Back to Journals » International Journal of Nanomedicine » Volume 13
Polyethyleneimine modification of aluminum hydroxide nanoparticle enhances antigen transportation and cross-presentation of dendritic cells
Authors Dong H ![]() , Wen ZF, Chen L, Zhou N, Liu H, Dong S, Hu HM, Mou YB
, Wen ZF, Chen L, Zhou N, Liu H, Dong S, Hu HM, Mou YB
Received 30 January 2018
Accepted for publication 9 April 2018
Published 7 June 2018 Volume 2018:13 Pages 3353—3365
DOI https://doi.org/10.2147/IJN.S164097
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Linlin Sun
Heng Dong,1,2,* Zhi-Fa Wen,2,3,* Lin Chen,1 Na Zhou,1 Hui Liu,1 Shiling Dong,1 Hong-ming Hu,2 Yongbin Mou1
1Central Laboratory, Nanjing Stomatological Hospital, Medical School of Nanjing University, Nanjing, China; 2Laboratory of Cancer Immunobiology, Robert W Franz Cancer Research Center, Earle A Chiles Research Institute, Providence Cancer Center, Portland, OR, USA; 3Department of Microbiology and Immunology, Medical School of Southeast University, Nanjing, China
*These authors contributed equally to this work
Background: The aim of this study was to explore the feasibility of delivering tumor antigens and enhancing the antigen cross-presentation of dendritic cells (DCs) by aluminum hydroxide nanoparticle with polyethyleneimine (PEI) modification (LV@HPA/PEI).
Materials and methods: The LV@HPA nanoparticles were modified by PEI first, then the influence of LV@HPA/PEI on DCs was examined. The distinct expression of ovalbumin (OVA) protein transported into DCs by LV@HPA/PEI was observed by flow cytometry and Western blot. The biocompatibility of LV@HPA/PEI, maturity and antigen cross-presentation of DCs was observed in vitro. Tumor derived autophagosomes (DRibbles) combined with LV@HPA/PEI were loaded into DCs, and DC vaccines were used to immunize mice. The percentage of CD3+CD8+IFN-γ+ T cells in immunized mice was determined by flow cytometry. Additionally, the functional properties of the LV@HPA/PEI-DRibble-DCs vaccine were examined in vivo in PancO2 tumor-bearing mice.
Results: In our study, we described how LV@HPA/PEI can be a functionalized antigen delivery system with notable antigen transport effect and negligible cytotoxicity. It was found that LV@HPA/PEI could be easily internalized into DCs to assist antigen release into the cytoplasm. In addition, DCs matured gradually after loading with LV@HPA/PEI-OVA, which increased significantly the cytokine IL-12 secretion and expression of surface molecules CD80 and CD86. Interestingly, DCs loaded with LV@HPA/PEI-DRibbles could promote the activation of tumor-specific T cells both in murine and in human T cells. In the following in vivo experiments, the vaccine of LV@HPA/PEI-DRibble-DCs significantly inhibited tumor growth and improved the survival rate of the PancO2 tumor-bearing mice.
Conclusion: We established a high-performance anti-tumor vaccine of DCs loaded with LV@HPA/PEI nanoparticles and tumor-associated antigens in autophagosomes (DRibbles), which could serve as a therapeutic strategy in cancer immunotherapy.
Keywords: aluminum hydroxide, antigen delivery, DRibbles, nano-adjuvant, cancer immunotherapy, autophagosome
Introduction
Cancer immunotherapy has been given more attention recently due to the approval of dendritic cell (DC)-based anti-tumor immunotherapy, such as Provenge® (sipuleucel-T) for metastatic prostate.1 DCs are the decisive professional antigen presenting cells (APCs) that can phagocytize antigens and activate a specific antigen immune response.2 DCs loaded with tumor antigens are a high-quality immunotherapeutic strategy to prevent or treat cancer. Effective anti-tumor DC vaccines should have the ability to induce antigen-specific T cell immune responses. After tumor antigens have been efficiently internalized, processed, and presented by DCs to naive T cells, it induces their proliferation and differentiation into effector cells such as CD8+ cytotoxic T lymphocyte cells (CTLs), to kill tumor cells.3
Many clinical trials indicated that DC-based vaccines could improve innate or nonspecific immune responses in cancer patients, but the dysfunction of antigen phagocytosis and lack of effective tumor antigens are usually the main reasons for the limited efficacy of anti-tumor DC vaccines.4–6 To overcome these barriers, tumor-associated antigens from tumor lysates as specific antigens have been recognized as a promising strategy in previous studies.7 Hence, many studies have searched for suitable carriers for transporting the tumor specific antigens.
