Back to Journals » International Journal of Nanomedicine » Volume 11
Polymer nanoparticles for cross-presentation of exogenous antigens and enhanced cytotoxic T-lymphocyte immune response
Received 18 April 2016
Accepted for publication 1 June 2016
Published 5 August 2016 Volume 2016:11 Pages 3753—3764
DOI https://doi.org/10.2147/IJN.S110796
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr Thomas Webster
Chanyoung Song,* Young-Woock Noh,* Yong Taik Lim
SKKU Advanced Institute of Nanotechnology (SAINT), School of Chemical Engineering, Sungkyunkwan University, Suwon, South Korea
*These authors contributed equally to this work
Abstract: Effective induction of an antigen-specific cytotoxic T lymphocyte (CTL) immune response is one of the key goals of cancer immunotherapy. We report the design and fabrication of polyethylenimine (PEI)-coated polymer nanoparticles (NPs) as efficient antigen-delivery carriers that can induce antigen cross-presentation and a strong CTL response. After synthesis of poly(d,l-lactide-co-glycolide) (PLGA) NPs containing ovalbumin (OVA) by the double-emulsion solvent-evaporation method, cationic-charged PLGA NPs were generated by coating them with PEI. In a methyl tetrazolium salt assay, no discernible cytotoxic effect of PEI-coated PLGA (OVA) NPs was observed. The capacity and mechanism of PEI-coated PLGA (OVA) NPs for antigen delivery and cross-presentation on dendritic cells (DCs) were determined by fluorescence microscopy and flow cytometry. PEI-coated PLGA (OVA) NPs were internalized efficiently via phagocytosis or macropinocytosis in DCs and induced efficient cross-presentation of the antigen on MHC class I molecules via both endosome escape and a lysosomal processing mechanism. The DCs treated with PEI-coated PLGA (OVA) NPs induced a release of IL-2 cytokine from OVA-specific CD8-OVA1.3 T cells more efficiently than DCs treated with PLGA (OVA) NPs. Therefore, the PEI-coated PLGA (OVA) NPs can induce antigen cross-presentation and are expected to be used for induction of a strong CTL immune response and for efficient anticancer immunotherapy.
Keywords: antigen delivery, dendritic cells, polymer NPs, vaccine, cross-presentation
Introduction
Polymeric nanoparticles (NPs) have attracted substantial attention in the pharmaceutical and biomedical fields, because of their high potential for controlled intracellular delivery of biomolecules and drugs.1–5 Particularly, various types of polymer NPs that can target and deliver specific antigens for immunotherapy have been shown to provide protective immunity against cancer and infectious diseases.6–9 As potent professional antigen-presenting cells, dendritic cells (DCs) play a major role in the production of antigen-specific cytotoxic T lymphocytes (CTLs) and in CTL-mediated cancer immunotherapy.10,11 CTLs require presentation of peptides derived from tumor-associated antigens via major histocompatibility complex (MHC) class I molecules on DCs. Therefore, delivery of exogenous tumor-associated antigens to DCs and activation of the MHC class I-presenting pathway, a process known as cross-presentation, constitute a crucial part of DC immunotherapy.12 In this regard, the intracellular delivery of antigens and induction of CTL responses through cross-presentation pathways have been realized in several NP systems, such as liposomes, polyelectrolytes, silicon particles, and poly(D,L-lactide-co-glycolide) (PLGA) particles.13–16
In this study, we propose polyethylenimine (PEI)-coated PLGA NPs as efficient antigen-delivery carriers that can induce antigen cross-presentation and a strong CTL response. PLGA is one of the most frequently used biodegradable polymers, because its use in humans has already been approved by the US Food and Drug Administration and provides long-term release of encapsulated materials in vitro and in vivo.1,17,18 However, the negatively charged surface of PLGA NPs limits their interactions with the negatively charged cell membrane and their intracellular uptake, which is the key step for antigen delivery into DCs.19 In this study, cationic-charged PEI was introduced in order to overcome this limitation and to deliver antigens effectively into DCs via the proton-sponge effect.20–25 We chose ovalbumin (OVA) as a model antigen, which has been well characterized for estimates of cross-presentation efficiency. In our study, we explored the effects of PEI-coated PLGA NPs containing OVA on the viability and phenotype of DCs, and analyzed the capacity and mechanism of PEI-coated PLGA NPs for antigen delivery and cross-presentation in DCs.
Materials and methods
Materials
PLGA (Resomer® RG502H, lactic:glycolic molar ratio 50:50) was purchased from Boehringer Ingelheim (Ingelheim, Germany). PEI (branched form, molecular weight 25 kDa), chicken-egg OVA, and polyvinyl alcohol (80% hydrolyzed, molecular weight 9,000–10,000 Da) were purchased from Sigma-Aldrich (St Louis, MO, USA). Fluorescein isothiocyanate (FITC)-conjugated OVA (FITC-OVA) was acquired from Thermo Fisher Scientific (Waltham, MA, USA).
Preparation of PLGA and PEI-coated PLGA NPs
PLGA NPs containing the OVA antigen were prepared by double-emulsion (W1/O/W2) methods. Briefly, 300 μL of aqueous solutions containing 1.8 mg of OVA and 200 μg of FITC-OVA were emulsified in 2 mL of a methylene chloride solution containing 60 mg of PLGA using a probe-type sonicator (VCX 750 Vibra-Cell™; Sonics and Materials Inc, Newton, CT, USA) at 24 W for 1 minute. The resulting primary emulsion was added to 10 mL of polyvinyl alcohol (2.5% w/w) and emulsified using a probe-type sonicator for 2 minutes. The resultant double emulsion was agitated using a magnetic stirrer at room temperature overnight until the methylene chloride evaporated completely. The PLGA NPs were collected by centrifugation (9,500×g, 20 minutes) at 4°C, washed three times with distilled water, freeze-dried, and stored at −20°C before use. To fabricate PEI-coated PLGA NPs, 5 mg of PEI that was dissolved in 5 mL distilled water (adjusted pH 8) was added to 5 mL of an aqueous solution containing 10 mg of PLGA NPs and mixed by stirring for 1 hour. The emulsion was washed three times with 10 mL of distilled water by centrifugation (6,500×g, 10 minutes) at 4°C, freeze-dried, and stored at −20°C before use. To quantify the OVA encapsulated in the PLGA NPs, 1 mg of OVA-containing NPs was dissolved in 0.1 N NaOH and 0.1% sodium dodecyl sulfate with stirring at room temperature overnight and then quantified by means of the Micro BCA™ protein-assay kit (Thermo Fisher Scientific).
Characterization of PLGA and PEI-coated PLGA NPs
The surface morphology of NPs was characterized using field-emission scanning electron microscopy (SEM; JSM-7000F; JEOL, Tokyo, Japan). The size and size distribution of the NPs were analyzed by a dynamic light-scattering technique using an electrophoretic light-scattering photometer (ELS-Z; Otsuka Electronics, Osaka, Japan). The surface charge of the NPs was determined by ζ-potential measurements using the ELS-Z.
