmacokinetics results and the in vitro release profile of QT-PBCA NPs may be caused by the possible enterohepatic recirculation.18
The result indicated that the relative bioavailability of QT-PBCA NPs and QT-PBCA+P-80 enhanced the QT bioavailability by 2.38- and 4.93-fold respectively as compared to that of free QT. The increased bioavailability of PBCA NPs may be a co-work of direct uptake of NPs through the GI tract. This is due to the increased permeability of the GI mucous membrane through surfactants. The decreased degradation and clearance for QT is due to the encapsulation by PBCA NPs. The plasma drug profiles of QT-PBCA NPs and QT-PBCA+P-80 showed an increase in oral bioavailability, exhibiting more sustained release when compared with free QT.
According to a recent study,35 oral administrations of QT self-emulsifying drug delivery systems showed a 4.95-fold increase in the AUC when compared to the AUC for a free QT solution. A previous report on the relative bioavailability of co-encapsulated QT showed that the oral bioavailability of QT increased 2.9-fold in comparison with free QT and its combination with free Tamoxifen-citrate.49 Elsewhere in the literature, puerarin-PBCA NPs caused a significant enhancement in oral bioavailability of puerarin by more than 5.5-fold to overcome enterohepatic recirculation.18 Other research groups have reported an improvement in bioavailability of both oral and intravenous administration by NP coupled to QT that has shown promising results.8,50
Figure 10 shows the QT concentration found in different tissues of rats after oral administration of free QT, QT-PBCA NPs, and QT-PBCA+P-80 over a period of 24 hours. The values for pharmacokinetics constants are tabulated in Table 3. The maximum concentrations of QT in the heart, liver, kidney, and spleen were 18.88±2.09, 21.44±3.97, 43.67±2.28, and 19.44±1.35 μg/mL respectively. However, the concentration of QT-PBCA NPs and QT-PBCA+P-80 in the heart, liver, kidney, and spleen were 17.45±3.71 and 15.76±0.43, 25.88±3.00 and 27.11±1.91, 32.49±2.81 and 30.95±2.31, and 26.21±1.17 and 25.83±2.67 μg/mL respectively. QT-PBCA NPs and QT-PBCA+P-80 show higher concentrations of QT in the liver and the spleen than in the case of free QT alone. This was caused by the reticuloendothelial system well known for the accumulation and metabolism of NPs with a sustained release of the drug from tissues. This result is in agreement with the data already reported.21,34 QT-PBCA NPs and QT-PBCA+P-80 were distributed less to the heart and kidney, resulting in their significantly lower concentration than that of free QT in this entire study. In addition, the most interesting observation was that the Cmax and AUCtotal of QT-PBCA NPs and QT-PBCA+P-80 NPs were significantly different (P<0.05) from that of free QT (Table 3). During the study period, no systemic toxicity, loss of body weight, fatalities, or other adverse effects were observed after the oral administration of QT-PBCA NPs and QT-PBCA+P-80 NPs in rats.