Nano-adjuvants may have the advantage of adsorbing soluble antigens by serving as antigen carriers and protecting the antigens from enzyme degradation, enabling delivery to DCs, and stimulating antigen presentation.8 Inorganic nanoparticles with antigens are controllable and simple antigen–adjuvant complexes, which can carry varieties of protein antigens into APCs, have been extensively used as antigen delivery systems for DC vaccines to induce antigen-specific CTL immune responses.9 Aluminum adjuvants were approved as inorganic nanoparticles by the US Food and Drug Administration for human use because of their favorable safety biocompatibility.10 Aluminum hydroxide nanoparticles have the advantage of high efficiency for adsorbing antigens and can be used as antigen depots and for slow release of antigens.11–13 Thus, we investigated whether aluminum hydroxide combined with tumor-associated antigens can facilitate phagocytosis by DCs in this study.
Rehydragel® (aluminium hydroxide@heparanase, LV@HPA),14,15 a specific aluminum hydroxide gel containing high purity aluminum hydroxide nanoparticles, has been used as an adjuvant for veterinary vaccines and human biological materials. LV@HPA nanoparticles possess a needle shape compare with other aluminum hydroxide adjuvants,16,17 but whether it can be used as immunologic adjuvant for DCs vaccine is still unknown. Studies have demonstrated that aluminum hydroxide nanoparticles are based on mineralized hydroxyapatite and can produce a high degree of protein adsorption performance.18,19 However, a previous study showed that it can only cause moderate intensity antigen-specific immune responses.16 Recently, extensive study of polyethyleneimine (PEI) found it to have a “proton sponge” effect when it was used as delivery carrier.20 LV@HPA nanoparticle possesses many advantages such as negligible cytotoxicity, large surface area, and highly effective drug loading.21 Owing to their excellent properties, LV@HPA nanoparticles modified with PEI (LV@HPA/PEI) are studied here. In particular, this study examines whether the LV@HPA/PEI-antigen can be used as a novel adjuvant for DCs in anti-tumor immunotherapy.
The efficient cross-presentation of tumor-associated antigens (TAAs) is pivotal for the success of DC vaccines. TAAs, including short-lived proteins (SLiPs) and defective ribosomal products (DRiPs), are ubiquitinated and degraded by proteasomes in tumor cells.22 A broad spectrum of SLiPs and DRiPs are shunted into autophagosomes named DRibbles (tumor derived autophagosomes).23 DRibbles, induced by accelerating autophagy, inhibiting the proteasome, and restraining the lysosome, can accumulate a large number of TAAs, which were used for enhancing the immune responses in a previous study.24 We demonstrated that DCs loaded with α-Al2O3-DRibbles could promote antitumor-specific immune responses and had great anti-tumor efficacy in a murine lung tumor model.25 Thus, we also examined how DRibbles combined with LV@HPA nanoparticles can promote the function of DC vaccines to obtain higher anti-tumor potency.
In this study, DCs with antigen cross-presenting functions were examined, including murine mutu DCs,26 murine bone marrow-derived DCs (BMDCs), and human DCs (Mutz-3).27 The abilities of LV@HPA/PEI nanoparticles to absorb antigen protein and enhance DC antigen cross-presentation were studied. Moreover, tumor-specific T cell responses were observed by LV@HPA/PEI-DRibbles activation and compared with LV@HPA-DRibbles and α-Al2O3-DRibbles after loading DCs. Furthermore, the effect of human tumor-specific T cells activation and anti-tumor abilities by LV@HPA/PEI-DRibble-DCs was detected.
Materials and methods
Preparation of LV@HPA/PEI
The LV@HPA/PEI nanoparticles were prepared by the co-incubation method.28 Briefly, 0.5 mL LV@HPA (5 mg/mL) was dripped dropwise into a 1.5 mL PEI (25 mg/mL) solution during stirring. After 3 hours of stirring, purification of the crude PEI-coated LV@HPA (LV@HPA/PEI) was performed twice at 750× g for 30 minutes, and the product was re-dispersed in PBS. LV@HPA was brought from Chemtrade Chemicals, Syracuse, NY, USA. HPA is a ubiquitous anionic linear polysaccharide. PEI MAX, Linear, MW 25,000 (PEI) was obtained from PolyScience (Catalog number 24765, Niles, IL, USA).
Mice
Specific pathogen-free, 8-week-old C57BL/6 and OT-1 mice were purchased from the Model Animal Research Center of Nanjing University. All research procedures were approved by the Animal Care and Use Committee of the Medical School of Nanjing University and conformed to the National Institutes of Health Guide for Care and Use of Laboratory Animals (Publication No 85-23, revised 1996).
