")
Back to Journals » International Journal of Nanomedicine » Volume 13
Poly(lactic-co-glycolic acid)-loaded nanoparticles of betulinic acid for improved treatment of hepatic cancer: characterization, in vitro and in vivo evaluations
Authors Kumar P, Singh AK, Raj V, Rai A, Keshari AK , Kumar D , Maity B, Prakash A, Maiti S, Saha S
Received 17 November 2017
Accepted for publication 23 December 2017
Published 16 February 2018 Volume 2018:13 Pages 975—990
DOI https://doi.org/10.2147/IJN.S157391
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr Thomas Webster
Pranesh Kumar,1 Ashok K Singh,1 Vinit Raj,1 Amit Rai,1 Amit K Keshari,1 Dinesh Kumar,2 Biswanath Maity,2 Anand Prakash,3 Sabyasachi Maiti,4 Sudipta Saha1
1Department of Pharmaceutical Sciences, Babasaheb Bhimrao Ambedkar University, Vidya Vihar, Lucknow, Uttar Pradesh, India; 2Centre of Biomedical Research, SGPGIMS Campus, Lucknow, Uttar Pradesh, India; 3Department of Biotechnology, Babasaheb Bhimrao Ambedkar University, Vidya Vihar, Lucknow, Uttar Pradesh, India; 4Department of Pharmacy, Indira Gandhi National Tribal University, Amarkantak, Anuppur, Madhya Pradesh, India
Purpose: The application of betulinic acid (B), a potent antineoplastic agent, is limited due to poor bioavailability, short plasma half-life and inappropriate tissue distribution. Thus, we aimed to prepare novel 50:50 poly(lactic-co-glycolic acid) (PLGA)-loaded B nanoparticles (BNP) and to compare its anti-hepatocellular carcinoma (HCC) activity with parent B.
Methods: BNP were synthesized and characterized using different methods such as scanning electron microscopy (SEM), fourier-transform infrared (FTIR) spectrometry and particle size analyses. Particle size of BNP was optimized through the application of the stabilizer, polyvinyl alcohol (PVA). The anti-HCC response was evaluated through in vitro cell line study using Hep-G2 cells, confocal microscopy, in vivo oral pharmacokinetics and animal studies. Further, quantitative reverse transcription polymerase chain reaction (qRT-PCR) analysis was conducted to observe the changes in the expression of specific genes.
Results: Particle size of BNP was optimized through the application of the stabilizer, polyvinyl alcohol. Physicochemical characterization exhibited particle size of 257.1 nm with zeta potential -0.170 mV (optimized batch B, BNP). SEM and FTIR analyses of BNP showed that cylindrical particles of B converted to spherical particles in BNP and there were no interaction between B and used polymers. The release study of optimized BNP was highest (≥80%) than any other formulation. Later, in vitro cell culture analysis using Hep-G2 cells and confocal microscopy studies revealed that BNP had the highest inhibition and penetration properties than parent B. Oral pharmacokinetics studies using albino Wistar rats at single 100 mg dose again exhibited BNP had the higher 50% of plasma concentration (t1/2), a higher maximum plasma concentration (Cmax) and took longer to reach the maximum plasma concentration (Tmax) than parent B. Next, our in vivo study using nitrosodiethyl amine (NDEA)-induced HCC model documented BNP decreased in number of nodules, restored body weight, oxidative stress parameters, liver marker enzymes and histological architecture than parent B. Lastly, qRT-PCR studies further demonstrated that anti-HCC properties of BNP may be due to over expression of antiapoptotic caspases i.e., caspase 3 and 8.
Conclusion: The prepared BNP showed a better therapeutic response against HCC and could be attributed as future candidate molecule for HCC treatment.
Keywords: betulinic acid, PLGA-loaded nanoparticles, HepG2 cells, hepatocellular carcinoma, caspase-3 and -8
Introduction
The development of drug delivery system of existing marketed drugs for better therapeutic index and reduced toxicological properties are the basic criteria of modern delivery research. This helps us to improve pharmacokinetics and biodistribution of drugs for better efficacy and effectiveness.1 Nowadays, preparation of nanoparticle-based therapeutics has drawn major attention among researchers due to their site-specific action.2 Commercially available polymers have important roles in the drug delivery system, especially in the nanocarrier system. Nanoparticulate carriers have always been attractive on account of their size and capacity of spatial and temporal-controlled delivery of bioactives.3
Cancer chemotherapy is the most challenging scenario in modern drug development where low toxicity with improved pharmacological responses and lower side effects are always a priority.1 Betulinic acid (B; Figure 1) is a pentacyclic triterpenoid obtained from the stem bark of Dillenia indica (family: Dilleniaceae), which has antimalarial, antiretroviral and anti-inflammatory properties.4–6 B has anticarcinogenic properties that generally trigger the apoptosis signaling cascade through the activation of caspase-3 and caspase-8.7,8 In addition, B induces direct mitochondrial damage and thereby overcomes the resistance that a tumor cell generally acquires.9–18
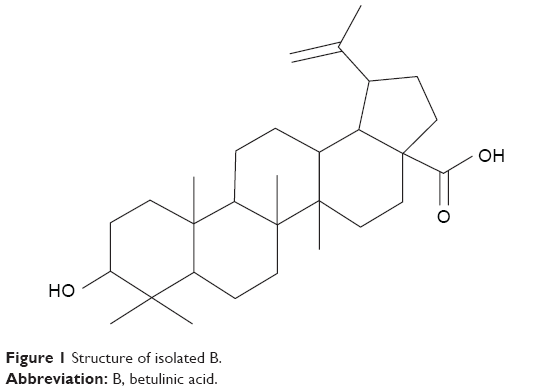 | Figure 1 Structure of isolated B. |
Hepatocellular carcinoma (HCC) is considered as the sixth most common malignancy and third most frequent leading cause of death worldwide.19 Synthetic chemotherapeutic agents have been proven efficacious for HCC treatment; however, researchers have achieved very little success due to their chemoresistance.20 It is, therefore, an urgent need to discover newer plant-derived anticancer agents for better treatment of cancer. Natural products have been used for hepatic cancer from ancient time but very few compounds isolated from natural origin are effective against hepatic cancer.21
The major drawback associated with natural products is poor oral absorption, which may lead to lower effectiveness. B is a class of natural products that has poor oral absorption and bioavailability.22 Recent investigation revealed that oral solubility and absorption may be enhanced via incorporation of drug into poly(lactic-co-glycolic acid) (PLGA).23,24 Therefore, this study focused on preparation of B nanoparticles (BNP) using PLGA, primarily, to improve oral bioavailability. Furthermore, both B and BNP were further screened for anti-HCC effect using both in vitro and in vivo models. Later, the molecular mechanism of the ameliorative potential of B and BNP was explored as apoptotic targets, ie, caspase-3 and -8.
Materials and methods
Isolation and characterization of B
D. indica stem bark (2 kg) was obtained from the coastal area of West Bengal during mid July and authenticated by Shakti College of Pharmacy, Balrampur, Uttar Pradesh, India. A voucher specimen has been deposited for future reference (SCOP/2017/829). The plant materials were air-dried under shade, powdered and defatted with petroleum ether (60°C–80°C) using Soxhlet apparatus by successive solvent extraction method. Later, extraction was performed using methanol:water (98:2), and the extracted samples were evaporated to dryness using a rotary vacuum evaporator (IKA, Königswinter, Germany). The white-colored material was again recrystallized with methanol. The final yield of B was 9.2%; melting point 298°C; Fourier-transform infrared (FTIR; KBr) 3,630, 2,980, 2,934, 1,638, 1,480, 1,362, 1,195, 1,123, 784 cm−1; 1H NMR (DMSO-d6) 0.73, 0.85, 0.95, 1.01 and 1.73 (5 s, 15H, all tertiary −CH3), 1.39 (m, 2H, H-21), 1.40 (m, 2H, H-16), 1.44 (m, 2H, H-20), 1.51 (m, 4H, H-18, H-19 and H-15), 2.19 (m, 3H, H-1 and H-9), 2.30 (m, 2H, H-14), 3.04 (s, 2H, H-2), 4.37 (s, 2H, H-7), 4.64 (2, 2H, H-11), 4.77 (s, 2H, H-12), 12.16 (s, 1H, −COOH); 13C NMR (DMSO-d6) 14.85, 16.21, 16.42, 18.45, 19.40, 20.33, 25.54, 27.62, 28.57, 28.78, 29.33, 31.54, 36.79, 37.19, 38.97, 40.33, 40.71, 42.47, 44.65, 48.98, 50.38, 55.34, 55.88, 61.80, 77.23, 110.13, 118.13, 150.79, 177.12 and electrospray ionization mass spectrometry (+ve mode) (M+H)+ m/e 457.8.
Preparation of BNP
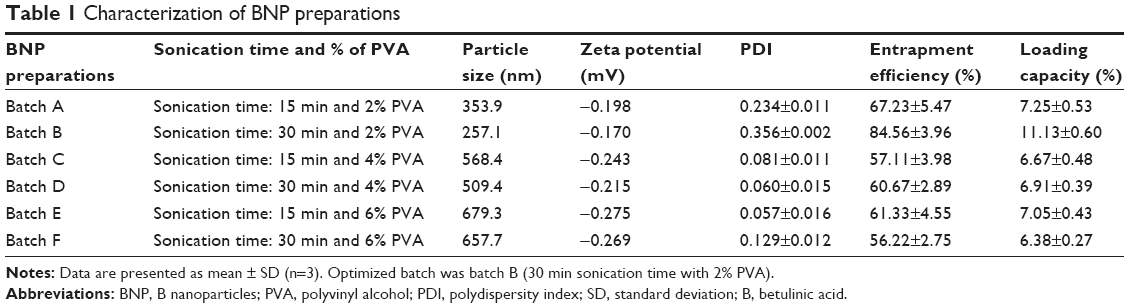
Emulsification and solvent evaporation techniques were adopted for the preparation of BNP. Small particle size was achieved through sonication and homogenization process. In all, 50 mg of PLGA and 2.5 mg of B were dissolved together in 3 mL of chloroform. This mixture was sonicated for 15 or 30 min with addition of 12 mL of different concentrations (w/v) of aqueous polyvinyl alcohol (PVA) at 10 kHZ (2% PVA formulations A and B with sonication times 15 and 30 min, 4% PVA formulations C and D with sonication times 15 and 30 min, 6% PVA formulations E and F with sonication times 15 and 30 min; Table 1). All the resultant mixtures were homogenized for 20 min at 20,000 rpm using a refrigerated centrifuge at 4°C. The resultant mixture was again stirred overnight to remove the organic solvent. Later, the BNP formed was harvested by ultracentrifugation at 30,000 rpm for 25 min at 4°C. BNP was washed with water, recentrifuged and preserved in vacuum desiccators at 4°C for further evaluations. Later, this procedure was followed for large-scale preparation where B concentration was 20 times higher than for the small-scale preparation.