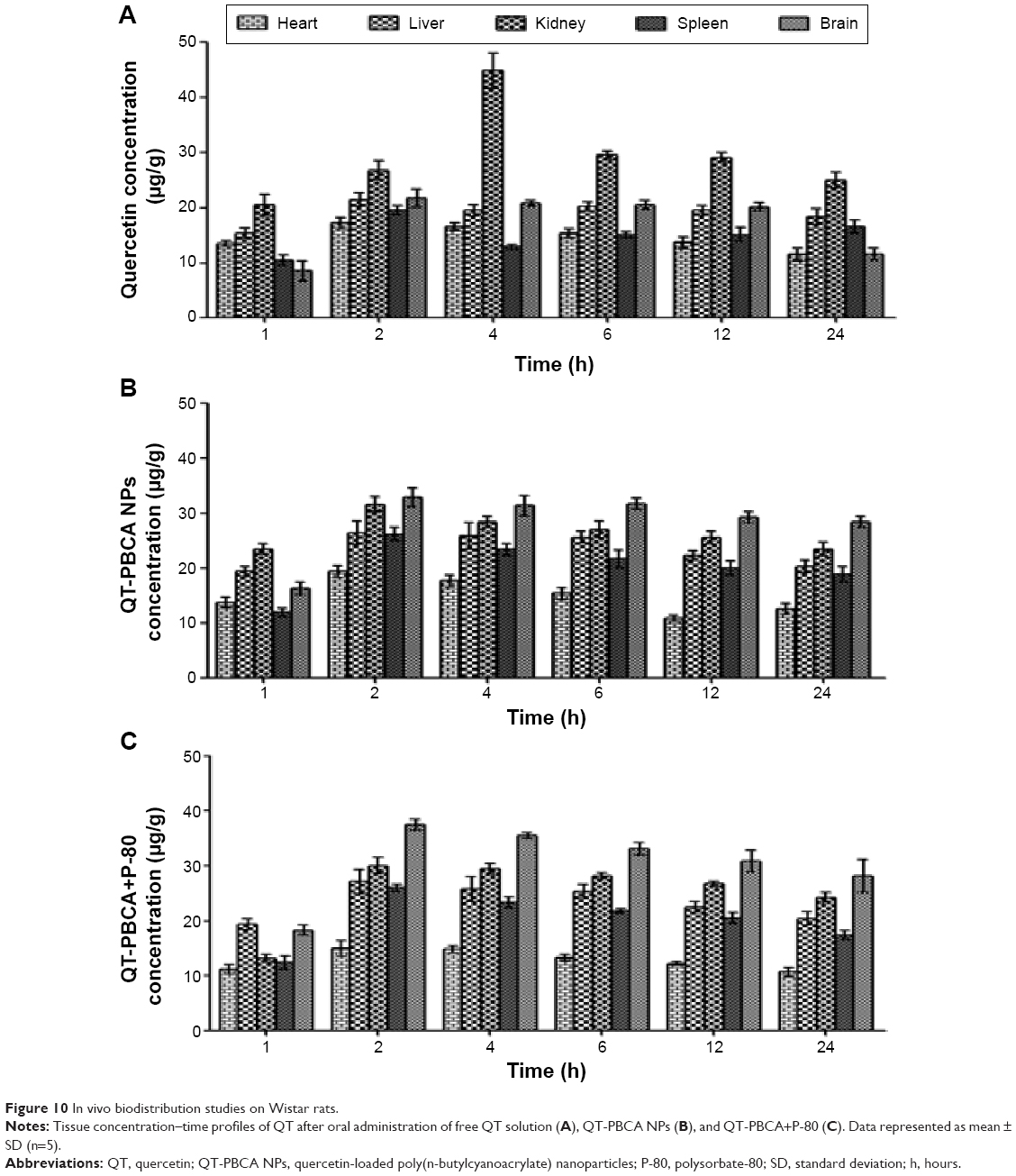 | Figure 10 In vivo biodistribution studies on Wistar rats. |
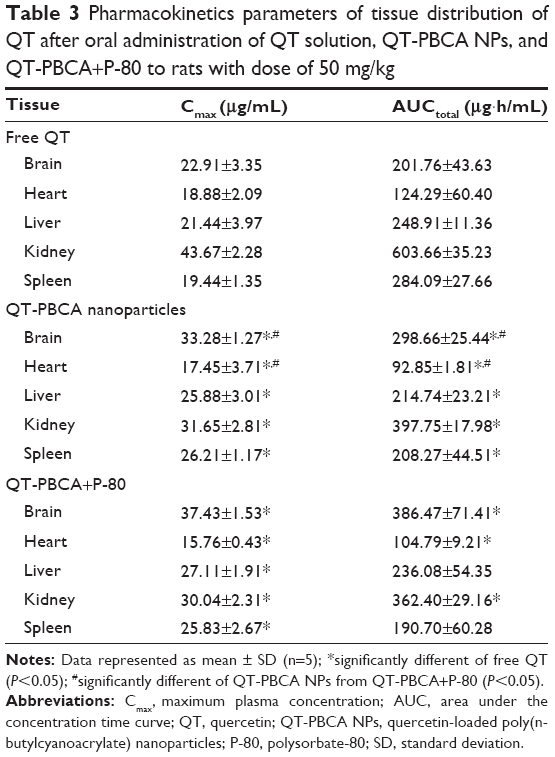 | Table 3 Pharmacokinetics parameters of tissue distribution of QT after oral administration of QT solution, QT-PBCA NPs, and QT-PBCA+P-80 to rats with dose of 50 mg/kg |
The AUCtotal and Cmax of QT in the brain delivered by QT-PBCA NPs and QT-PBCA+P-80 NPs were about 1.4- and 1.9-folds, and 1.5- and 1.7-folds higher respectively when compared to that of free QT. It indicates that polysorbate-coated NPs were able to deliver QT to the brain. According to the literature, the size (nm) of NPs plays an important role in drug delivery to the brain and it (especially the particle size ranging from 70 to 345 nm) might be able to overcome BBB.27 From this observation, QT-PBCA NPs coated with P-80 were able to enhance the biodistribution of drugs into the brain when compared to free QT and uncoated QT-PBCA NPs. Previous reports show that P-80, coated with PBCA NPs that mimic low-density lipoprotein, may absorb apolipoproteins such as apolipoproteins B and E or apolipoproteins A-I and IV after being injected into the blood stream.17,19,22 As far as the mechanism is concerned, receptor-mediated endocytosis P-80 acts mainly as an anchor for apolipoprotein-overcoated NPs. QT-PBCA would mimic the lipoprotein particles and interact with the brain capillary endothelial cells and later be taken up by the same endothelial system.15,17 PBCA NPs have been intensively investigated for decades, showing that coating the NPs with a nonionic surfactant such as P-80 makes them cross the BBB.22 It has been reported that the drug methotrexate, bound to 1% P-80 coated NPs, could significantly increase the drug levels in the brain.22,51 Based on the above results, it can be concluded that QT levels, due to PBCA NPs coated with P-80, are significantly higher than that of the free QT solution at all the time points in the plasma and the brain.