Cells culture
BMDCs were generated from bone marrow precursors of C57BL/6 mice. Briefly, femur bones were removed from C57BL/6 mice, and bone marrow was flushed out with RPMI 1640 medium (Thermo Fisher Scientific, Waltham, MA, USA). The cells (1×106 cell/well) were washed twice with PBS and then cultured in complete RPMI 1640 medium supplemented with murine granulocyte-macrophage colony-stimulating factor (10 ng/mL, GM-CSF; Gibco), and murine IL-4 (1 ng/mL, PeproTech, Rocky Hill, NJ, USA) for 5 days. Half medium was gently replaced on day 2 and day 4. Murine mutu DCs were cultured in DMEM supplemented with 10% fetal bovine serum (FBS), 2 mM L-glutamine, 100 units/mL penicillin, and 100 mg/mL streptomycin (Invitrogen, Thermo Fisher Scientific). Human acute myeloid leukemia cell line Mutz-3 was maintained in α-minimum essential medium (α-MEM) supplemented with 10% FBS and 10 ng/mL GM-CSF. Murine pancreas cancer cell line, including PancO2 and PancO2-ovalbumin (OVA), were cultured in complete RPMI 1640 medium supplemented with 10% FBS, 2 mM L-glutamine, and 100 units/mL penicillin.29 Melanoma1383, a human tumor cell line, was cultured in RPMI 1640 medium supplemented with 10% FBS, 2 mM L-glutamine, and 100 units/mL penicillin. TIL 1383I cells (HLA-A2 restricted, tyrosinase: 368–376 reactive) were cultured with RPMI 1640 medium supplemented with 10% heat-inactivated pooled human AB serum (Valley Biomedical, Winchester VA, USA), 100 units/mL penicillin, 2 mM L-glutamine, and 6,000 IU/mL recombinant human IL-2 (Cetus, Berkley, CA, USA) as previously described.30
The cell lines, PancO2, PancO2-OVA, murine mutu DCs, and B3Z, were provided by Prof Hong-ming Hu, Providence Medical Center. Mutz-3 cells were provided by Dr Reeneke von der Ven, Department of Pathology, VU University Medical Center. Melanoma1383 were provided by Dr Rosenberg, NCI, and TIL 1383I were provided by Micheal Nishimura, Loyola University Medical Center. All cell lines were approved by Ethics committee of the Medical School of Nanjing University.
Properties of adsorbing protein
The adsorption of OVA protein by nanoparticles was carried out by mixing them in solution. Briefly, 250 μg/mL OVA protein solution was added into tubes followed by the addition of LV@HPA and LV@HPA/PEI (final concentrations were 1, 2.5, 5, 10, 20, and 30 μg/mL). The tubes were shaken at 4°C for 2 hours and then centrifuged at 750× g for 30 minutes. Thereafter, the supernatant liquid was collected and the OVA protein content was detected using bicinchoninic acid (BCA) Protein Assay kit (Thermo Fisher Scientific). The supernatant protein was subtracted from the total amount of protein to determine the adsorbed OVA protein.
Cell counting kit-8 (CCK-8) assay
According to precious protocol, nano-adjuvants were co-cultured with mutu DCs for 6 hours and then co-cultured with B3Z T cells for another 24 hours.31 Hence, mutu DCs (50 μL, 2×104 cell/well) were loaded with either 50 μL LV@HPA/PEI-OVA or LV@HPA-OVA (final concentrations were 1, 2.5, 5, 10, 20 and 30 μg/mL) for 24 hours. Cell viability was detected by CCK-8 kit (Sigma-Aldrich, St Louis, MO, USA) assay, and the absorbance was read at 450 nm using a microplate reader (Synergy HT, BioTek, Winooski, VT, USA) according to the manufacturer’s instructions.
Transmission electron microscopy (TEM) and surface charges of nano-adjuvant
The mutu DCs (50 μL, 2×106 cell/well) were incubated with the 50 μL nano-adjuvants (10 μg/mL, either LV@HPA-OVA or LV@HPA/PEI-OVA) for 2 hours.31 After fixing, they were cut into ultra-thin sections (90 nm) and stained with osmium tetroxide (1%) and potassium ferrocyanide (0.8%) in 100 mM sodium cacodylate buffer for 2 hours. Then, samples were rinsed, stained, dehydrated, and infiltrated with a 1:1 mix of acetone and Epon 812 (EMS cat#14120) overnight with rotation. After that, the 1:1 mixture was replaced with Epon 812 and allotted to polymerize overnight at 60°C. Thin sections obtained from the block face were imaged at 80 kV on an FEI-Tecnai 12 system interfaced to a digital camera and analyzed with the associated software (Advanced Microscopy Techniques, Danvers, MA).