 | Table 1 Characterization of BNP preparations |
Particle size, polydispersity index (PDI) and zeta potential
The particle size and PDI of various batches of BNP were determined in NanoPlus zeta/nano particle analyzer against a 4 mW helium–neon (He–Ne) laser beam (633 nm) and a back scattering angle of 173° by photon correlation spectroscopy (PCS) in triplicate. Zeta potentials were measured using the NanoPlus zeta/nano particle analyzer using disposable zeta cells. Aliquots from each preparation type were injected in electrophoretic zeta cells, and zeta potentials were analyzed using the Smoluchowski equation.23
BNP entrapment efficiency (%) and loading capacity (%)
BNP entrapment efficiency was performed using Shimadzu LC-20AD (Kyoto, Japan) equipped with a photodiode array detector. The mobile phase consisted of a gradient elution of acetonitrile (ACN):water (H2O) of 9:1, and the flow rate was 1 mL/min. The separation was performed using reversed phase (RP)-C18 column (5.0 μ particle size, 4.6 mm internal diameter and 250 mm length) with  max of 230 nm, and the temperature were maintained at 40°C throughout the experiment. Total run time was 15 min. The column was washed after every run in an elution solvent ratio (ACN:H2O =50:50) with the injection of blank ACN. A peak area (y) versus concentration (x) graph for B was prepared first; y =211.97x +3.99, R2=0.9987. This equation was used to detect the concentration of B in each solution. Mass of B before and after nanoparticulation in supernatant (obtained by centrifugation using a refrigerated centrifuge at 4°C and 20,000 rpm for 20 min) was determined in high-performance liquid chromatography (HPLC) experiments, for calculation. The encapsulation efficiency and the actual loading capacity expressed in percentage were calculated using the following formulae:
max of 230 nm, and the temperature were maintained at 40°C throughout the experiment. Total run time was 15 min. The column was washed after every run in an elution solvent ratio (ACN:H2O =50:50) with the injection of blank ACN. A peak area (y) versus concentration (x) graph for B was prepared first; y =211.97x +3.99, R2=0.9987. This equation was used to detect the concentration of B in each solution. Mass of B before and after nanoparticulation in supernatant (obtained by centrifugation using a refrigerated centrifuge at 4°C and 20,000 rpm for 20 min) was determined in high-performance liquid chromatography (HPLC) experiments, for calculation. The encapsulation efficiency and the actual loading capacity expressed in percentage were calculated using the following formulae:
|

|
In vitro release studies
In vitro drug release of all six batches was measured using BNP equivalent to ~2 mg of B dissolved in 1 mL of phosphate buffer (100 mM, pH 7.4), and the dispersion was carefully washed into a dialysis bag (molecular weight cutoff [MWCO] 10 kDa; Thermo Fisher Scientific, Waltham, MA, USA). The dialysis bags were placed in glass vials containing 10 mL of phosphate buffer and maintained at 37°C in a shaker bath. At predetermined time intervals, the release medium of 2 mL in vials was taken out and replaced with the same volume of fresh buffer medium. The amount of B released per timed sample was estimated spectrophotometrically. The in vitro releases of all batches were completed in triplicate at each time point, and the cumulative percentage release over time was plotted.23,24 The Korsmeyer–Peppas model was applied to determine n and K values and was calculated using GraphPad Prism 5.0 software (trial version; GraphPad Software, Inc., La Jolla, CA, USA).
FTIR and scanning electron microscopy (SEM) studies
FTIR studies were carried out to evaluate any interaction between B and using polymers. Separately, FTIR spectra were recorded of PVA, PLGA, B and BNP in this experiment. In all, 5 mg of compounds were mixed with 200 mg of KBr and were pelletized at 1,000 psi. Finally, FTIR data were recorded and analyzed through Nicolet™ 6700 (Thermo Fischer Scientific) over a wave number range of 4,000–400 cm−1. SEM images were taken using JSM-6490LV (JEOL, Tokyo, Japan).
Cell culture and sulforhodamine B (SRB) assay
HepG2 cell line was commercially purchased from National Cell Repository (NCCS, Pune, India) and later cultured in our laboratory facility for in vitro testing. HepG2 cells were grown in the Roswell Park Memorial Institute (RPMI)-1640 medium containing 10% fetal bovine serum and 2 mM L-glutamine in a T-75 flask at 37°C, 5% CO2, 95% air and 100% relative humidity for 24 h. After growing, 100 μL cells containing media were inoculated into 96 well plates at a concentration of 5×103 cells/well. Separately, all the compounds to be tested were solubilized in dimethyl sulfoxide at 100 mg/mL and diluted to 1 mg/mL using water and stored frozen prior to use. Next day, 100 μL of compounds containing the medium were added into each well (10, 20, 40 and 80 μg/mL) and incubated at standard conditions for 48 h. To terminate the reaction, 50 μL of the cold 30% trichloroacetic acid (TCA) was added and incubated at 4°C for 1 h. The supernatant was discarded; the plates were washed five times with tap water and air-dried. Furthermore, 50 μL of SRB solution at 0.4% (w/v) in 1% acetic acid was added to each of the wells and incubated for 20 min at room temperature. After staining, the residual dye was removed by washing five times with 1% acetic acid and the plates were air-dried. The bound stain was subsequently eluted with 10 mM Trizma base, and the absorbance was read on a plate reader at a wavelength of 540 nm with 690 nm reference wavelength. The results were obtained in triplicate on separate plates, and finally, the average values were determined from these three experiments. Using the seven absorbance measurements (time zero [Tz], control growth (C) and test growth in the presence of drug at the five concentration levels [Ti]), the percentage growth was calculated at each of the drug concentration levels. Percentage growth inhibition was calculated as:
|

|

GI50 was calculated from [(Ti − Tz)/(C − Tz)] ×100=50, which is the drug concentration resulting in a 50% reduction in the net protein increase (as measured by SRB staining) in control cells during the drug incubation.25
Confocal laser scanning microscopy
HepG2 cells were seeded on Lab-Tek chambered cover glasses (Nalge Nunc International, Naperville, IL, USA) and incubated at 37°C in 95% air and 5% CO2 environment until cells were ~70% confluent. On the day of experiment, the growth medium was replaced by Hank’s buffered salt solution (HBSS; pH 7.4). After equilibration with HBSS at 37°C for 30 min, the buffer was replaced with fluorescein isothiocyanate (FITC)-loaded BNP suspension (250 mg/mL in HBSS) and then the monolayers were further incubated for 1 or 2 h. At the end of the experiment, the monolayers were washed three times with fresh prewarmed transport buffer to eliminate excess nanoparticles that were not attached to the cells. Cells were then fixed with 70% ethanol, and the samples were mounted in the fluorescent mounting medium (Dako Denmark A/S, Glostrup, Denmark) until examination was performed using the confocal laser scanning microscope (LSM 410; Zeiss, Berlin, Germany) equipped with an imaging software, FluoView FV300 (Olympus corporation, Tokyo, Japan).26
In vivo pharmacokinetic studies
Determination of B and BNP in rat plasma
In a separate experiment, both B and BNP were administered orally at 100 mg/kg body weight to albino Wistar rats (Ref No SDCOP&VS/AH/CPCSEA/01/0027) and blood was collected from retro-orbital plexus at 0.083, 0.25, 0.5, 1, 2, 4, 8, 12, 16, 24 and 48 h. After collection, blood was centrifuged at 10,000 rpm for 10 min and serum was separated and kept at −20°C for further HPLC.27,28
Preparation of plasma standards and test samples
In all, 1 mg/mL stock solution of B was prepared in ACN. A total of 0.02, 0.1, 0.2, 0.5, 1, 2, 5, 10, 50, 100 μg/mL of working solutions were prepared from this stock solution in ACN. In all, 100 μL of blank plasma and 100 μL of working solutions were taken in separate tubes and vortexed for 30 min. After that, the tubes were centrifuged at 13,000 rpm for 5 min and supernatant was taken into other tubes and dried at 40°C overnight. Later, the tubes were reconstituted with 50 μL of ACN, vortexed for 10 min and 20 μL was injected for HPLC analysis. The final concentrations of working solutions were 0.01, 0.05, 0.1, 0.25, 0.5, 1, 2.5, 5.0 and 25.0 and 50.0 μg/mL, respectively. Quality control (QC) samples were also prepared at concentrations of 0.005, 0.5 and 40.0 μg/mL, representing low quality control (LQC), medium quality control (MQC) and high quality control (HQC) samples, respectively. Triplicate calibrants and QC samples were prepared at each concentration.
For test samples, 100 μL of test plasma and 100 μL of ACN were taken in a test tube and vortexed for 30 min. The procedure was similar to the previously described method.
HPLC conditions
For the determination of B and BNP in plasma samples, chromatographic separations were performed in a Waters Shimadzu LC-20AD equipped with a photodiode array detector. The mobile phase consisted of a gradient elution of ACN:H2O of 9:1, and flow rate was 1 mL/min. The separation was performed using RP-C18 column (5.0 μ particle size, 4.6 mm internal diameter and 250 mm length) with max of 230 nm, and the temperature was maintained at 40°C throughout the experiment. The total run time was 15 min. The column was washed after every run in an elution solvent ratio (ACN:H2O =50:50) with the injection of blank ACN. The final data were calculated using WinNonlin version 1.5.3 software.
In vivo pharmacodynamic studies
Experimental animals
Healthy male Wistar albino rats, weighing 80–120 g, were obtained from the central animal house facility of SD College of Pharmacy & Vocational Studies, Muzaffarnagar, India, in association with Babasaheb Bhimrao Ambedkar University, Lucknow, India, and were housed in polypropylene cages with rice husks for bedding. Animal welfare protocol and experiments were performed as per CPCSEA guidelines for laboratory animals and ethics, Department of Animal Welfare, Government of India. The Institutional Animal Ethical Committee (IAEC) of SD College of Pharmacy & Vocational Studies approved this study (Ref No SDCOP&VS/AH/CPCSEA/01/0027). All methods were followed in accordance with the relevant guidelines and regulations of IAEC. Animals were acclimatized under standard laboratory conditions (at 25°C±5°C, relative humidity of 44%–56%, 12 h light:dark cycle) with free access to standard rat chow and water ad libitum. Animals were kept for 1 week before starting the experiment.