Mice and cell lines
C57BL/6 mice (females, 6–8 weeks old) were purchased from Orient Bio (Seongnam, South Korea) and maintained under pathogen-free conditions. This animal study was reviewed and approved by the institutional animal care and use committee (IACUC) of Sungkyunkwan University School of Medicine (Permission No 14-38), which is an accredited facility by the Association for Assessment and Accreditation of Laboratory Animal Care International (AAALAC International) and abides by the Institute of Laboratory Animal Resources (ILAR) guide. CD8-OVA1.3 T cells, an OVA SIINFEKL peptide-specific CD8+ T-cell hybridoma, were maintained in Roswell Park Memorial Institute (RPMI) 1640 medium supplemented with 10% heat-inactivated fetal bovine serum, 5×10−5 M 2-mercaptoethanol, 50 IU/mL penicillin, and 50 mg/mL streptomycin (Thermo Fisher Scientific).
Generation of bone marrow-derived dendritic cells from mice
Bone marrow-derived DCs (BMDCs) were isolated from the bone marrow cells of C57BL/6 (H-2b) mice.26 Briefly, bone marrow was collected from the tibias and femurs by flushing with RPMI 1640 using a 1 mL syringe with a 26 G needle (BD Biosciences, San Jose, CA, USA). After one wash (490×g, 5 minutes) in RPMI 1640 medium, the cells were incubated with 5 mL of 0.83 M NH4Cl buffer (Sigma-Aldrich) to lyse red blood cells at 37°C for 5 minutes. Then, cells were centrifuged twice in RPMI 1640 medium at 150×g. The bone marrow cells (2×106 cells) were collected and cultured in 100 mm petri dishes (BD, Franklin Lakes, NJ, USA) containing 10 mL of the RPMI 1640 medium supplemented with 10% of heat-inactivated fetal bovine serum, 50 IU/mL penicillin, 50 mg/mL streptomycin, and 20 ng/mL mouse recombinant granulocyte-macrophage colony-stimulating factor (R&D Systems, Minneapolis, MN, USA). After 7 days, nonadherent and loosely adherent cells (immature DCs) were harvested, washed, and used for in vitro experiments.
Cell-viability assay
BMDCs (5×104 cells/100 μL per well) were treated with PLGA and PEI-coated PLGA NPs for 24 hours in flat-bottomed 96-well plates (Corning Incorporated, Corning, NY, USA). For the methyl tetrazolium salt (MTS) assay, the CellTiter 96 aqueous one-solution kit (Promega Corporation, Fitchburg, WI, USA) was used, following the manufacturer’s protocol. Briefly, the MTS reagent was added (10 μL per well), and the plates were incubated at 37°C for 3 hours. Absorbance was determined at 490 nm on a microplate reader (VersaMax; Molecular Devices, Sunnyvale, CA, USA).
Fluorescence-activated cell-sorting analysis
For analysis of DC surface molecules, 5×105 DCs were treated with soluble OVA (20 μg/mL), PLGA NPs (20 μg/mL OVA), PEI-coated PLGA NPs (20 μg/mL OVA), or 1 μg/mL lipopolysaccharide (LPS) for 6 hours. DCs were stained at 4°C for 30 minutes using FITC-conjugated rat anti-CD54, CD80, CD86, and MHC I monoclonal antibodies (Pharmingen; BD Biosciences). After being washed with phosphate-buffered saline (PBS), the cells were analyzed using an Accuri C6 cytometer and CFlow software (BD Biosciences).
Analysis of cellular uptake
To determine the intracellular delivery capacity and localization of the PLGA (OVA) and PEI-coated PLGA (OVA) NPs, freeze-dried NPs were suspended in RPMI medium. Then, BMDCs were treated with NPs (20 μg/mL OVA with 10% FITC-OVA) in a μ-Slide eight-well microscopy chamber (Ibidi GmbH, Planegg, Germany) at a density of 2×104 per well at 37°C for 3 hours. After removal of the culture medium by aspiration and gentle washing with PBS, the cells were fixed in 4% paraformaldehyde for 20 minutes, and stained with 2 μg/mL Hoechst 33342 (trihydrochloride, trihydrate; Thermo Fisher Scientific) in PBS for 10 minutes. Fluorescent images were obtained by using a high-resolution fluorescence microscope (DeltaVision PD; GE Healthcare, Little Chalfont, UK). To analyze the intracellular localization of the OVA antigens in BMDCs, DC lysosomes were visualized with 50 nM LysoTracker red DND-99 (Thermo Fisher Scientific) for 30 minutes at 37°C. After being washed, the cells were treated with PLGA (OVA) or PEI-coated PLGA (OVA) NPs (20 μg/mL OVA with 10% FITC-OVA) for 1 hour at 37°C. The cells were washed twice with RPMI medium and incubated without NPs for an additional 12 hours. The cells were then washed, fixed, and examined by DeltaVision PD as described earlier.
For fluorescence-activated cell-sorting analysis, BMDCs (106 cells/well) were treated with approximately 0.125–0.5 mg/mL NPs (5–20 μg/mL OVA) in six-well plates for 3 hours at 37°C. After being washed with PBS, the cells were analyzed using the Accuri C6 cytometer and CFlow software. The effect of phagocytosis inhibition on NP uptake by the BMDCs was measured with cytochalasin D pretreatment. The cells were pretreated with cytochalasin D (20–40 μM; Sigma-Aldrich) at 37°C for 30 minutes and washed with the culture medium, followed by addition of PEI-coated PLGA (OVA) NPs (20 μg/mL OVA with 10% FITC-OVA). The cells were then incubated at 37°C for 1 hour. After cells had been washed, their fluorescence was measured by fluorescence-activated cell-sorting analysis, as described earlier.
Cross-presentation analysis
For cross-presentation analysis, BMDCs were treated with soluble OVA (20 μg/mL) or PEI-coated PLGA (OVA) NPs (20 μg/mL OVA) for 6 hours at 37°C. Next, the BMDCs were collected and washed twice in PBS and stained with PEI-conjugated antimouse H-2Kb bound to OVA257–264 peptide (SIINFEKL, clone 25-D1.16; eBioscience, San Diego, CA, USA). Fluorescence was measured by the Accuri C6 cytometer and CFlow software. For the cytokine assay, BMDCs (105 cells/well) were seeded in a 96-well round-bottomed plate and cultured for 12 hours at 37°C. Each well was washed twice, and then the cells were pulsed with soluble OVA (20 μg/mL), PLGA (OVA), and PEI-coated PLGA (OVA) NPs (20 μg/mL OVA), or 5 μg/mL OVA257–264 MHC class I epitope peptide (InvivoGen, San Diego, CA, USA) for 6 hours. Next, the cells were washed twice and fixed with 0.05% glutaraldehyde (Sigma-Aldrich) for 20 minutes. The cells were then washed and cocultured with 105 CD8-OVA1.3 T cells for 18 hours at 37°C. The response of stimulated CD8-OVA1.3 T cells was assessed by determining the amount of IL-2 released into the culture by an enzyme-linked immunosorbent assay (ELISA) using OptEIA ELISA kits (BD Biosciences) according to the manufacturer’s instructions. Cytokine concentrations were quantified using the VersaMax microplate reader. For the inhibition assay, BMDCs were pretreated with 10 μM MG-132 (Sigma-Aldrich) and 40 μM leupeptin (Sigma-Aldrich) for 1 hour at 37°C before antigen pulsing.