Conclusion
In the present study, QT was successfully entrapped in PBCA NPs for oral delivery by using an anionic emulsion polymerization method with an entrapment efficiency of 79.86%±0.45% and 74.58%±1.44% for QT-PBCA and QT-PBCA+P-80 respectively. The particle size distribution pattern of QT-PBCA NPs and QT-PBCA+P-80 indicated an average size of 161.1±0.44 nm and 166.6±0.33 nm respectively, and the particles were spherical in shape under TEM. It could be concluded from FTIR, DSC, and XRD analyses that QT-PBCA NPs were in an amorphous form. The in vitro release of QT from QT-PBCA NPs and QT-PBCA+P-80 NPs showed a sustained release when compared with free QT. The plasma pharmacokinetics data of Cmax, AUCtotal, t1/2, and MRT values of QT-PBCA NPs and QT-PBCA+P-80 NPs on rats were higher than those of free QT. The relative bioavailabilities of QT-PBCA and QT-PBCA+P-80 NPs were enhanced by more than 2.38- and 4.93-folds respectively when compared to free QT. The biodistribution study in rats showed that a higher concentration of QT was found in the brain when NPs are coating with P-80. It can be concluded that PBCA NPs coated with P-80 show the potential drug carrier ability of poorly water soluble drugs by improving their oral bioavailability and by enhancing the transportation ability of the drug to the brain that in turn may prove to be a promising aspect in clinical application.
Acknowledgments
The authors acknowledge VIT University, Vellore, India for providing laboratory facilities and funds for proceeding with the research work. A special thanks for proofreading this research article to Dr Shahila Zafar and Ms Jancy Nandhini Feleciya A, School of Social Sciences and Languages, VIT University, Vellore.
Disclosure
The authors report no conflicts of interest in this work.