Experimental design
Six-week-old experimental Wistar albino rats were randomly allocated to seven groups bearing eight animals in each group (n=8), and the groups were classified as follows: Group 1 (normal control group): 0.25% carboxy methyl cellulose (2 mL/kg), Group 2 (carcinogen control): N-nitrosodiethylamine (NDEA; 100 mg/kg, intraperitoneal (i.p.) once a week for 6 weeks),29–31 Group 3 (positive control group): NDEA + 5-fluorouracil (5-FU; 10 mg/kg, i.p. for 15 days after the induction of HCC), Group 4: NDEA + B (50) (50 mg/kg, orally for 15 days after the induction of HCC), Group 5: NDEA + B (100) (100 mg/kg, orally for 15 days after the induction of HCC), Group 6: NDEA + BNP (50) (50 mg/kg, orally for 15 days after the induction of HCC), Group 7: NDEA + BNP (100) (100 mg/kg, orally for 15 days after the induction of HCC).
Recent reports30,31 have documented that HCC is induced to Wistar albino rats by i.p. injection of NDEA at a dose of 100 mg/kg body weight once a week for 6 weeks. By adopting this protocol, NDEA was administrated to all animals of Groups 2–6 after adaptive inhabitation to experimental conditions for an initial 1 week. After the 6 weeks completion of NDEA administration, 5-FU, B and BNP were given to animals for 15 days as a curative agent against hepatic injury. At the end of treatment, animals were sacrificed by cervical decapitation and livers were excised immediately, rinsed in ice-cold saline and stored at −80°C for further morphological studies, oxidative parameter analysis and exploring gene, as well as protein expression at the molecular level.
Calculation of various physiological parameters
Changes in body weight were measured at the initial and final days of the experiment, and % weight gains were calculated. The number of carcinogenic nodules and their % incidence were also calculated to monitor the cytotoxic effects between treated and untreated groups.
Estimation of various enzymes and antioxidant markers
The levels of enzymes such as aspartate aminotransferase (AST), alanine aminotransferase (ALT), alkaline phosphatase (ALP)32 and lactate dehydrogenase (LDH) were estimated in serum using a commercially available kit. On the other hand, the oxidative stress parameters like catalase (CAT), protein carbonyl (ProC), superoxide dismutase (SOD), glutathione (GSH) and thiobarbituric acid reactive substances (TBARS)32–35 were estimated in liver tissue.
Estimation of catabolic by-products
The procedures adopted for determination of catabolic by-products (bilirubin and biliverdin)36 in liver tissue have been described in a previous paper.
Histopathological and SEM analysis of liver tissue
The procedures adopted for histopathology and SEM36 have been described in a previous paper.
Estimation of cytokines by enzyme-linked immunosorbent assay (ELISA)
Elevated levels of pro-inflammatory cytokines such as IL-2, IL-6, IL-10 and IL-1β in hepatic tissue were examined by ELISA as per manufacturer’s instructions. Furthermore, the levels of caspase-3 and -8 were also examined by ELISA as per manufacturer’s protocol.37
Quantitative reverse transcription polymerase chain reaction (qRT-PCR) analysis
To explore the expression of mRNA for the target gene, 10 mg of tissue samples of each group were taken and total mRNA was isolated using TRIzol reagent and RNeasy Mini Kit was applied to purify the mRNA. cDNA was prepared according to the manufacturer’s protocol for GeneSure First Strand cDNA Synthesis Kit (Genetix Biotech Asia Pvt. Ltd., New Delhi, India). Finally, qRT-PCR was performed in Agilent Stratagene Mx3000P series (Thermo Fisher Scientific) using SYBR@ Green PCR Master Mix. The mRNA was normalized with housekeeping control GAPDH. ΔCt values were normalized with untreated control samples for all compounds (ΔCt = Ct gene of interest − Ct housekeeping gene). Relative changes in the expression level of one specific gene were calculated in terms of 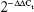 (ΔΔCt = ΔCt test − ΔCt control).35 The primer sequences were as follows: GAPDH, 5-TGATGGGTTTCCCATTGATGA-3 (forward) and 5-TGATTCTACCCACGGCAAGTT-3 (reverse);37 caspase-3, 5-GGTATTGAGACAGACAGTGG-3 (forward) and 5-CATGGGATCTGTTTCTTTGC-3 (reverse)38 and caspase-8, 5′-GATGAGGCAGACTTTCTGCT-3′ (forward) and 5′-CATAGTTCACGCCAGTCAGGAT-3′ (reverse).39
(ΔΔCt = ΔCt test − ΔCt control).35 The primer sequences were as follows: GAPDH, 5-TGATGGGTTTCCCATTGATGA-3 (forward) and 5-TGATTCTACCCACGGCAAGTT-3 (reverse);37 caspase-3, 5-GGTATTGAGACAGACAGTGG-3 (forward) and 5-CATGGGATCTGTTTCTTTGC-3 (reverse)38 and caspase-8, 5′-GATGAGGCAGACTTTCTGCT-3′ (forward) and 5′-CATAGTTCACGCCAGTCAGGAT-3′ (reverse).39
Statistical data analysis
Statistical data analysis was performed using GraphPad Prism 5.0. All results were expressed as mean ± standard deviation (SD). The data were analyzed by one-way analysis of variance (ANOVA) followed by the Bonferroni multiple comparison test. Statistical significant differences were considered with respect to the carcinogen control group (***p<0.001, **p<0.01 and *p<0.05).
Results
Limited solubility and poor oral bioavailability are the major drawbacks for B, and therefore, it has limited therapeutic index during pharmacological responses.8–14 This problem may be overcome through enhancing the solubility in biological matrixes and increasing the oral bioavailability. Here, we tried to increase the solubility of B via formation of nanoparticles (BNP). B has limited solubility in a polar solvent, and it is soluble in a medium polar solvent, ie, chloroform, due to being lipophilic in nature. Six batches of BNP were prepared with various concentrations of PVA and sonication times, with homogenization (Table 1). A clear stable dispersion of BNP in water was obtained for each batch with smaller particle size of batch B.
Nanoparticle size, zeta potential and PDI
All the prepared batches of BNP were further characterized by particle size, zeta potential and PDI (Table 1). A Small concentration of PVA (2%) had a distinctive role in smaller particle size BNP formation (batch B) with perfect Gaussian particle distribution. Batch B contained smaller particle size (257.1 nm) than batch A (353.9 nm) with less zeta potential (−0.170 mV). A higher concentration of PVA (4 and 6%) had an oscillating effect, and therefore, particle size of BNP could not decrease to a lesser extent. PDI is a measure of the distribution of molecular mass/size in a given polymer sample. For all formulations, it was observed that PDI was within the range of 0–1.
Nanoparticle entrapment efficiency and loading capacity
Effectiveness of drug delivery is dependent on entrapment efficiency, also termed as payload efficiency. It is an important parameter for the batch optimization process. As per data obtained from Table 1, we found that batch B has the highest entrapment efficiency (84.56±3.96) and loading capacity (11.13±0.60).
In vitro drug release and reaction kinetics
Figure 2A shows the average percentage cumulative release versus time of all six batches of prepared BNP. The release responses of all formulations were biphasic in nature with faster initial release of 300 min, indicating surface adsorption of BNP. However, all batches released faster up to 1,440 min (24 h). The release rate depended on particle size of all batches, and batches A and B showed a higher release rate than the remaining batches. Batch B had the highest release rate (~80%), and batch A followed a similar trend with a slightly lesser release rate (~70%). This action demonstrated the roles of PVA and sonication during formation of nanoparticles, and 2% PVA helped in the formation of a lower particle size than in other batches. Therefore, batch B had a superior effect on particle size, Gaussian particle distribution and sustained release for a prolonged period. However, all batches followed zero-order kinetics, consistent with a prolonged-release drug delivery system. The Korsmeyer–Peppas model (Mt/Mα = Ktn) was applied to show the release kinetics in this experiment, where Mt is the mass released at time point “t”, Mα the total mass load, n the release exponent and K the constant accommodating the structural and geometric features.
 | Figure 2 Drug release profile, SEM imaging and FTIR spectrum. |
SEM and FTIR studies
SEM analysis of batch B was carried out to find out the final structure of BNP particles. The cylindrical particles of B converted to spherical particles after BNP formation (Figure 2B). FTIR studies revealed that both B and BNP had O−H stretching vibrations at 2,900 cm−1. Both had C−O stretching between 1,625 and 1,650 cm−1, which shows that no interaction was found between B and the used polymers. PLGA and PVA had not shown any characteristic peaks for interaction (Figure 2C).