Statistics
Differences between groups were analyzed by Student’s t-test using GraphPad Prism 5.0 software (GraphPad Software, San Diego, CA, USA). Differences with P-values <0.05 and <0.01 were regarded as significant and highly significant, respectively.
Results and discussion
Characterization of PEI-coated PLGA NPs
In this study, we tested whether surface-modified PLGA NPs can act as antigen-delivery carriers that improve antigen presentation to CD8+ T cells via the MHC class I pathway. PLGA NPs containing the model antigen, OVA, were synthesized using the double-emulsion technique, and their surface charge was modified by coating with PEI (Figure 1). The microscopic morphology of PEI-coated PLGA NPs was examined by SEM. On SEM images, we could see that the spherical type of PLGA NPs and PEI-coated PLGA NPs were successfully fabricated (Figure 2). The size of PLGA NPs and PEI-coated PLGA NPs was found to be in the range 150–250 nm (diameter 173±67.3 and 208.8±66.8 nm, respectively [mean ± standard deviation]; Table 1). By ζ-potential analysis, we confirmed that the negative surface charge of PLGA NPs (−27.2±1.9 mV) was switched to positive values in the case of PEI-coated PLGA NPs (25.6±1.2 mV). These empirical results suggested that PLGA NPs were successfully coated with PEI (Table 1). The amount of OVA protein encapsulated in each PLGA NP was then determined. This protein was encapsulated in PEI-coated PLGA NPs at 32.6±3.7 μg/mg of NPs (Table 1). In fact, no significant changes in the encapsulation amount (P>0.1) were observed in comparison with PLGA NPs (41.3±5 μg/mg, Table 1).
 | Figure 1 Schematic illustrations of the synthesis of PEI-coated PLGA NPs as antigen carriers. |
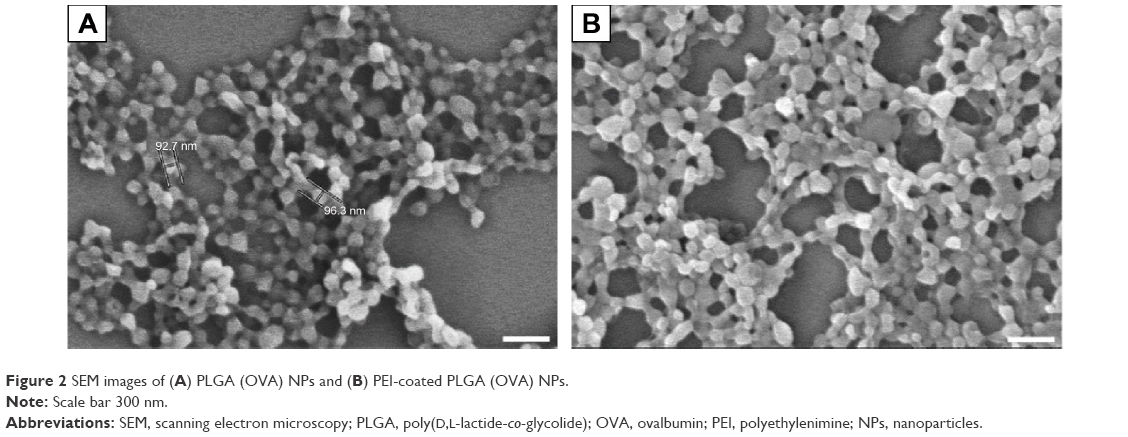 | Figure 2 SEM images of (A) PLGA (OVA) NPs and (B) PEI-coated PLGA (OVA) NPs. |
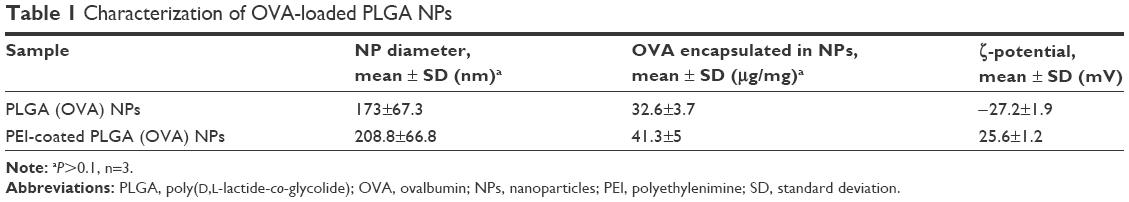 | Table 1 Characterization of OVA-loaded PLGA NPs |
Effects of PEI-coated PLGA NPs on DC viability and maturation
In general, it is important to evaluate safety and toxicity of NPs on target cells before using the NPs in a functional study. Here, we examined the possible effects of PEI-coated PLGA NPs on DC viability and maturation. To evaluate the possible adverse effects of the PEI-coated PLGA NPs on the survival rate of cells, DCs were treated with different concentrations of PEI-coated PLGA NPs for 24 hours. As shown in Figure 3A, the cytotoxicity of PEI-coated PLGA NPs was nearly identical to that of PLGA NPs, which are generally considered safe and have been approved as drug carriers. These empirical results suggested that the cytotoxicity of the PEI-coated PLGA NPs was negligible, even though the NPs had a positive charge. To determine the effects of PEI-coated PLGA NPs on DC maturation, we assessed the relative expression level of maturation markers (CD54, CD80, CD86, and MHC I) on immature DCs treated with PEI-coated PLGA (OVA) NPs (20 μg/mL OVA), and LPS (1 μg/mL) for 6 hours. As shown in Figure 3B, these surface markers were expressed at similar levels in DCs treated or not treated with NPs compared with DCs stimulated with LPS (Figures 3B and S1). These results suggested that the uptake of PEI-coated PLGA (OVA) NPs (containing 20 μg/mL OVA) by DCs did not induce DC maturation, but enhanced antigen delivery.