The zeta potentials of the LV@HPA, LV@HPA/PEI, and LV@HPA/PEI-OVA were measured using a Zetasizer Nano ZS90 potential analyzer (Malvern, UK).
Uptake of LV@HPA/PEI-OVA into DCs
LV@HPA/PEI were incubated with OVA-fluorescein isothiocyanate (FITC) (Sigma-Aldrich) for 2 hours and then co-cultured with mutu DCs (5×104 cell/well) at 37°C under 5% CO2 for 6 hours. Then, the fluorescence intensity of mutu DCs was detected by flow cytometry.
OVA protein contained in mutu DCs cell lysates was detected by Western blot. In brief, samples of cell total lysates were incubated with the antibody of anti-OVA (1:1,000) (Cell Signaling Technology, Danvers, MA, USA) at 4°C overnight and secondary antibody labeled with horseradish peroxidase (1:1,000) was incubated for another 2 hours. Thereafter, the Western blots were visualized using an enhanced chemiluminescence system (EMD Millipore, Billerica, MA, USA), and the density of glyceraldehyde-3-phosphate dehydrogenase (GAPDH) served as an internal loading control.
Analysis of surface markers by flow cytometry
For analyzing the changes of surface markers, mutu DCs were incubated with either 10 μg/mL LV@HPA/PEI-OVA, and LV@HPA-OVA for 6 hours. Then, cells were harvested, thoroughly washed twice and incubated with fluorochrome-conjugated specific antibodies including CD80 and CD86 (eBioscience, Thermo Fisher Scientific) at recommended dilutions for 30 minutes at 4°C. After washing, the surface-marker analyses were performed by FACSCalibur (BD, San Jose, CA, USA), and the results were analyzed using FlowJo software (vX.0.7, Tree Star, Ashland, OR, USA).
Chlorophenol red-β-D-galactopyranoside (CPRG) assay detected antigen cross-presentation in vitro
As B3Z T cells can specifically recognize OVA-major histocompatibility complex I (MHC-I) molecular compounds on the DC surface in association with H-2Kb, the activation of B3Z T cells could be quantified by CPRG assay. First, mutu DCs (2×104 cell/mL) were loaded with LV@HPA/PEI-OVA for 5 hours. Second, the cultures were washed to remove the suspended cells and then added to B3Z T cells (2×105 cell/mL) overnight. OVA alone (100 μg/mL), PBS, and SIINFEKL (1 μg/mL) were used as control. The activated response of B3Z T cells was measured by CPRG (Sigma-Aldrich) and the absorbance of 595 nm in this assay indicates the antigen cross-presentation ability of DCs.
Carboxy-fluoresceindiacetate succinimidylester (CFSE) dilution assay in vitro
For a DC cross-presentation assay, the splenocytes were obtained from OT-1 mice. BMDCs (2×105 cell/mL) were incubated with either LV@HPA/PEI-OVA or LV@HPA-OVA (10 μg/mL) for 6 hours and then co-cultured with CFSE-labeled primed OT-1 splenocytes (2×106 cell/mL) for another 72 hours. Proliferation of CFSE-labeled T cells was determined by flow cytometry.
Preparation of DRibbles and nanoparticle-DRibbles
DRibbles were prepared as described previously.32 Briefly, murine PancO2, PancO2-OVA, or human 1383 melanoma cells were treated with 100 nM Rapamycin (Enzo Life Sciences, Farmingdale, NY, USA), 100 nM Bortezomib (Millennium Pharmaceuticals, Cambridge, MA, USA), and 10 mM ammonium chloride (Sigma-Aldrich) in complete RPMI 1640 medium for 24 hours. Cells were harvested and centrifuged at 300× g for 5 minutes and the supernatant was then centrifuged at 12,000× g for 15 minutes to collect the DRibbles of the tumor cells. Tumor cell lysates (Lysates) were prepared through three freeze-thaw cycles, and the supernatants were collected by centrifugation at 12,000× g for 10 minutes. The LV@HPA/PEI-DRibbles were prepared by the co-incubation method. Briefly, DRibbles were prepared by undertaking three freeze-thaw cycles and the supernatants were collected by centrifugation at 12,000× g for 10 minutes. The obtained supernatants of DRibbles were dripped into the nanoparticle solution during stirring at 4°C for 2 hours.