In vitro cytotoxicity of B and BNP on the HepG2 cell line and confocal microscopy
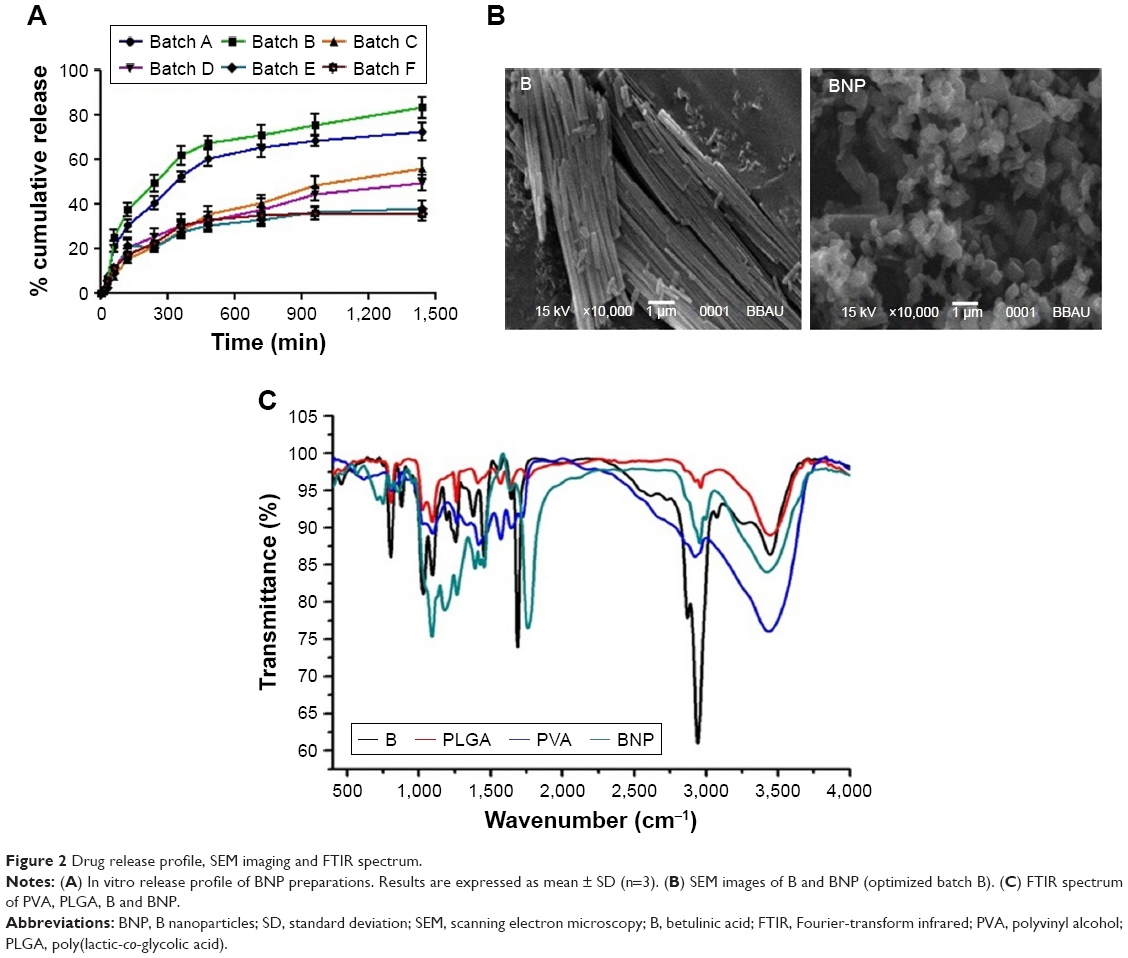
All the compounds, PLGA, PVA, B and BNP, were screened against human hepatoma cancer cell lines (HepG2 cells) using the reference drug Adriamycin (ADR). Effects of these compounds and ADR on HepG2 cell lines are demonstrated in Figure 3A. The results of the study demonstrated that PLGA and PVA did not show any considerable in vitro cytotoxicity against HepG2 cells. However, PLGA–PVA-loaded nanoparticle (BNP) exhibited enhanced cytotoxicity (GI50 <10 μg/mL) to that of the parent compound B (GI50 =10 μg/mL) toward the HepG2 cell line. Activity results proved that BNP had more anticancer properties than parent B.
 | Figure 3 Growth curve of HepG2 cells and Confocal microscopy. |
Figure 3B shows the confocal images of drug particles that penetrate inside the HepG2 cells after incubation for 2 h. It was obvious that BNP penetrated and entered inside the cells more than B due to lower particle size.
Determination of plasma concentration of B and BNP using HPLC
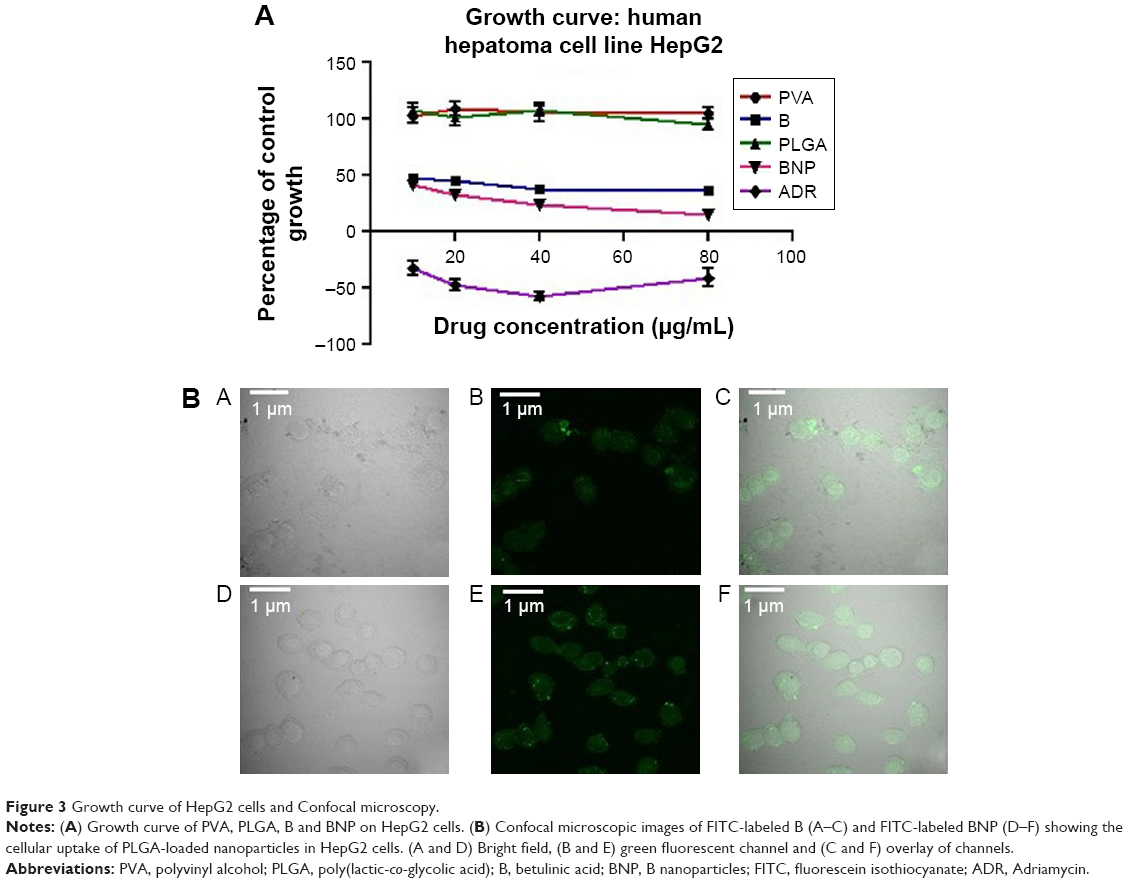
A linear regression performed over a range of 0.01–50 μg/mL yielded a correlation coefficient (r2) of >0.9987 for B. The accuracy of the assay was found to be within 83%–94%, and recovery of the samples was 75–81%. The retention time (RT) for B was 8.87 min (Figure 4A). As depicted in Table 2 and Figure 4B, the maximum plasma concentration (Cmax) and time required to reach the maximum plasma concentration (Tmax) were 10.34 μg/mL and 4 h for B and 47.43 μg/mL and 8 h for BNP, respectively. The plasma concentration reached 50% (t1/2) at 10.73 and 11.11 h for B and BNP, respectively. The total area under the curve (AUC) after 48 h was 133.81 and 838.43 μg·h/mL for B and BNP, respectively.
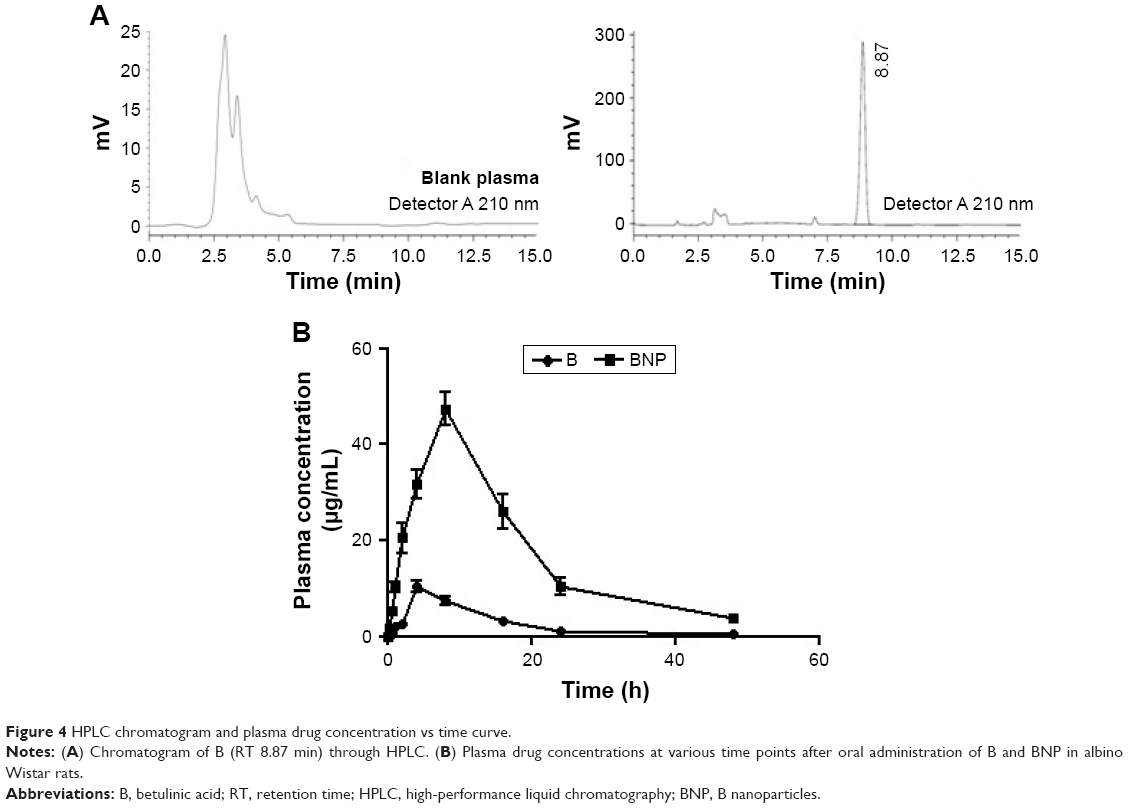 | Figure 4 HPLC chromatogram and plasma drug concentration vs time curve. |
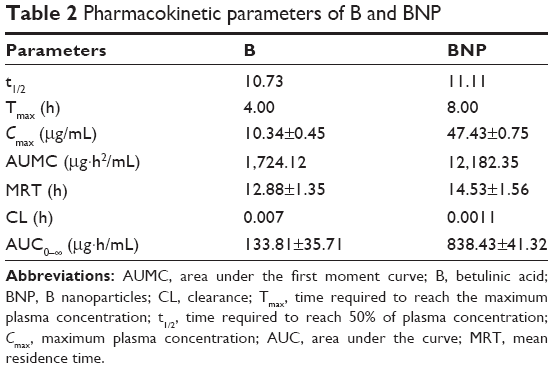 | Table 2 Pharmacokinetic parameters of B and BNP |
Calculation of physiological and biochemical parameters in liver
The protective effects of B and BNP were estimated through various physiological parameters, including body weight, liver weight and tumor incidence number in the rat liver. The body weight variation in the carcinogen control group was more prominent than that in the normal control group. Treatment with B, BNP and 5-FU successfully normalized the body weight variation as seen in the carcinogen control group. An increase in liver weight and tumor incidence number were noticed in the carcinogen control group. Treatment with B, BNP and 5-FU caused reduction in liver weight and tumor incidence number when compared to the carcinogen control group. The overall effect of BNP at 100 mg/kg was higher than the commercially available chemotherapeutics, such as 5-FU (Table 3).