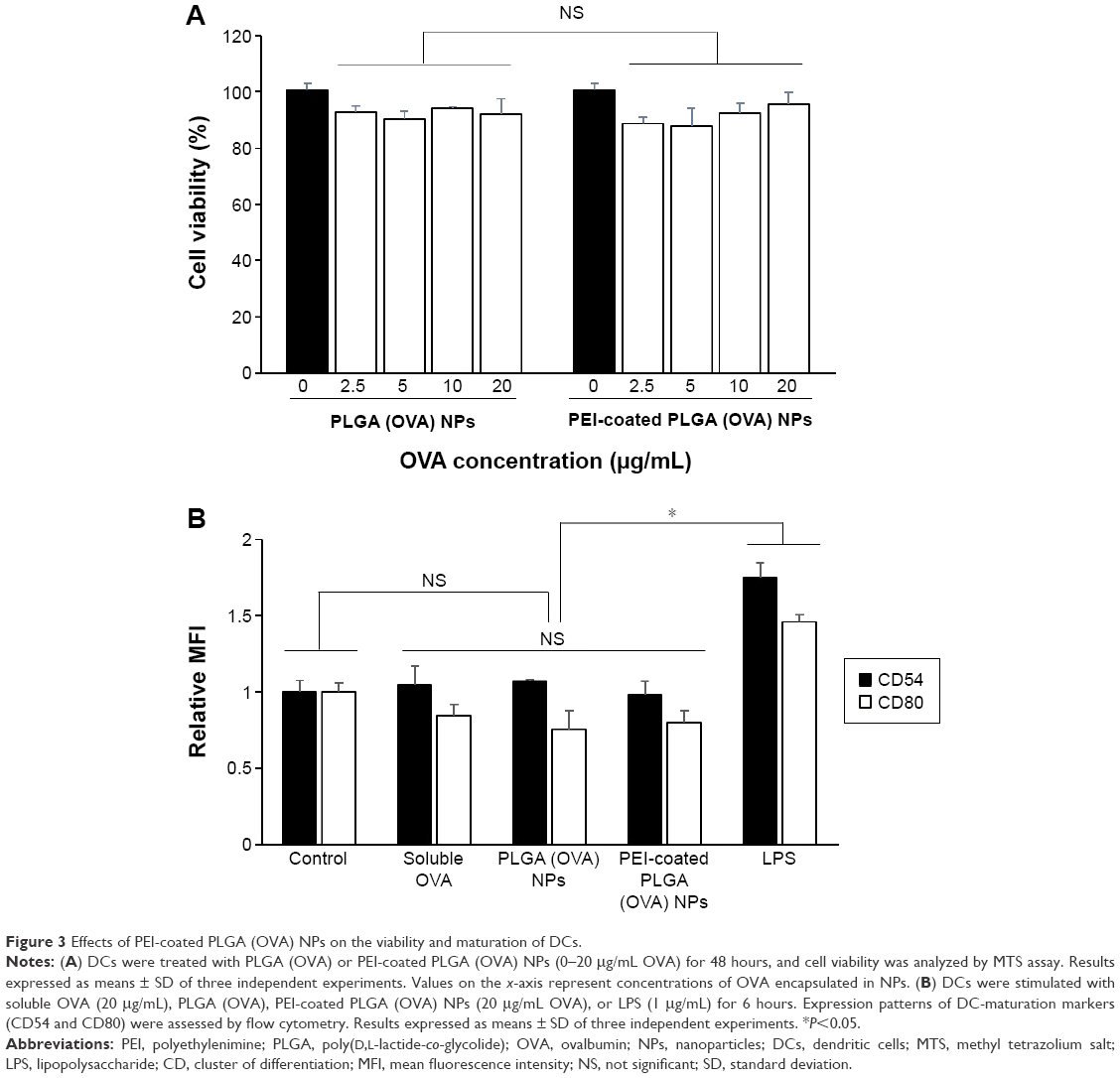 | Figure 3 Effects of PEI-coated PLGA (OVA) NPs on the viability and maturation of DCs. |
Uptake of PEI-coated PLGA NPs by DCs in vitro
In order to evaluate the cellular uptake of PEI-coated PLGA NPs by DCs, BMDCs were treated with soluble OVA (20 μg/mL with 10% FITC-OVA), PLGA NPs, or PEI-coated PLGA NPs (20 μg/mL OVA with 10% FITC-OVA) for 3 hours. As shown in fluorescence-microscopy images, OVA protein (FITC-OVA, green fluorescence) was internalized more efficiently in DCs treated with PEI-coated PLGA (OVA) NPs compared with DCs treated with PLGA (OVA) NPs (Figure 4A). To further evaluate the efficiency of the uptake, DCs were treated with different concentrations of NPs (5–20 μg/mL OVA with 10% FITC-OVA) for 1 hour, and fluorescence intensity of the cells was analyzed by flow cytometry. As shown in Figure 4B, a significant dose-dependent increase in fluorescence intensity was observed in all experimental groups (5–20 μg/mL of OVA). PEI-coated PLGA (OVA) NPs were taken up much more efficiently by DCs at the same concentration (Figure 4B). These results revealed that PEI-coated PLGA (OVA) NPs delivered the OVA antigen into the DCs efficiently. To determine the uptake mechanism of DCs, we performed phagocytosis-inhibition studies. Phagocytosis can be blocked by cytochalasin D due to the destruction of the actin cytoskeleton, which can have a strong impact on the transport of NPs into the cells.27 DCs were pretreated with the phagocytosis/macropinocytosis inhibitor cytochalasin D for 30 minutes, followed by incubation with PEI-coated PLGA (OVA) NPs for 1 hour, and were assessed by flow cytometry. The cell entry of PEI-coated PLGA (OVA) NPs (20 μg/mL OVA with 10% FITC-OVA) was clearly inhibited by cytochalasin D (Figures 4C and S2). These results suggested that phagocytosis or macropinocytosis was one of the main endocytic mechanisms for the intracellular delivery of PEI-coated PLGA (OVA) NPs into DCs.