Scanning electron microscopy (SEM)
The DRibbles were collected, and the sample for SEM was added to 4% paraformaldehyde to fix at 4°C overnight. Thereafter, the sample was dripped on a small piece of aluminum foil, which was used as the counter electrode, and dried in air. The surface morphology of the sample was observed with an Ultra Plus Field Emission scanning electron microscope (Zeiss, Oberkochen, Germany) operating at 1.00 kV. The image was acquired with a CCD camera (Evolution MP 5.0; Media Cybernetics, Rockville, MD, USA).
T cell activation
C57BL/6 mice (n=6) were injected PancO2 tumor cells (2×105 cell/mL) for 2 weeks, then the splenocytes were isolated and expanded with α. CD3 antibody (10 ng/mL, eBioscience) and IL-2 (1,000 U/mL, Endogen, Thermo Fisher Scientific) for 3 days. DRibbles, obtained from PancO2 cells, were incubated with 10 μg/mL LV@HPA/PEI for 1 hour and then co-cultured with the mutu DCs for 6 hours. After the splenocytes were incubated with LV@HPA/PEI-DRibble-DCs vaccine for another 6 hours, Brefeldin A (5 μg/mL, Sigma-Aldrich) was added into the cell medium to stop the Golgi transportation of intracellular cytokines and then the activated T cells were stained with the antibodies of CD3, CD8, and interferon-gamma (IFN-γ) (eBioscience). Next, the percentages of CD3+, CD8+, and IFN-γ+ T cells were determined by flow cytometry and analyzed by FlowJo software (Tree Star, Ashland, OR, USA).
Mice were subcutaneously vaccinated with LV@HPA/PEI-DRibbles, LV@HPA-DRibbles, LV@HPA/PEI-Lysates, LV@HPA-Lysates and PBS (containing 5 μg/mL PancO2 DRibbles and 10 μg/mL nanoparticles). The same vaccination was performed on the 7th day from the first injection. On the 14th day, the mice were sacrificed and their splenocytes were taken out. Then the splenocytes were cultured with PancO2 tumor cells for 24 hours. The immune response was evaluated by IFN-γ releasing using mouse IFN-γ enzyme-linked immunosorbent assay (ELISA) Ready-SET-Go! kit (eBioscience).
For the measurement of cytokine production by TIL 1383I, Mutz-3 DCs were treated with LV@HPA/PEI-1383 DRibbles for 6 hours and then incubated with TIL 1383I for 24 hours. The cytokine production of IFN-γ and GM-CSF was detected by ELISA detection after 24 hours. ELISAs used Human IFN-γ ELISA Ready-SET-Go! kit and Human GM-CSF ELISA Ready-SET-Go! kit (eBioscience).
Animal assay
To investigate the anti-tumor efficacy of LV@HPA/PEI-DRibble-DCs in vivo, PancO2 cells (2×105 cell/mL) were subcutaneously injected (s.c.) into C57BL/6 mice (n=24 mice) in the right flank on day 0. The mice were randomly distributed into four groups (n=6) 7 days later. Vaccines of either DRibble-DCs or LV@HPA/PEI-DRibble-DCs were administered into the mice by s.c. on day 7 and into the inguinal lymph nodes on day 14. Thereafter, the tumor volumes were measured every 3 days with Vernier caliper and calculated using the formula V = 1/2 × a × b2, where a is the largest diameter (length) and b is the smallest diameter (width) of the tumor. The mice were sacrificed on day 54.
Statistical analysis
All statistical analyses were performed using SPSS (version 11.0, SPSS Inc., Chicago, IL, USA). Data were expressed as mean ± SD. Differences between control and test groups were assessed by one-way analysis of variance or two-tailed Student’s t-tests. Survival rates were assessed by the Kaplan–Meier method. The level of statistical significance was set at P<0.05.