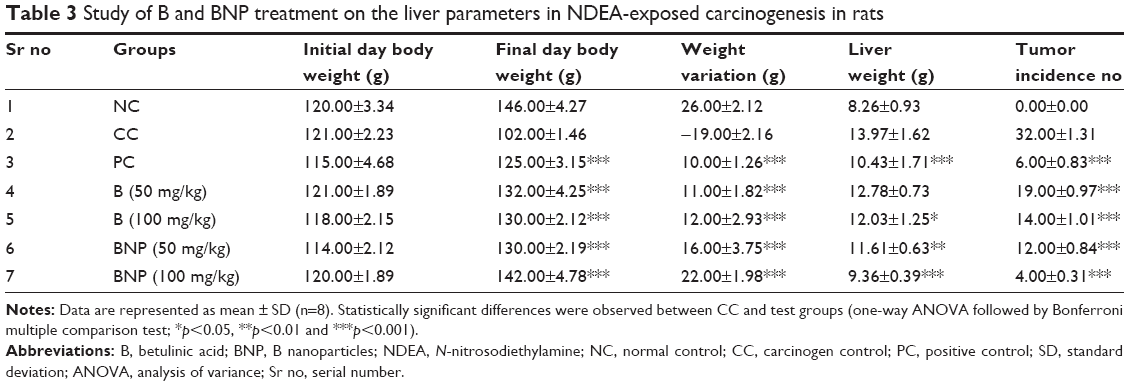 | Table 3 Study of B and BNP treatment on the liver parameters in NDEA-exposed carcinogenesis in rats |
Furthermore, we noted that the carcinogen control group exhibited a marked increase in hepatic damage marker enzyme levels of AST, ALT, ALP and LDH in rat serum when compared to the normal control group (Figure 5A). The treatment with B, BNP and 5-FU showed their ability to significantly restore (p<0.001) these markers to normal. The effect shown by BNP at 100 mg/kg was equivalent to 5-FU and more pronounced than B. Furthermore, the effects of B and BNP on catabolic pigments (bilirubin and biliverdin) in liver tissues were also analyzed (Figure 5B). The treatments with B and BNP reflected a significant (p<0.001) normalization in the levels of these pigments that were elevated more than twofold in the carcinogen control group. The effect of BNP was almost similar to that of the standard chemotherapeutics, such as 5-FU.
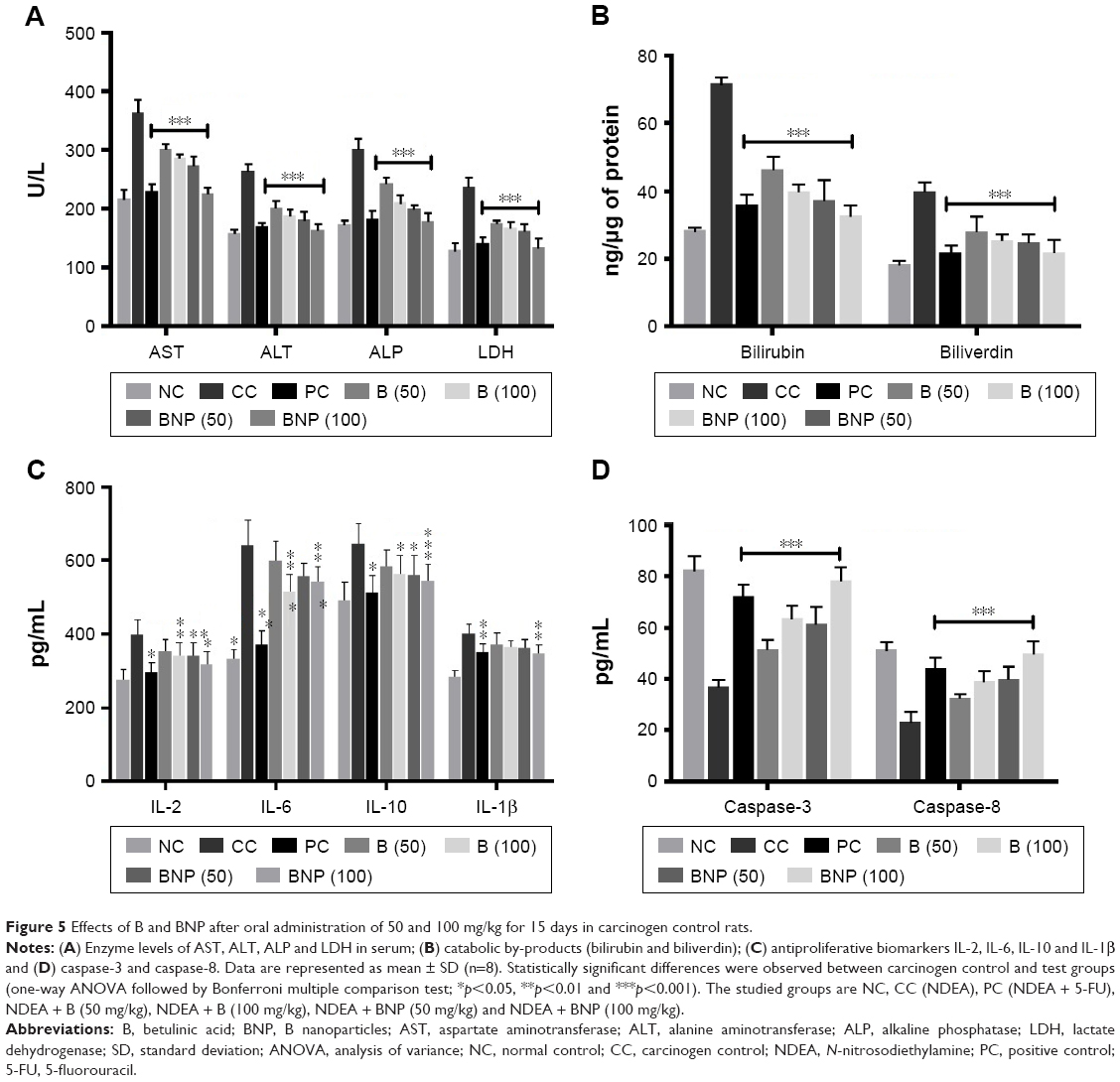 | Figure 5 Effects of B and BNP after oral administration of 50 and 100 mg/kg for 15 days in carcinogen control rats. |
Calculation of oxidative stress parameters in liver tissue
The liver antioxidant potential was assessed due to the functional involvement of oxidative stress in cancer pathogenesis. Table 4 depicts the effects of treatments on the oxidative stress markers (SOD, CAT, malondialdehyde, ProC and GSH). It was noticed that SOD and CAT enzymes were substantially reduced in the carcinogen control group as compared to the normal control group, whereas only a trivial reduction was observed in B-, BNP- and 5-FU-treated groups. The concentration of GSH was reduced up to fivefold to sixfold in carcinogen control rats, and was significantly (p<0.001) restored after treatment with BNP and 5-FU, but not with B. To the contrary, the level of ProC was increased up to twofold in the carcinogen control group, which successfully restored after treatment with B, BNP and 5-FU. Similarly, the level of MDA was increased up to twofold in the carcinogen control group, which significantly (p<0.001) restored after treatment with BNP at 100 mg/kg and 5-FU.
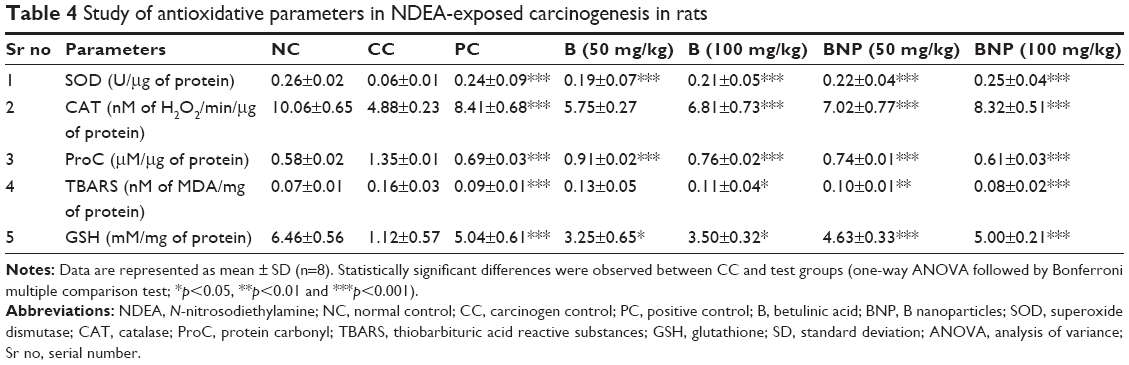 | Table 4 Study of antioxidative parameters in NDEA-exposed carcinogenesis in rats |
Effects of B and BNP on proinflammatory (IL-2, IL-6, IL-10 and IL-1β) and apoptotic mediators (caspase-3 and -8)
In order to explore the effects of B and BNP on cancer-associated inflammatory and apoptotic events, we conducted ELISA and predicted the concentration of proinflammatory cytokines (IL-2, IL-6, IL-10 and IL-1β; Figure 5C) and apoptotic mediators (caspase-3 and -8) in rat liver (Figure 5D). The concentrations of all these inflammatory markers were elevated in the liver of the carcinogen control group. To the contrary, all the apoptotic markers were reduced up to twofold in the carcinogen control group. The reduction in caspase-3 and -8 levels was more pronounced than the elevations in all the cytokines. Both B and BNP restored the levels of caspase-3 and -8 more efficiently than those of the tested cytokines. Although both compounds, B and BNP, showed their ability to elevate the reduced levels of caspase-3 and -8, the restoring capability of BNP was more prominent than that of B and 5-FU.
qRT-PCR analysis
As per evidence through ELISA, B and BNP exerted comparatively more impact over the regulation of caspase-3 and -8 levels than the tested cytokines. Thus, we hypothesized that anti-HCC action of B and BNP may be due to the overexpression of caspase-3 and -8 genes. In order to explore the molecular mechanism underlying the action of B and BNP, qRT-PCR analysis was conducted to scrutinize whether they have ability to upregulate the reduced gene expression of caspase-3 and -8 during carcinogen control group condition. Our findings suggested that the levels of caspase-3 and -8 were substantially downregulated in the carcinogen control group when compared to the normal control group. However, the treatment with B and BNP significantly elevated (p<0.001) the reduced levels of caspase-3 and -8. The ability of BNP at 100 mg/kg was found to be almost fourfold better than commercially available chemotherapeutics, such as 5-FU, at 10 mg/kg (Figure 6A and B).