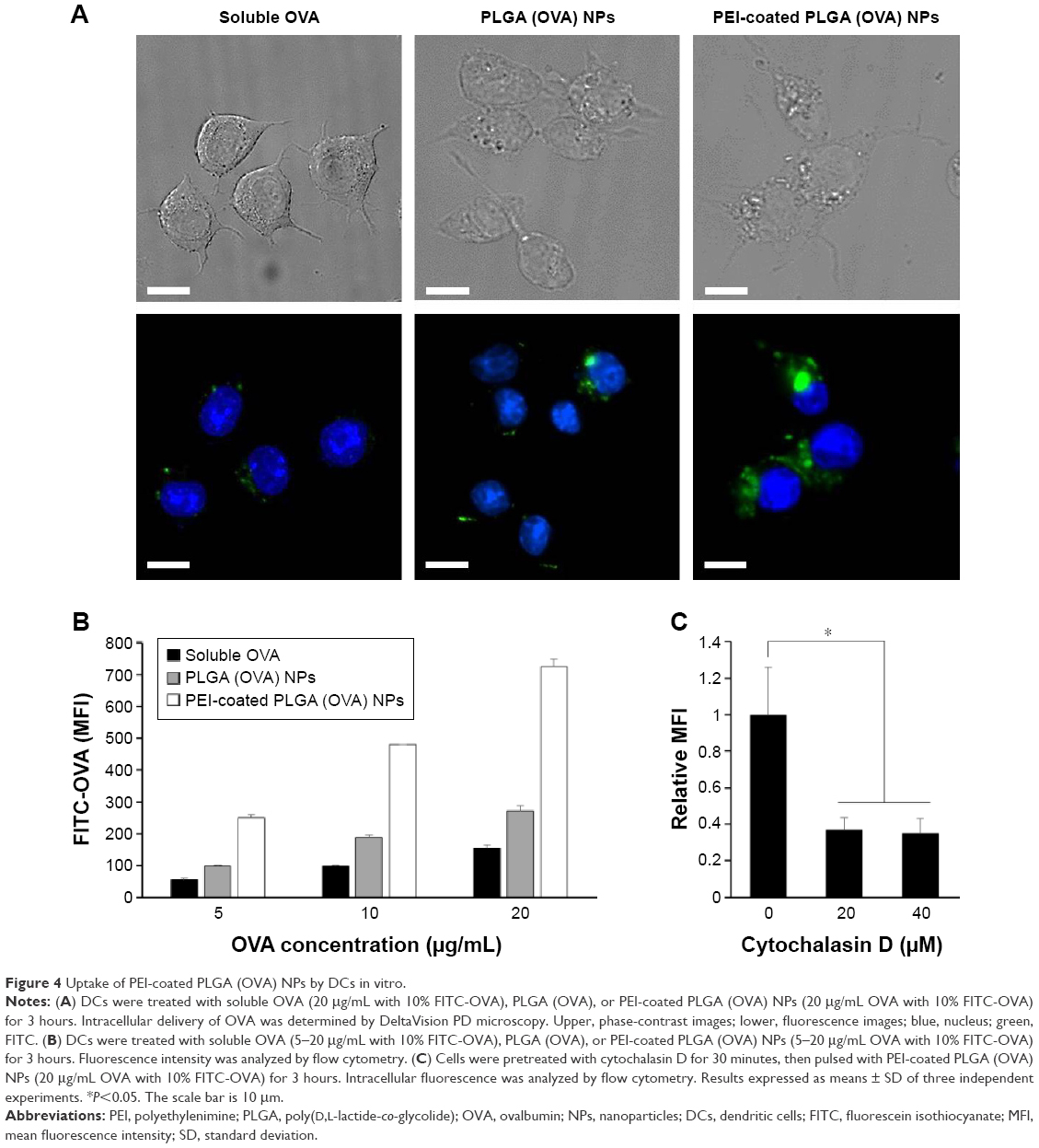 | Figure 4 Uptake of PEI-coated PLGA (OVA) NPs by DCs in vitro. |
Localization of antigen-loaded PEI-coated PLGA NPs
The efficient endosomal escape of an exogenous antigen after entering a cell is essential for antigen cross-presentation.12 We assessed the subcellular trafficking and localization of OVA in DCs using fluorescence microscopy. DCs were treated with PLGA (OVA) NPs or PEI-coated PLGA (OVA) NPs (loaded with 20 μg/mL OVA) for 1 hour, followed by extensive washing, incubation without NPs for an additional 12 hours, and the lysosomes being stained with a lysosome-specific dye. As shown in Figure 5, OVA was internalized in endosomes/lysosomes under the influence of PLGA (OVA) NPs. The colocalization (yellow) of red and green fluorescence indicated that the OVA loaded in PLGA (OVA) NPs ended up in endosomes/lysosomes (Figures 5 and S3, upper panel). In contrast, green and red fluorescence were separated significantly when the cells were treated with PEI-coated PLGA (OVA) NPs (Figures 5 and S3, lower panel). The results suggested that PEI-coated PLGA (OVA) NPs can enter the cells, escape from endosomes/lysosomes, and finally deliver OVA to the cytoplasm.
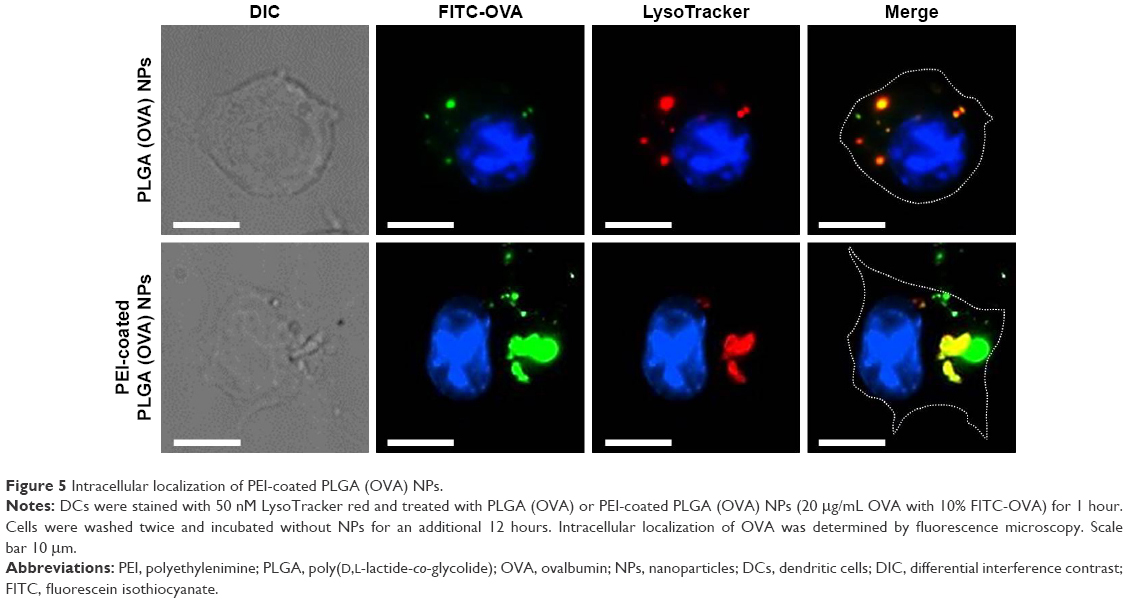 | Figure 5 Intracellular localization of PEI-coated PLGA (OVA) NPs. |
Cross-presentation of antigen-loaded PEI-coated PLGA NPs by DCs
We examined the ability of PEI-coated PLGA (OVA) NPs to mediate the antigen cross-presentation in DCs using flow cytometry. Because the processed OVA antigen is carried to the surface of DCs by MHC class I molecules, we quantified the processed antigen peptide as an indicator of the efficiency of antigen cross-presentation using the 25-D1.16 monoclonal antibody, which specifically recognizes the complex of the OVA257–264 peptide with MHC class I (SIINFEKL-H2-Kb).28 In this experiment, a higher percentage of 25-D1.16-positive cells was found among DCs treated with PEI-coated PLGA (OVA) NPs (2.1%) than among DCs treated with PLGA (OVA) NPs (0.9%) or soluble OVA (0.4%) (Figures 6A and S4). We next studied the effect of OVA-specific CD8+ T-cell priming to confirm the cross-presentation. As shown in Figure 6B, the DCs treated with PEI-coated PLGA (OVA) NPs (20 μg/mL OVA) induced a release of IL-2 cytokine from OVA-specific CD8-OVA1.3 T cells more efficiently than DCs treated with PLGA (OVA) NPs or soluble OVA (20 μg/mL).