Results
Uptake of nanoparticle-OVA by DCs
The surface charge of LV@HPA/PEI nanoparticles was assessed by measuring zeta potential. It was found that LV@HPA nanoparticles exhibited negative zeta potential (pH 7.2–7.4) and positive charge after modification by PEI (Figure 1A). To successfully construct LV@HPA/PEI-OVA with stable positive charge, the optimized concentration of LV@HPA/PEI:OVA mass ratio was 1:10, which was used for further experiments. The result of the BCA assay confirmed that the protein adsorption abilities of LV@HPA/PEI were greater than those of LV@HPA in different protein concentrations (Figure 1B). The cytotoxicity assay of nanoparticles demonstrated that LV@HPA/PEI-OVA and LV@HPA-OVA did not show obvious cytotoxicity against DCs compared with the DCs alone up to 10 μg/mL, indicating an outstanding in vitro cellular biocompatibility of both LV@HPA-OVA and LV@HPA/PEI-OVA (Figure 1C). The ultrastructure of DCs labeled with either LV@HPA-OVA or LV@HPA/PEI-OVA was observed under TEM. It can be seen that the nanoparticle–OVA complexes were either internalized through cell membrane retraction or stayed as vesicles in the cytoplasm. DCs treated with LV@HPA/PEI-OVA displayed a higher number of vesicles as compared to LV@HPA-OVA, in which the needle-like nanoparticles, as shown by the red arrows, were internalized by cell membrane retraction and trapped in the cytoplasm (Figure 1D). Furthermore, OVA-FITC was devoured by phagocytosis, and the fluorescence intensity of FITC was only about 7.0%. With the combination of OVA-FITC and LV@HPA in the DCs, the fluorescence intensity reached about 17.7%. Interestingly, the fluorescence intensity of FITC reached about 25.5% in the intracellular parts of DCs after treatment with LV@HPA/PEI-OVA-FITC, suggesting that LV@HPA/PEI could make the transition of OVA-FITC into the DCs more effectively (Figure 1E and F). The results of Western blot also showed that the content of OVA in both LV@HPA/PEI and LV@HPA groups was higher than that in OVA alone, and a notable higher content of OVA was detected in the LV@HPA/PEI group than in the LV@HPA group, indicating that LV@HPA/PEI could be designed to more effectively facilitate the assimilation of OVA protein in DCs (Figure 1G).