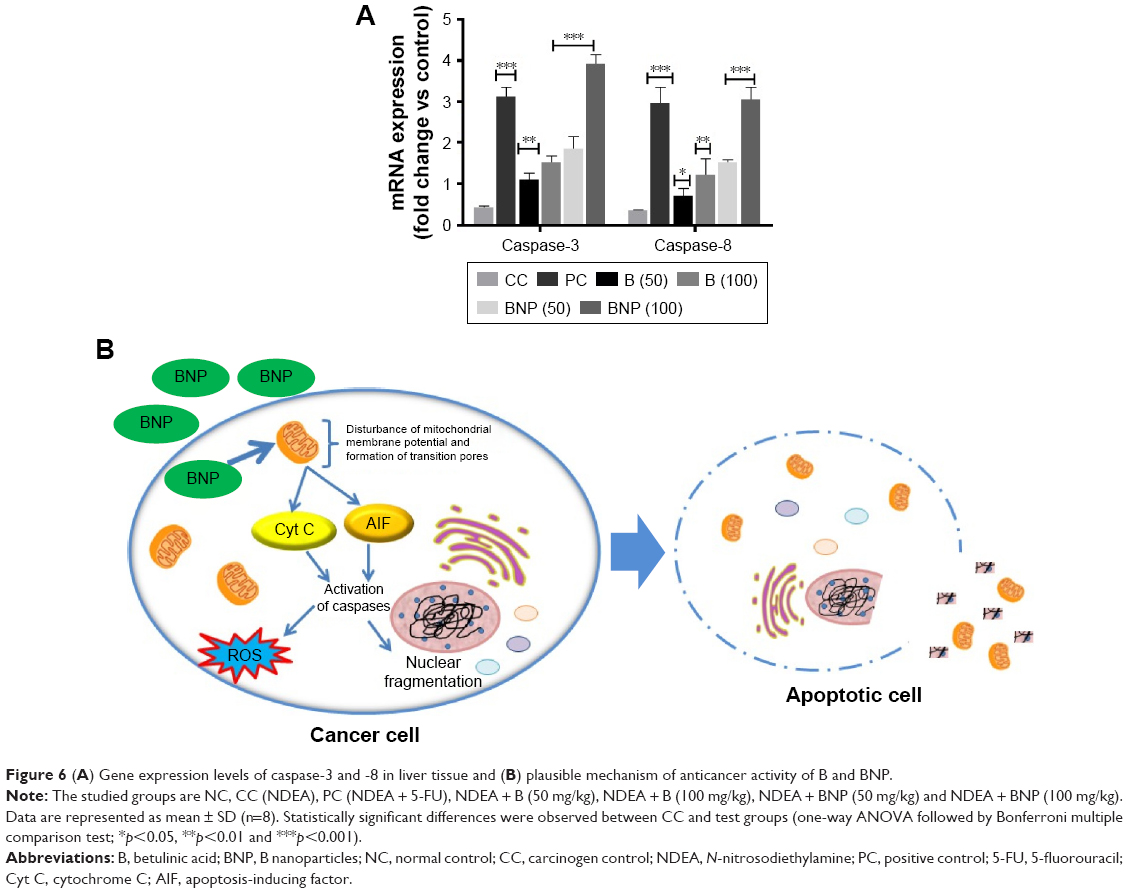 | Figure 6 (A) Gene expression levels of caspase-3 and -8 in liver tissue and (B) plausible mechanism of anticancer activity of B and BNP. |
Morphology, histopathology and SEM analysis
Morphological changes were observed using intact liver of the various groups. There was a clear visual difference in the number of carcinogenic nodules among B, BNP and carcinogen control groups (Figure 7A). Furthermore, histological changes in the liver tissue were observed in normal and various treated groups using hematoxylin and eosin (H&E) staining. In the normal control group, the section showed normal architecture of cells with the presence of nuclei within the cells. However, the liver of the carcinogen control group showed loss of architecture and presence of binucleate, enlarged, polygonal hepatocytes. Particularly, the liver section of carcinogenic animals exhibited irregular sinusoids, degenerated nucleus (dN), tumor anaplastic cells (TA), ruptured hepatic cells (RC) of Kupffer cells (K) and tumoral vacuoles. A marked improvement in gross microscopic appearance of hepatic tissue was observed after the treatment with B and BNP at 50 and 100 mg/kg (Figure 7B). Furthermore, SEM analysis also followed a similar pattern where loss of architecture and degenerated tumor cells were less prominent in B-, BNP- and 5-FU treated rats compared to the carcinogen control group (Figure 7C).
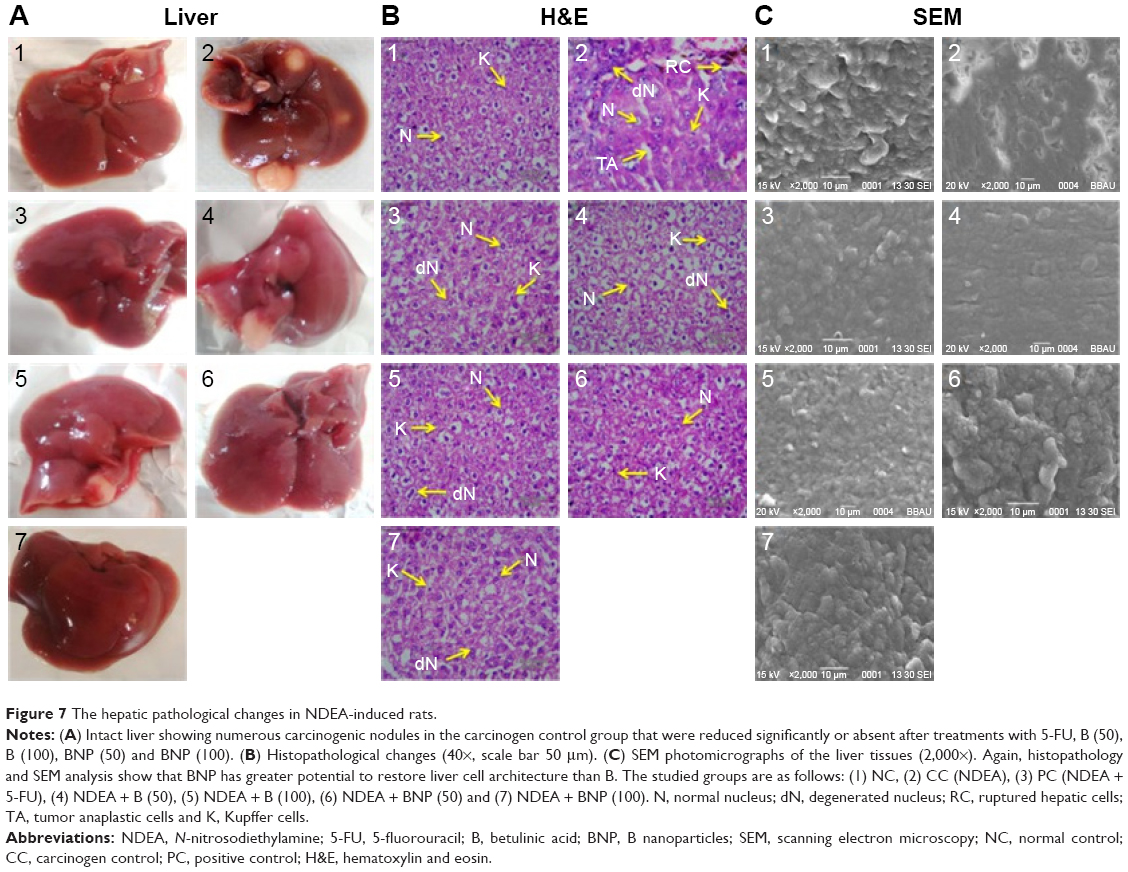 | Figure 7 The hepatic pathological changes in NDEA-induced rats. |
Discussion
B has drawn major research interest in anticancer drug discovery due to its strong cytotoxicity. In spite of having an interesting cytotoxicity on a variety of tumor cells, the normal cells are rarely affected by B, enhancing its therapeutic window.10,11,16 Its tumor selectivity was identified by comparison of different cell line studies where normal cells of different origins have been found to be much more resistant to B than cancer cells, pointing to its tumor selectivity.10,11,16 B is known to provoke an event of direct cell death and bypass the resistance to normal chemotherapeutics. However, the most challenging aspect of its biomedical use is low water solubility, which subsequently causes poor bioavailability;40,41 hence, there is an immense need to develop a delivery system that can enhance the aqueous solubility of B. In view of this, we have developed PLGA-loaded nanoparticles of B, namely BNP, for the purpose of improvement in anticancer efficacy.
Here, we tried to reduce the particle size of B through the nanoparticulate approach, ie, BNP using PLGA and PVA. PLGA and PVA are safe biopolymers that were used previously for andrographolide nanoparticles.23 PVA was used to reduce the interfacial tension and stabilization of nanoparticles.23,24 PVA-induced stabilization effect was reported previously,42,43 and a similar procedure was followed for our experiment. In all, 2% PVA and 30 min sonication time gave a lower particle size of B, ie, BNP (batch B). The entrapment efficiency, loading capacity and release studies demonstrated that BNP had ~84% entrapment efficiency, ~11% loading capacity and ~80% drug release. This finding was because of the increase in viscosity of the dispersed phase by minimizing its size which allows rapid solidification and diminishes the leaching capacity of B to the continuous phase. FTIR data represented no interaction between B and used copolymers. Later, this procedure was followed in large scale, and a larger amount of BNP was prepared for characterization and in vivo studies.