References
Nijveldt RJ, Nood E, Van Hoorn DEC, Boelens PG, Van Norren K, Van Leeuwen PA. Flavonoids: a review of probable mechanisms of action and potential applications. Am J Clin Nutr. 2001;74(4):418–425. | ||
Erlund I. Review of the flavonoids quercetin, hesperetin, and naringenin. Dietary sources, bioactivities, bioavailability, and epidemiology. Nutr Res. 2004;24(10):851–874. | ||
Kim GN, Jang HD. Protective mechanism of quercetin and rutin using glutathione metabolism on H2O2-induced oxidative stress in HepG2 cells. Ann N Y Acad Sci. 2009;1171:530–537. | ||
Formica JV, Regelson W. Review of the biology of quercetin and related bioflavonoid. Food Chem Toxicol. 1995;33(12):1061–1080. | ||
Priprem A, Watanatorn J, Sutthiparinyanont S, Phachonpai W, Muchimapura S. Anxiety and cognitive effects of quercetin liposomes in rats. Nanomedicine. 2008;4(1):70–78. | ||
Huebbe P, Wagner AE, Boesch-Saadatmandi C, Sellmer F, Wolffram S, Rimbach G. Effect of dietary quercetin on brain quercetin levels and the expression of antioxidant and Alzheimer’s disease relevant genes in mice. Pharmacol Res. 2010;61(3):242–246. | ||
Kumari A, Yadav SK, Pakade YB, Singh B, Yadav SC. Development of biodegradable nanoparticles for delivery of quercetin. Colloids Surf B Biointerfaces. 2010;80(2):184–192. | ||
Dhawan S, Kapil R, Singh B. Formulation development and systematic optimization of solid lipid nanoparticles of quercetin for improved brain delivery. J Pharm Pharmacol. 2011;63(3):342–351. | ||
Hariharan S, Bhardwaj V, Bala I, Sitterberg J, Bakowsky U, Ravi Kumar MNV. Design of estradiol loaded PLGA nanoparticulate formulations: a potential oral delivery system for hormone therapy. Pharm Res. 2006;23(1):184–195. | ||
LI SD, Huang L. Pharmacokinetics and biodistribution of nanoparticles. Mol Pharm. 2008;5(4):496–504. | ||
Date AA, Nagarsenker MS, Patere S, et al. Lecithin based novel cationic nanocarriers (LeciPlex) II: improving therapeutic efficacy of quercetin on oral administration. Mol Pharm. 2011;8(3):716–726. | ||
Zhang Y, Yang Y, Tang K, Hu X, Zou G. Physicochemical characterization and antioxidant activity of quercetin-loaded chitosan nanoparticles. J Appl Polym Sci. 2008;107(2):891–897. | ||
Gang W, Jie WJ, Ping ZL, et al. Liposomal quercetin: evaluating drug delivery in vitro and biodistribution in vivo. Expert Opin Drug Deliv. 2012;9(6):599–613. | ||
Pool H, Quintanar D, Figueroa JD, et al. Antioxidant effects of quercetin and catechin encapsulated into PLGA nanoparticles. J Nanomat. 2012: Article ID 145380: 1–12. | ||
Kreuter J. Nanoparticulate systems for brain delivery of drugs. Adv Drug Deliv Rev. 2001;47(1):65–81. | ||
Yordanov G. Poly(alkyl cyanoacrylate) nanoparticles as drug carriers: 33 years later. Bulg J Chem. 2012;1:61–73. | ||
Sun M, Gao Y, Guo C, et al. Enhancement of transport of curcumin to brain in mice by poly(n-butylcyanoacrylate) nanoparticles. J Nanopart Res. 2010;12:3111–3122. | ||
Zhao L, Liu A, Sun M, et al. Enhancement of oral bioavailability of puerarin by polybutylcyanoacrylate nanoparticles. J Nanomater. 2011:1–8. | ||
Wilson B, Samanta MK, Santhi K, Sampath Kumar KP, Paramakrishnan N, Suresh B. Poly(n-butylcyanoacrylate) nanoparticles coated with polysorbate 80 for the targeted delivery of Rivastigmine into the brain to treat Alzheimer’s disease. Brain Res. 2008;1200:159–168. | ||