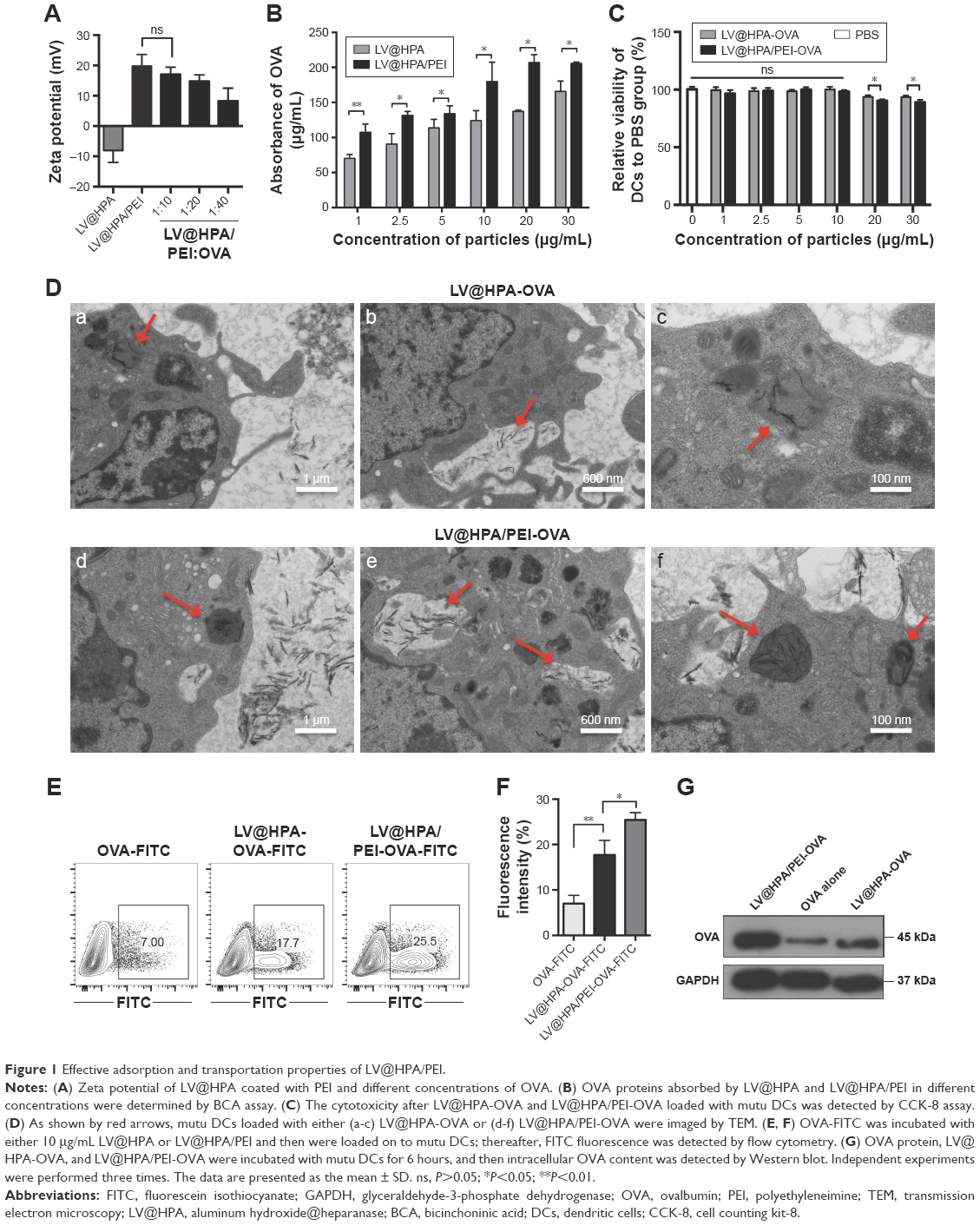 | Figure 1 Effective adsorption and transportation properties of LV@HPA/PEI. |
LV@HPA/PEI-OVA activates DCs and promotes cross-presentation
Both LV@HPA/PEI-OVA and LV@HPA-OVA adjuvants could induce the release of IL-12 by DCs. With the concentration of adjuvants increased, the secretion of IL-12 was increased in both LV@HPA/PEI-OVA and LV@HPA-OVA groups, but that in the former group was higher than in the latter one. Secretion of IL-12 in LV@HPA/PEI-OVA reached maximum values and remained stable beyond 10 μg/mL (Figure 2A). In addition, there were changes in the expression of surface co-stimulatory molecules (CD80 and CD86) after DCs were loaded with 10 μg/mL nanoparticle–OVA complexes (Figure 2B and C). The results indicated that aluminate adjuvants could promote the maturation and activation of DCs, especially with PEI modification.
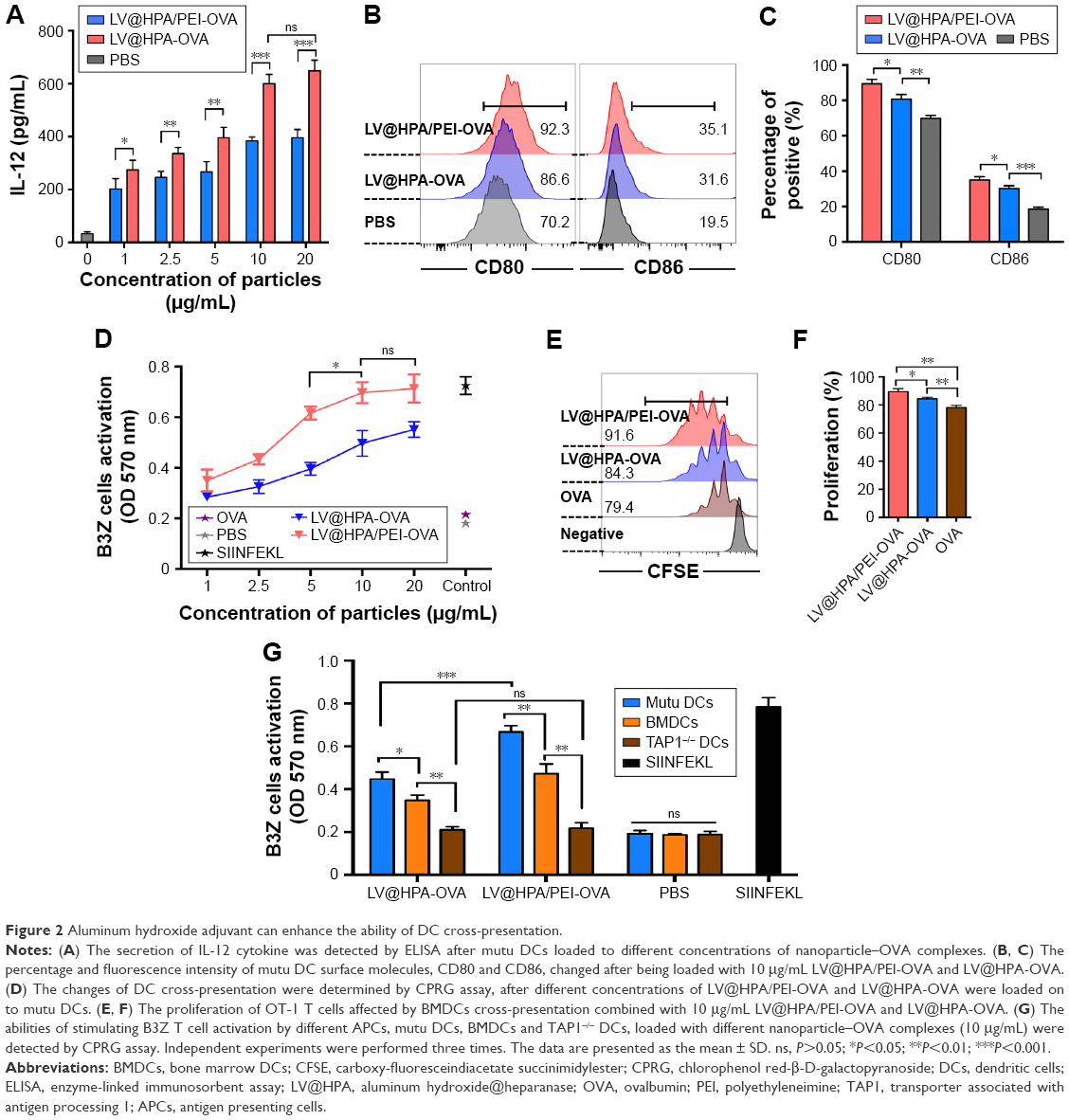 | Figure 2 Aluminum hydroxide adjuvant can enhance the ability of DC cross-presentation. |
To further investigate the effect of T-cell activation in a murine system, B3Z T cells were co-cultured with DCs and the activation of T cells was determined by measuring the production of β-galactosidase through a CPRG assay. Both LV@HPA/PEI-OVA and LV@HPA-OVA yielded a sufficient response to antigen cross-presentation. At each concentration, the effect of cross-presentation of LV@HPA/PEI-OVA was much better than LV@HPA-OVA and remained stable at 10 μg/mL. Meanwhile, OVA protein alone as negative control group showed no effect on cross-presentation to T cells by DCs (Figure 2D). Furthermore, the flow cytometry was adopted to determine the CD8+ T cell proliferation from OT-1 mice and it was found that both LV@HPA/PEI-OVA and LV@HPA-OVA can significantly promote CD8+ OT-1 T cell proliferation compared to OVA alone. The proliferation peak of LV@HPA/PEI-OVA was shifted to the left side, indicating that its proliferation rate was faster and stronger than that of LV@HPA-OVA (Figure 2E and F). Moreover, in order to investigate the