Furthermore, growing evidence suggested that B induces apoptosis in cancer cells through the activation of the mitochondrial (intrinsic) pathway and not the death receptor (extrinsic) pathway.44,45 This evidence has clearly been supported by a study where B was recognized to induce the process of apoptosis by releasing cytochrome c (Cyt C) and/or apoptosis-inducing factor (AIF) through the mitochondrial membrane.46 Some other studies proved that B generally induces apoptosis through Cyt C and AIF release, which causes the activation of caspase-3 and -8 and nuclear fragmentation; however, its action is totally independent of Bcl-2 family proteins, such as Bax/Bak.47,48 It is also evidenced by the fact that the apoptosis is probably due to the activation of caspase-3 and -8 signaling cascade.7,8
To investigate further, we performed in vivo antitumor activity of B and BNP in the NDEA-induced HCC model to investigate the underlying mechanism associated with the anticancer effects of these compounds and accumulated some noteworthy evidence in favor of the enhanced efficacy of BNP compared to that of B, which is discussed further.
Primarily, compounds B and BNP were screened against human hepatoma (HepG2) cancer cell lines using the standard drug ADR. Although the parent compound B showed a good cytotoxic potential (GI50 =10 μg/mL) against the HepG2 cell line, BNP however exhibited excellent selectivity (GI50 <10 μg/mL) toward this cell line. Activity results proved that PLGA–PVA nanoparticles loaded with B play a crucial role in imparting the anticancer activity; however, PLGA and PVA did not show any considerable in vitro antitumor effect. Interestingly, the growth curve of in vitro data suggested that, at 10 μg/mL concentration of BNP, the control growth rates are 50% or <50%, but they do not fall in the negative region of % control growth rate. Thus, it might be expected that BNP has a potent ability to kill the cancerous cells while minimizing the normal cell death. This action was further substantiated through confocal images where we found that BNP had good penetration power and entered inside the HepG2 cell to a greater extent than parent B. Both these experiments suggested that BNP had more antineoplastic potential than parent B.
It is also necessary to investigate in vivo pharmacokinetics of prepared BNP after oral administration. In vivo pharmacokinetic studies demonstrated that BNP had a higher volume of distribution in plasma than parent B. This action may be due to the lower particle size of BNP, which was ultimately absorbed to a greater extent after oral administration. AUC results obtained from pharmacokinetic studies clearly demonstrated that BNP had approximately sevenfold higher plasma distribution than B. This remarkable observation encouraged us to evaluate anti-HCC potential of BNP in the experimental hepatocarcinogenic rat model.
After finding notable in vitro efficacy and acceptable pharmacokinetic properties, these compounds were subjected to in vivo study. The primary investigations of this study suggested that the treatment with BNP at 100 mg/kg significantly restored the body weight, liver weight and tumor incidence number in the rat liver. The functional role of oxidative stress in tumor progression has long been a hotly debated topic. It has previously been reported that there is an increase in oxidative stress through the generation of reactive oxygen species (ROS) and defects in redox defense mechanisms.49 This study revealed the biochemical alterations associated with NDEA-induced oxidative stress that were evidenced through decreased levels of SOD, CAT and GSH and, conversely, increased formation of MDA and ProC. Alterations in these biochemical parameters confirmed the onset of hepatic oxidative stress during HCC conditions. Generation of excess ROS during oxidative stress condition crucially involves HCC pathogenesis through biochemical perturbation, upregulation of intracellular signaling pathways and overexpression of oncogenes.50 Treatment with B and BNP enhanced the antioxidative physiological processes by retrieving the altered levels of GSH, SOD, CAT, MDA and ProC. Furthermore, bilirubin and biliverdin are the catabolic by-products of red blood corpuscles and variation in the levels of these biomarkers can indicate hepatic disease state.51 It was observed that NDEA exposure elevated the levels of bilirubin and biliverdin and they were significantly normalized after B and BNP treatments. In addition, enhanced levels of AST and ALT are a symptom of cirrhosis and hepatic liver damage.52 Among the profile of liver function tests, ALP is an independent factor for HCC patient survival, and its preoperative level can be used to monitor the recurrence in high-risk HCC patients.53 Thus, the raised levels observed in the liver function enzymes (AST, ALT and ALP) reflect the advancement of carcinogenesis during HCC condition. Treatment with BNP significantly lowered these enzymes to normal which indicated the reduction in hepatic injury.54 Similarly, a considerable increase in serum LDH level shows nonspecific alteration in the cell membrane integrity and liver metastases of HCC.55 A high level of serum LDH was observed in the NDEA-exposed group, which could be responsible for liver damage and occurrence of preneoplastic lesions. Treatment with BNP led to the abatement of hepatic injury and alleviation of preneoplastic lesions, probably by its capacity to improve LDH enzyme levels and thereby potentiating its anti-HCC activity.
The serum cytokines are key regulators involved in pathogenesis of inflammation and cancer progression.56 B generally exerts anticancer action either by triggering the mitochondrial pathway of apoptosis or by inducing cellular stress, such as cytokine withdrawal and DNA damage.7–18 Thus, we investigated the altered levels of cytokines and caspases among different groups through ELISA and screened the effects of B and BNP treatments over the carcinogen control group. All these inflammatory cytokines were elevated in NDEA-exposed rats and were decreased to a certain extent after B and BNP treatments. In contrast, caspase-3 and -8 were quickly reduced in the carcinogen control group and were significantly elevated back to normal levels after B and BNP treatments. It was noticed that unlike the tested cytokines, the levels of caspase-3 and -8 in the NDEA-exposed group were quickly restored to normal after administration of B and BNP, and this effect was more pronounced in the BNP-treated group than the B-treated group. This observation again substantiated that caspase expression plays an imperative role in the development of the HCC condition and our newly discovered BNP nanoformulation can act predominantly through the overexpression of caspase-3 and -8 at the liver-specific cancer site. Furthermore, qRT-PCR analysis revealed that mRNA expressions of caspase-3 and -8 were rapidly reduced in the NDEA-exposed group, providing similarity of the trend appearing in ELISA. Interestingly, BNP treatments, predominantly at 100 mg/kg, gave spectacular return of reduced caspase-3 and -8 gene expressions back to normal, which probably gives strong support in favor of BNP action through the overexpression of caspase-3 and -8 genes.
There was a clear visual difference in the number of carcinogenic nodules between BNP and carcinogen control groups. The architecture of liver tissue in NDEA-exposed rats was highly altered as evidenced through the appearance of multiple degenerated tumor cells, RC, TA and loss of architecture and tumoral vacuoles in H&E staining. Nevertheless, treatment with B and BNP reduced the incidence of these alterations through the restoration of regeneration of liver parenchymal cells. Almost normal appearance and unaltered morphology were observed particularly in the BNP-treated group, which further corroborated well with previously discussed biochemical aspects and revealed the improved antitumor efficacy of BNP. Later, SEM analysis also followed a similar pattern where loss of architecture and degenerated tumor cells were least prominent in the BNP-treated group.
Conclusion
A newer formulation of BNP was successfully developed, and oral pharmacokinetics and anti-HCC properties were established. BNP had higher plasma t1/2, Cmax and Tmax than the parent B. BNP restored various physiological, biochemical and oxidative stress parameters to normal during HCC condition. These anti-HCC properties of BNP were due to overexpression of caspases, ie, caspase-3 and -8 at tumorigenic sites. The study established various biochemical, physiological and molecular links associated with enhanced efficacy of BNP as compared to B. Thus, the study can provide evidence of the preventive action of BNP against HCC.
Acknowledgments
Dr Sudipta Saha would like to express his thanks to the University Grants Commission (UGC), New Delhi, India, for UGC-MRP grant, Project No 42-680/2013(SR), and Department of Science and Technology (DST), India, for DST-SERB project, Ref No DST/SB/EMEQ-320/2014. Pranesh Kumar acknowledges the DST and Government of India for the award of DST INSPIRE Fellowship, Ref No DST/INSPIRE Fellowship/2016/IF160364.
Disclosure
The authors report no conflicts of interest in this work.