Gulyaev AE, Gelperina SE, Skidan IN, Antropov AS, Kivman GY, Kreuter J. Significant transport of doxorubicin into the brain with polysorbate 80-coated nanoparticles. Pharm Res. 1999;16(10):1564–1569. | ||
Yordanov G. Influence of the preparation method on the physicochemical properties of econazole-loaded poly(butyl cyanoacrylate) colloidal nanoparticles. Colloids Surf A Physicochem Eng Asp. 2012;413:260–265. | ||
Kreuter J, Alyautdin RN, Kharkevich DA, Ivanov AA. Passage of peptides through the blood–brain barrier with colloidal polymer particles (nanoparticles). Brain Res. 1995;674(1):171–174. | ||
Friese A, Seiller E, Quack G, Lorenz B, Kreuter J. Increase of the duration of the anticonvulsive activity of a novel NMDA receptor antagonist using poly(butylcyanoacrylate) nanoparticles as a parenteral controlled release system. Eur J Pharm Biopharm. 2000;49(2):103–109. | ||
Page-Clisson ME, Pinto-Alphandary H, Ourevitch M, Andremont A, Couvreur P. Development of ciprofloxacin-loaded nanoparticles: physicochemical study of the drug carrier. J Control Release. 1998;56(1–3):23–32. | ||
Miyazaki S, Takahashi A, Kubo W, Bachynsky J, Loebenberg R. Poly n-butylcyanoacrylate (PNBCA) nanocapsules as a carrier for NSAIDs: in vitro release and in vivo skin penetration. J Pharm Pharm Sci. 2003;6(2):238–245. | ||
Kuo YC, Chen HH. Effect of nanoparticulate polybutylcyanoacrylate and methylmethacrylate–sulfopropylmethacrylate on the permeability of zidovudine and lamivudine across thein vitroblood–brain barrier. Int J Pharm. 2006;327(1–2):160–169. | ||
Wohlfart S, Khalansky AS, Gelperina S, Begley D, Kreuter J. Kinetics of transport of doxorubicin bound to nanoparticles across the blood–brain barrier. J Control Release. 2011;154:103–107. | ||
Gao K, Jiang X. Influence of particle size on transport of methotrexate across blood–brain barrier by polysorbate 80-coated polybutylcyanoacrylate nanoparticles. Int J Pharm. 2006;310(1–2):213–219. | ||
Joshi SA, Chavhan SS, Sawant KK. Rivastigmine-loaded PLGA and PBCA nanoparticles: preparation, optimization, characterization, in vitro and pharmacodynamic studies. Eur J Pharm Biopharm. 2010;76(2):189–189. | ||
Wilson B, Sampath KP, Santhi K, Kumar KPS, Paramakrishnan N, Suresh B. Targeted delivery of tacrine into the brain with polysorbate 80-coated poly(n-butylcyanoacrylate) nanoparticles. Eur Pharm Biopharm. 2008;70:75–84. | ||
Pimple S, Manjappa AS, Ukawala M, Murthy RSR. PLGA nanoparticles loaded with etoposide and quercetin dihydrate individually: in vitro cell line study to ensure advantage of combination therapy. Cancer Nanotechnol. 2012;3:25–36. | ||
Das D, Lin S. Double-coated poly(butylcyanoacrylate) nanoparticulate delivery systems of brain targeting of dalargin via oral administration. J Pharm Sci. 2005;94(6):1343–1353. | ||
Bose S, Duc Y, Takhistov P, Michniak-Kohn B. Formulation optimization and topical delivery of quercetin from solid lipid based nanosystems. Int J Pharm. 2013;441(1–2):56–66. | ||
Tian X, Lin X, Wei F, et al. Enhanced brain targeting of temozolomide in polysorbate-80 coated polybutylcyanoacrylate nanoparticles. Int J Nanomedicine. 2011;6:445–452. | ||