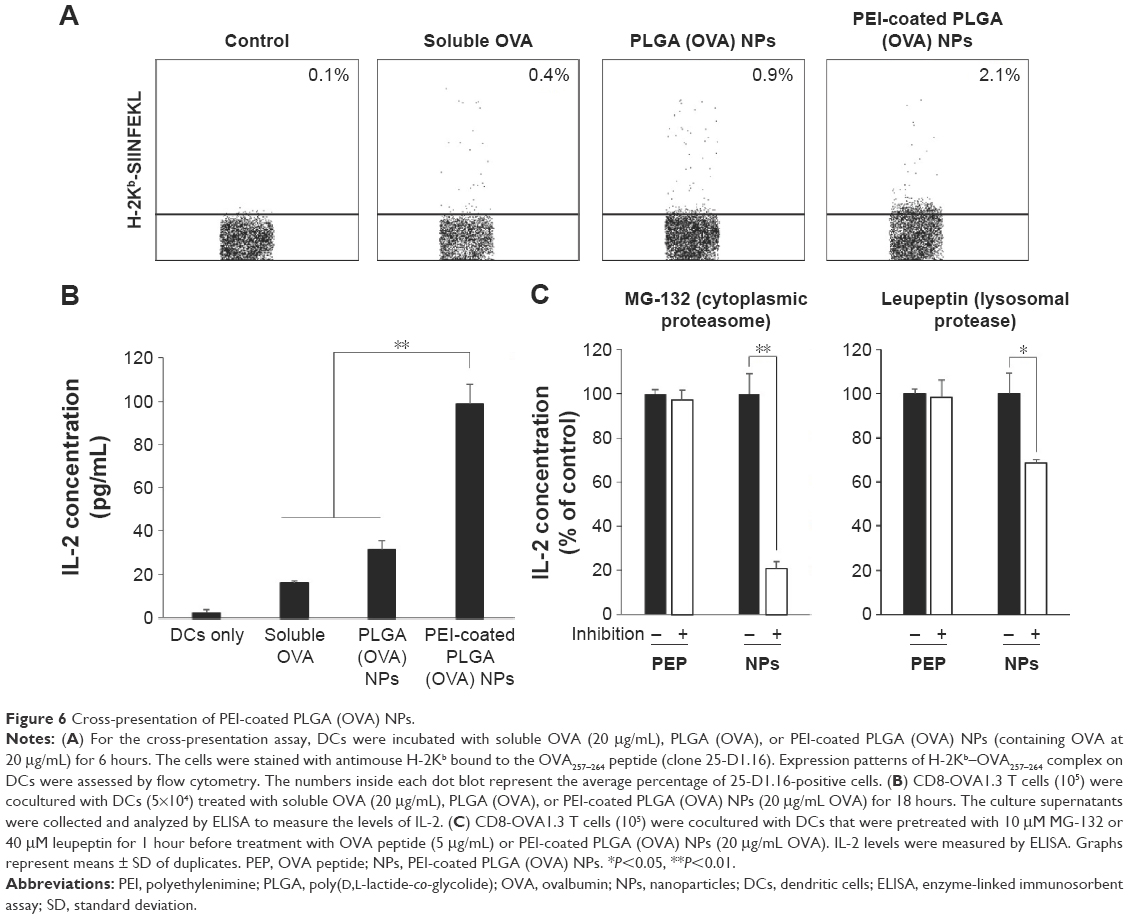 | Figure 6 Cross-presentation of PEI-coated PLGA (OVA) NPs. |
To identify the pathway of cross-presentation induced by PEI-coated PLGA (OVA) NPs, we used inhibitors of cytoplasmic proteasomes and lysosomal proteases, which are related to the antigen-processing pathway for MHC class I antigen presentation. DCs were pretreated with 10 μM MG-132 (cytoplasmic proteasome inhibitor) or 40 μM leupeptin (lysosomal protease inhibitor) for 1 hour before the addition of PEI-coated PLGA (OVA) NPs or OVA peptide (SIINFEKL, 5 μg/mL), and the OVA-specific CD8+ T-cell response was measured. The CD8+ T-cell response induced by PEI-coated PLGA (OVA) NP treatment was strongly suppressed by the proteasome inhibitor (P<0.01; Figure 6C, left graph) and lysosomal protease inhibitor (P<0.05; Figure 6C, right graph). In contrast, the response induced by OVA peptide was not inhibited by all inhibitors (Figure 6C). These results indicated that the internalized PEI-coated PLGA (OVA) NPs (through phagocytosis) escaped from endosomes or lysosomes, entered the cytoplasm, and released antigens. The released antigen was processed by the proteasome and presented by MHC class I molecules via a classical endogenous antigen-presentation pathway. In addition, the antigens delivered into endosomes were also processed by lysosomal protease and cross-presented in an endosome or lysosome (Figure 7).29
 | Figure 7 Schematic illustrations of the predicted mechanism of cross-presentation and CD8+ T-cell response induced by PEI-coated PLGA (OVA) NPs. |
Conclusion
In this study, we designed and developed surface charge-controlled polymer NPs as antigen carriers that could improve CD8+ T-cell priming via the cross-presentation pathway. PEI-coated PLGA NPs carrying a model antigen had no significant effect on cell viability or spontaneous activation, and were easily taken up by antigen-presenting cells through phagocytosis and macropinocytosis. Via endosome escape and a lysosomal processing mechanism, PEI-coated PLGA NPs induced efficient cross-presentation of an antigen on MHC class I molecules for strong antigen-specific CTL induction, which is essential for the development of an immunotherapeutic cancer vaccine. Therefore, PEI-coated PLGA NPs are expected to be useful for cancer immunotherapy as efficient antigen-delivery carriers.
Acknowledgments
This work was supported by a grant from the National Research Foundation of Korea (NRF), funded by the Korean government (MEST; grant 2014R1A2A1A10049960 and 2015R1A2A1A15051980), and a grant from the Korea Health Technology R&D Project, provided through the Korea Health Industry Development Institute, funded by the Ministry of Health and Welfare (grant HI14C2680).
Disclosure
The authors report no conflicts of interest in this work.