References
Davaran S, Rashidi MR, Pourabbas B, Dadashzadeh M, Haghsheans NM. Adriamycin release from poly(lactide-co-glycolide)-polyethylene glycol nanoparticles: synthesis and in vitro characterization. Int J Nanomed. 2006;1(4):535–539. | ||
Adibkia K, Javadzadeh Y, Dastnakchi S, Mohammadi G, Niri FK, Beirami MA. Naproxen eudragit RS 100 nanoparticles: preparation and physiochemical characterization. Colloids Surf B. 2011;83:155–159. | ||
Nahar M, Mishra D, Dubey V, Jain NK. Development characterization and toxicity evaluation of amphotericin B loaded gelatine nanoparticles. Nanomedicine. 2008;4:252–261. | ||
Armstrong B, Doll R. Environmental factors and cancer incidence and mortality in different countries, with special reference to dietary practices. Int J Cancer. 1975;15(4):617–631. | ||
Fujioka T, Kashiwada Y, Kilkuskie RE, et al. Anti-AIDS agents, 11. Betulinic acid and platanic acid as anti-HIV principles from Syzigium claviflorum, and the anti-HIV activity of structurally related triterpenoids. J Nat Prod. 1994;57(2):243–247. | ||
Mayaux JF, Bousseau A, Pauwels R, et al. Triterpene derivatives that block entry of human immunodeficiency virus type 1 into cells. Proc Natl Acad Sci U S A. 1994;91(9):3564–3568. | ||
Adamson CS, Waki K, Ablan SD, Salzwedel K, Freed EO. Impact of human immunodeficiency virus type 1 resistance to protease inhibitors on evolution of resistance to the maturation inhibitor bevirimat (PA-457). J Virol. 2009;83:4884–4894. | ||
Fulda S. Betulinic acid for cancer treatment and prevention. Int J Mol Sci. 2008;9:1096–1107. | ||
Wick W, Grimmel C, Wagenknecht B, Dichgans J, Weller M. Betulinic acid-induced apoptosis in glioma cells: a sequential requirement for new protein synthesis, formation of reactive oxygen species, and caspase processing. J Pharmacol Exp Ther. 1999;289(3):1306–1312. | ||
Fulda S, Jeremias I, Steiner HH, Pietsch T, Debatin KM. Betulinic acid: a new cytotoxic agent against malignant brain-tumor cells. Int J Cancer. 1999;82(3):435–441. | ||
Zuco V, Supino R, Righetti SC, et al. Selective cytotoxicity of betulinic acid on tumor cell lines, but not on normal cells. Cancer Lett. 2002;175(1):17–25. | ||
Thurnher D, Turhani D, Pelzmann M, et al. Betulinic acid: a new cytotoxic compound against malignant head and neck cancer cells. Head Neck. 2003;25(9):732–740. | ||
Noda Y, Kaiya T, Kohda K, Kawazoe Y. Enhanced cytotoxicity of some triterpenes toward leukemia L1210 cells cultured in low pH media: possibility of a new mode of cell killing. Chem Pharm Bull (Tokyo). 1997;45(10):1665–1670. | ||
Ehrhardt H, Fulda S, Fuhrer M, Debatin KM, Jeremias I. Betulinic acid-induced apoptosis in leukemia cells. Leukemia. 2004;18(8):1406–1412. | ||
Gopal DR, Narkar AA, Badrinath Y, Mishra KP, Joshi DS. Betulinic acid induces apoptosis in human chronic myelogenous leukemia (CML) cell line K-562 without altering the levels of Bcr-Abl. Toxicol Lett. 2005;155(3):343–351. | ||
Selzer E, Pimentel E, Wacheck V, et al. Effects of betulinic acid alone and in combination with irradiation in human melanoma cells. J Invest Dermatol. 2000;114(5):935–940. | ||
Fulda S. Betulinic acid: a natural product with anticancer activity. Mol Nutr Food Res. 2009;53(1):140–146. | ||
Rieber M, Strasberg RM. Induction of p53 without increase in p21WAF1 in betulinic acid mediated cell death is preferential for human metastatic melanoma. DNA Cell Biol. 1998;17:399–406. | ||
WHO. Burden: Mortality, Morbidity and Risk Factors. 9–31. Available from: http://www.who.int/nmh/publications/ncd_report_chapter1.pdf. Accessed January 8, 2018. | ||
Newman DJ, Cragg GM, Snader KM. Natural products as sources of new drugs over the period 1981–2002. J Nat Prod. 2003;66(7):1022–1037. | ||
Cragg GM, Newman DJ, Snader KM. Natural products in drug discovery and development. J Nat Prod. 1997;60(1):52–60. | ||
Xu H, Ren X, Du Y, et al. Study on absorption kinetics of betulic acid in rat’s intestines. Zhongguo Zhong Yao Za Zhi. 2012;37(3):377–380. | ||
Roy P, Das S, Bera T, Mondol S, Mukherjee A. Andrographolide nanoparticles in leishmaniasis: characterization and in vitro evaluations. Int J Nanomed. 2010;5:1113–1121. | ||
El-Say KM. Maximizing the encapsulation efficiency and the bioavailability of controlled-release cetirizine microspheres using Draper–Lin small composite design. Drug Des Devel Ther. 2016;10:825–839. | ||
Keshari AK, Singh AK, Raj V, et al. p-Tsa-promoted syntheses of 5h-benzo [h] thiazolo [2, 3-b] quinazoline and indeno [1, 2-d] thiazolo [3, 2-a] pyrimidine analogs: molecular modeling and in vitro antitumor activity against hepatocellular carcinoma. Drug Des Devel Ther. 2017;11:1623–1642. | ||
Roy P, Das S, Mondal A, Chatterji U, Mukherjee A. Nanoparticle engineering enhances anticancer efficacy of andrographolide in MCF-7 cells and mice bearing EAC. Curr Pharm Biotechnol. 2012;13(15):2669–2681. | ||
Bhandari R, Kaur IP. A sensitive HPLC method for determination of isoniazid in rat plasma, brain, liver and kidney. J Chromatogr Sep Tech. 2012;3:1–5. | ||
Conte JE, Lin E, Zurlinden E. High-performance liquid chromatographic determination of pyrazinamide in human plasma, bronchoalveolar lavage fluid, and alveolar cells. J Chromatogr Sci. 2000;38(1):33–37. | ||
Matsuzaki T, Murase N, Yagihashi A, et al. Liver transplantation for diethylnitrosamine-induced hepatocellular carcinoma in rats. Transplant Proc. 1992;24(2):748–751. | ||
Shiota G, Harada KI, Ishida M, et al. Inhibition of hepatocellular carcinoma by glycyrrhizin in diethylnitrosamine-treated mice. Carcinogenesis. 1999;20(1):59–63. | ||
Furuta K, Sato S, Miyake T, et al. Anti-tumor effects of cimetidine on hepatocellular carcinomas in diethylnitrosamine-treated rats. Oncol Rep. 2008;19(2):361–368. | ||
Keshari AK, Kumar G, Kushwaha PS, et al. Isolated flavonoids from Ficus racemosa stem bark possess antidiabetic, hypolipidemic and protective effects in albino Wistar rats. J Ethnopharmacol. 2016;181:252–262. | ||
Lodhi RL, Maity S, Kumar P, Saraf SA, Kaithwas G, Saha S. Evaluation of mechanism of hepatotoxicity of leflunomide using albino wistar rats. Afr J Pharm Pharmacol. 2013;7(24):1625–1631. | ||
Kushwaha PS, Raj V, Singh AK, et al. Antidiabetic effects of isolated sterols from Ficus racemosa leaves. RSC Adv. 2015;5(44):35230–35237. | ||
Saha S, Chan DS, Lee CY, et al. Pyrrolidinediones reduce the toxicity of thiazolidinediones and modify their anti-diabetic and anti-cancer properties. Eur J Pharmacol. 2012;697(1–3):13–23. | ||
Kumar P, Singh AK, Raj V, et al. 6, 7-dimethoxy-1, 2, 3, 4-tetrahydro-isoquinoline-3-carboxylic acid attenuates heptatocellular carcinoma in rats with NMR-based metabolic perturbations. Future Sci OA. 2017;3(3):FSO202. | ||
Keshari AK, Singh AK, Kumar U, et al. 5H-benzo [h] thiazolo [2, 3-b] quinazolines ameliorate NDEA-induced hepatocellular carcinogenesis in rats through IL-6 downregulation along with oxidative and metabolic stress reduction. Drug Des Devel Ther. 2017;11:2981–2995. | ||
Liu W, Wang G, Yakovlev AG. Identification and functional analysis of the rat caspase-3 gene promoter. J Biol Chem. 2002;277(10):8273–8278. | ||
Li CP, Li JH, He SY, Li P, Zhong XL. Roles of Fas/Fasl, Bcl-2/Bax, and Caspase-8 in rat nonalcoholic fatty liver disease pathogenesis. Genet Mol Res. 2014;13(2):3991–3999. | ||
Jäger S, Laszczyk MN, Scheffler A. A preliminary pharmacokinetic study of betulin, the main pentacyclic triterpene from extract of outer bark of birch (Betulae alba cortex). Molecules. 2008;13(12):3224–3235. | ||
Drag-Zalesinska M, Kulbacka J, Saczko J, et al. Esters of betulin and betulinic acid with amino acids have improved water solubility and are selectively cytotoxic toward cancer cells. Bioorganic Med Chem Lett. 2009;19(16):4814–4817. | ||
Vandervoort J, Ludwig A. Biocompatible stabilizers in the preparation of PLGA nanoparticles: a factorial design study. Int J Pharm. 2002;238(1–2):77–92. | ||
Murakami H, Kawashima Y, Niwa T, Hino T, Takeuchi H, Kobayashi M. Influence of the degrees of hydrolyzation and polymerization of poly (vinylalcohol) on the preparation and properties of poly (DL-lactide-co-glycolide) nanoparticle. Int J Pharm. 1997;149(1):43–49. | ||
Mullauer FB, Kessler JH, Medema JP. Betulin is a potent anti-tumor agent that is enhanced by cholesterol. PLoS One. 2009;4(4):e1. | ||
Zhang X, Hu J, Chen Y. Betulinic acid and the pharmacological effects of tumor suppression (Review). Mol Med Rep. 2016;14(5):4489–4495. | ||
Drąg-Zalesińska M, Drąg M, Poręba M, Borska S, Kulbacka J, Saczko J. Anticancer properties of ester derivatives of betulin in human metastatic melanoma cells (Me-45). Cancer Cell Int. 2017;17(1):1–7. | ||
Liu Y, Luo W. Betulinic acid induces Bax/Bak-independent cytochrome c release in human nasopharyngeal carcinoma cells. Mol Cells. 2012;33(5):517–524. | ||
Suresh C, Zhao H, Gumbs A, Chetty CS, Bose HS. New ionic derivatives of betulinic acid as highly potent anti-cancer agents. Bioorganic Med Chem Lett. 2012;22(4):1734–1738. | ||
Liu HT, Huang YC, Cheng SB, Huang YT, Lin PT. Effects of coenzyme Q10 supplementation on antioxidant capacity and inflammation in hepatocellular carcinoma patients after surgery: a randomized, placebo-controlled trial. J Nutr. 2016;15(1):1–9. | ||
Newman DJ. Natural products as leads to potential drugs: an old process or the new hope for drug discovery? J Med Chem. 2008;51(9):2589–2599. | ||
Makos BK, Youson JH. Tissue levels of bilirubin and biliverdin in the sea lamprey, Petromyzon marinus L., before and after biliary atresia. Comp Biochem Physiol A Physiol. 1988;91(4):701–710. | ||
Green RM, Flamm S. AGA technical review on the evaluation of liver chemistry tests. Gastroenterology. 2002;123(4):1367–1384. | ||
Yu MC, Chan KM, Lee CF, et al. Alkaline phosphatase: does it have a role in predicting hepatocellular carcinoma recurrence? J Gastrointest Surg. 2011;15(8):1440–1449. | ||
Ilamathi M, Prabu PC, Ayyappa KA, Sivaramakrishnan V. Artesunate obliterates experimental hepatocellular carcinoma in rats through suppression of IL-6-JAK-STAT signalling. Biomed Pharmacother. 2016;82:72–79. | ||
Xu HN, Kadlececk S, Profka H, Glickson JD, Rizi R, Li LZ. Is higher lactate an indicator of tumor metastatic risk? A pilot MRS study using hyperpolarized 13 C-pyruvate. Acad Radiol. 2014;21(2):223–231. | ||
Wu H, Li N, Jin R, et al. Cytokine levels contribute to the pathogenesis of minimal hepatic encephalopathy in patients with hepatocellular carcinoma via STAT3 activation. Sci Rep. 2016;6:1–9. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.