Jain S, Jain AK, Pohekar M, Thanki K. Novel self-emulsifying formulation of quercetin for improved in vivo antioxidant potential: implications for drug-induced cardiotoxicity and nephrotoxicity. Free Radic Biol Med. 2013;65:117–130. | ||
Bhandari R, Kaur IP. Pharmacokinetics, tissue distribution and relative bioavailability of isoniazid-solid lipid nanoparticles. Int J Pharm. 2013;441(1–2):202–212. | ||
Reddy LH, Murthy RR. Influence of polymerization technique and experimental variables on the particle properties and release kinetics of methotrexate from poly(butylcyanoacrylate) nanoparticles. Acta Pharm. 2004;54:103–118. | ||
Behan N, Birkinshaw C. Mechanism of polymerization of butylcyanoacrylate in aqueous dispersions. Macromol Rapid Commun. 2000;21:884–886. | ||
Behan N, Birkinshaw C, Clarke N. Polyn-butyl cyanoacrylate nanoparticles: a mechanistic study of polymerisation and particle formation. Biomaterials. 2001;22(11):1335–1344. | ||
Yordanov G, Skrobanska R, Evangelatov A. Colloidal formulations of etoposide based on poly(butyl cyanoacrylate) nanoparticles: preparation, physicochemical properties and cytotoxicity. Colloids Surf B Biointerfaces. 2013;101:215–222. | ||
Huang J, Si L, Jiang L, Fan Z, Qiu J, Li G. Effect of pluronic F68 block copolymer on P-glycoprotein transport and CYP3A4 metabolism. Int J Pharm. 2008;356:351–353. | ||
Duan J, Zhang Y, Han S, et al. Synthesis and in vitro/in vivo anti-cancer evaluation of curcumin-loaded chitosan/poly(butyl cyanoacrylate) nanoparticles. Int J Pharm. 2010;400(1–2):211–220. | ||
Muller RH, Mader K, Gohla S. Solid lipid nanoparticles (SLN) for controlled drug delivery-are view of the state of the art. Eur J Pharm Biopharm. 2000;50:161–177. | ||
Wu T, Yen F, Lin L, Tsai T, Lin C, Cham T. Preparation, physicochemical characterization, and antioxidant effect of quercetin nanoparticles. Int J Pharm. 2008;346(1–2):160–168. | ||
Souza MP, Vaz AFM, Correia MTS, Serqueira MA, Vicente AA, Carneiro-da-cunha MG. Quercetin-loaded lecithin/chitosan nanoparticles for functional food applications. Food Bioprocess Technol. 2014;7(4):1149–1159. | ||
Mulik R, Mahadik K, Paradkar A. Development of curcuminoids loaded poly(butyl) cyanoacrylate nanoparticles: physicochemical characterization and stability study. Eur J Pharm Sci. 2009;37(3–4):395–404. | ||
Graefe EU, Wittig J, Mueller S, et al. Pharmacokinetics and bioavailability of quercetin glycosides in humans. J Clin Pharmacol. 2001;41(5):492–499. | ||
Manach C, Texier O, Morand C, et al. Comparison of the bioavailability of quercetin and catechin in rats. Free Radic Biol Med. 1999;27(11–12):1259–1266. | ||
Jain AK, Thanki K, Jain S. Co-encapsulation of tamoxifen and quercetin in polymeric nanoparticles: implications on oral bioavailability, antitumor efficacy, and drug-induced toxicity. Mol Pharm. 2013;10(9):3459–3474. | ||
Ghosh A, Mandal AK, Sarkar S, Panda S, Das N. Nanoencapsulation of quercetin enhances its dietary efficacy in combating arsenic-induced oxidative damage in liver and brain of rats. Life Sci. 2009;84:75–80. | ||
Alyautdin RN, Tezikov EB, Ramge P, Kharkevich DA, Begley DJ, Kreuter J. Significant entry of tubocurarine into the brain of rats by adsorption to polysorbate 80-coated polybutylcyanoacrylate nanoparticles: an in situ brain perfusion study. J Microencapsul. 1998;15(1):67–74. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.