References
Danhier F, Ansorena E, Silva JM, Coco R, Le Breton A, Préat V. PLGA-based nanoparticles: an overview of biomedical applications. J Control Release. 2012;161(2):505–522. | ||
Irvine DJ, Hanson MC, Rakhra K, Tokatlian T. Synthetic nanoparticles for vaccines and immunotherapy. Chem Rev. 2015;115(19):11109–11146. | ||
Shao K, Singha S, Clemente-Casares X, Tsai S, Yang Y, Santamaria P. Nanoparticle-based immunotherapy for cancer. ACS Nano. 2014;9(1):16–30. | ||
Vasir JK, Labhasetwar V. Biodegradable nanoparticles for cytosolic delivery of therapeutics. Adv Drug Deliv Rev. 2007;59(8):718–728. | ||
Yih TC, Al-Fandi M. Engineered nanoparticles as precise drug delivery systems. J Cell Biochem. 2006;97(6):1184–1190. | ||
Friede M, Aguado MT. Need for new vaccine formulations and potential of particulate antigen and DNA delivery systems. Adv Drug Deliv Rev. 2005;57(3):325–331. | ||
Heo MB, Lim YT. Programmed nanoparticles for combined immunomodulation, antigen presentation and tracking of immunotherapeutic cells. Biomaterials. 2014;35(1):590–600. | ||
Noh YW, Hong JH, Shim SM, et al. Polymer nanomicelles for efficient mucus delivery and antigen-specific high mucosal immunity. Angew Chem Int Ed Engl. 2013;52(30):7684–7689. | ||
Serda RE. Particle platforms for cancer immunotherapy. Int J Nanomedicine. 2013;8:1683–1696. | ||
Caminschi I, Shortman K. Boosting antibody responses by targeting antigens to dendritic cells. Trends Immunol. 2012;33(2):71–77. | ||
Gilboa E. DC-based cancer vaccines. J Clin Invest. 2007;117(5):1195–1203. | ||
Lin ML, Zhan Y, Villadangos JA, Lew AM. The cell biology of cross-presentation and the role of dendritic cell subsets. Immunol Cell Biol. 2008;86(4):353–362. | ||
Shen H, Ackerman AL, Cody V, et al. Enhanced and prolonged cross-presentation following endosomal escape of exogenous antigens encapsulated in biodegradable nanoparticles. Immunology. 2006;117(1):78–88. | ||
Uto T, Wang X, Sato K, et al. Targeting of antigen to dendritic cells with poly(γ-glutamic acid) nanoparticles induces antigen-specific humoral and cellular immunity. J Immunol. 2007;178(5):2979–2986. | ||
Tanaka Y, Taneichi M, Kasai M, Kakiuchi T, Uchida T. Liposome-coupled antigens are internalized by antigen-presenting cells via pinocytosis and cross-presented to CD8 T cells. PLoS One. 2010;5(12):e15225. | ||
Xia X, Mai J, Xu R, et al. Porous silicon microparticle potentiates anti-tumor immunity by enhancing cross-presentation and inducing type I interferon response. Cell Rep. 2015;11(6):957–966. | ||
Mundargi RC, Babu VR, Rangaswamy V, Patel P, Aminabhavi TM. Nano/micro technologies for delivering macromolecular therapeutics using poly(D,L-lactide-co-glycolide) and its derivatives. J Control Release. 2008;125(3):193–209. | ||
Guedj AS, Kell AJ, Barnes M, et al. Preparation, characterization, and safety evaluation of poly(lactide-co-glycolide) nanoparticles for protein delivery into macrophages. Int J Nanomedicine. 2015;10:5965–5979. | ||
Yu D, Zhang Y, Zhou X, Mao Z, Gao C. Influence of surface coating of PLGA particles on the internalization and functions of human endothelial cells. Biomacromolecules. 2012;13(10):3272–3282. | ||
Fay F, Quinn DJ, Gilmore BF, McCarron PA, Scott CJ. Gene delivery using dimethyldidodecylammonium bromide-coated PLGA nanoparticles. Biomaterials. 2010;31(14):4214–4222. | ||
Han R, Zhu J, Yang X, Xu H. Surface modification of poly(D,L-lactic-co-glycolic acid) nanoparticles with protamine enhanced cross-presentation of encapsulated ovalbumin by bone marrow-derived dendritic cells. J Biomed Mater Res A. 2011;96(1):142–149. | ||
Intra J, Salem AK. Characterization of the transgene expression generated by branched and linear polyethylenimine-plasmid DNA nanoparticles in vitro and after intraperitoneal injection in vivo. J Control Release. 2008;130(2):129–138. | ||
Kim SH, Jeong JH, Chun KW, Park TG. Target-specific cellular uptake of PLGA nanoparticles coated with poly(L-lysine)-poly(ethylene glycol)-folate conjugate. Langmuir. 2005;21(19):8852–8857. | ||
Gómez JM, Csaba N, Fischer S, et al. Surface coating of PLGA microparticles with protamine enhances their immunological performance through facilitated phagocytosis. J Control Release. 2008;130(2):161–167. | ||
Zhu L, Mahato RI. Lipid and polymeric carrier-mediated nucleic acid delivery. Expert Opin Drug Deliv. 2010;7(10):1209–1226. | ||
Noh YW, Jang YS, Ahn KJ, Lim YT, Chung BH. Simultaneous in vivo tracking of dendritic cells and priming of an antigen-specific immune response. Biomaterials. 2011;32(26):6254–6263. | ||
Goddette DW, Frieden C. Actin polymerization: the mechanism of action of cytochalasin D. J Biol Chem. 1986;261(34):15974–15980. | ||
Porgador A, Yewdell JW, Deng Y, Bennink JR, Germain RN. Localization, quantitation, and in situ detection of specific peptide-MHC class I complexes using a monoclonal antibody. Immunity. 1997;6(6):715–726. | ||
Ackerman AL, Cresswell P. Cellular mechanisms governing cross-presentation of exogenous antigens. Nat Immunol. 2004;5(7):678–684. |
Supplementary materials
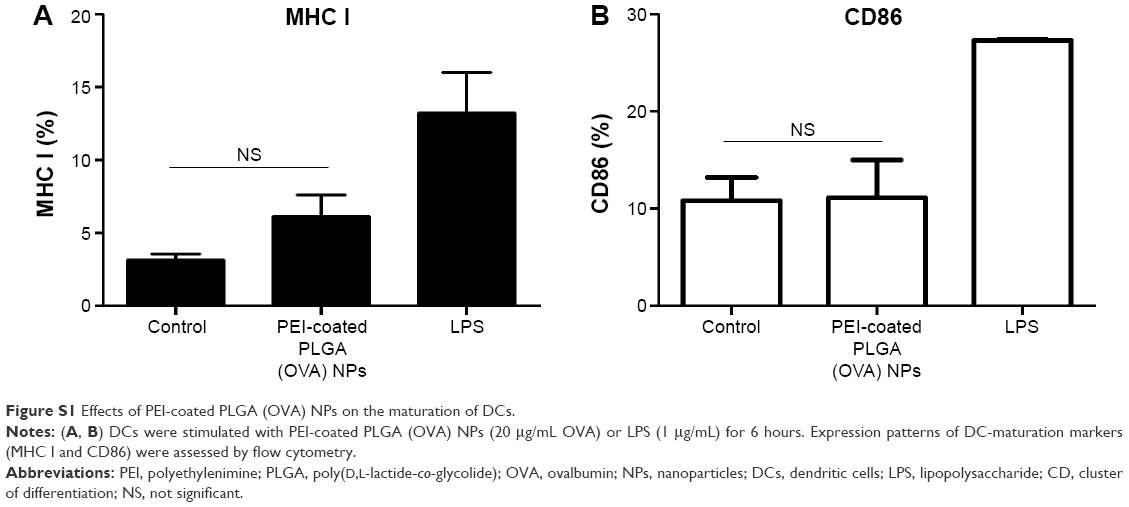 | Figure S1 Effects of PEI-coated PLGA (OVA) NPs on the maturation of DCs. |
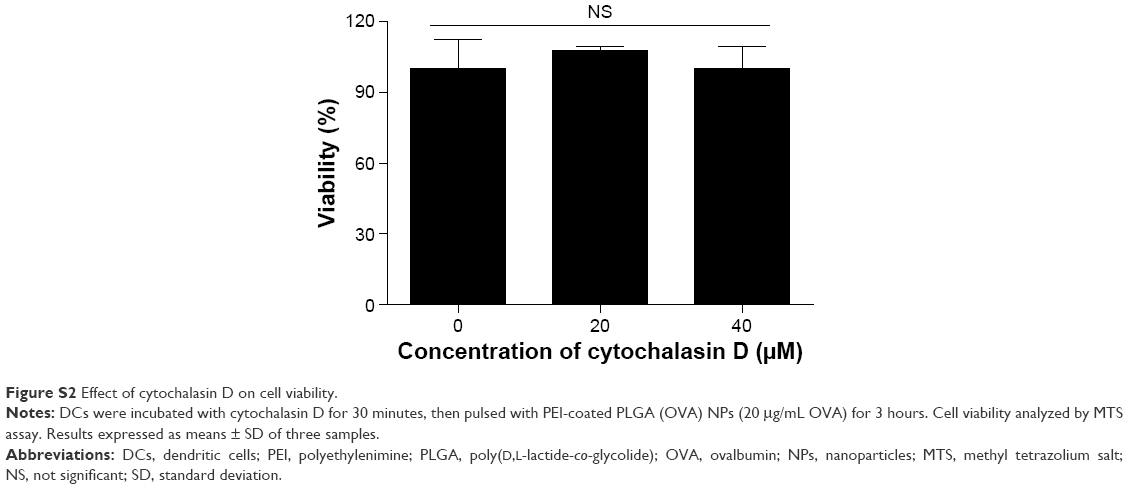 | Figure S2 Effect of cytochalasin D on cell viability. |
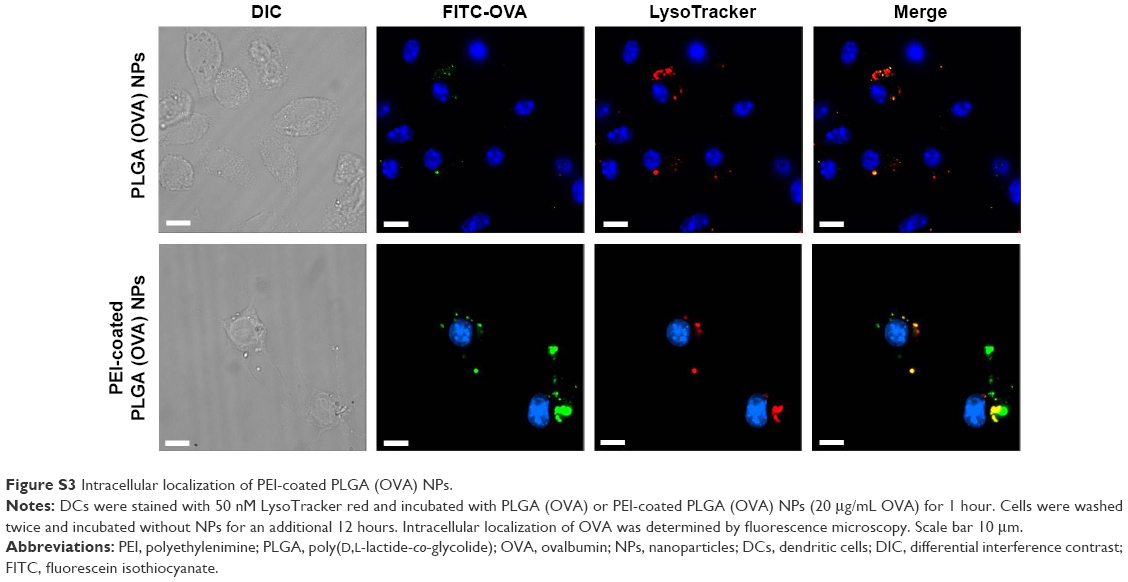 | Figure S3 Intracellular localization of PEI-coated PLGA (OVA) NPs. |
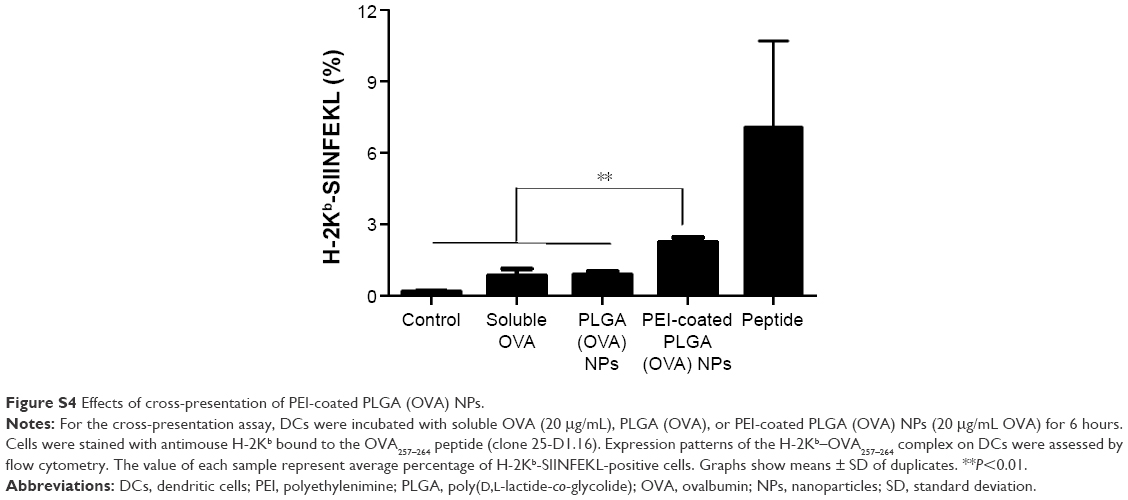 | Figure S4 Effects of cross-presentation of PEI-coated PLGA (OVA) NPs. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.