mechanism of DCs cross-presentation, DCs were generated from TAP1 knockout mice and co-cultured with either LV@HPA/PEI-OVA or LV@HPA-OVA. TAP1−/− DCs loaded with complexes could not effectively stimulate the B3Z T cell activation, which means that TAP1 molecule in DCs was absolutely necessary for cross-presentation of nanoparticle-OVA complexes (Figure 2G).
The efficiency of cross-presentation increased by the combination of DRibbles with nanoparticles
SEM revealed the surface morphology and diameters of the DRibbles, which exhibited relatively spherical structures and smooth surfaces (Figure 3A). DRibbles, autophagosome-containing vesicles isolated from tumor cells, which contain TAAs including SLiPs and DRiPs, were combined with LV@HPA/PEI to load DCs (Figure 3B). DCs, loaded with different concentrations of DRibbles, and combined with nano-adjuvant, could present antigens to effectively active B3Z T cells (Figure 3C). Furthermore, we found that LV@HPA/PEI-DRibbles could significantly stimulate IFN-γ secretion by tumor-specific CD3+CD8+ T cells, which were separated from PancO2 tumor-bearing mice and incubated with different DC vaccines (Figure 3D and E). After re-stimulation with PancO2 tumor cells, splenocytes from mice vaccinated with LV@HPA/PEI-DRibbles produced a higher level of IFN-γ than splenocytes from mice vaccinated with LV@HPA-DRibbles, LV@HPA/PEI-Lysates, and LV@HPA-Lysates (Figure 3F). In order to validate whether the adjuvant could be applied to human DC vaccines in the future, 1383 DRibbles were extracted from Melanoma1383, combined with aluminum adjuvants, and then loaded to the DCs. T cells transferred 1383i antigen-specific T cell receptor (TIL 1383I) were incubated with different DCs vaccines. We found that LV@HPA/PEI-1383 DRibbles and LV@HPA-1383 DRibbles could significantly increase the secretion of IFN-γ and GM-CSF in the supernatant compared with the 1383 DRibbles alone, and the effect of LV@HPA/PEI-1383 DRibbles was superior to that of LV@HPA-1383 DRibbles. It was further demonstrated that PEI-modified LV@HPA combined with DRibbles could be regarded as a high-quality DC adjuvant (Figure 3G and H).
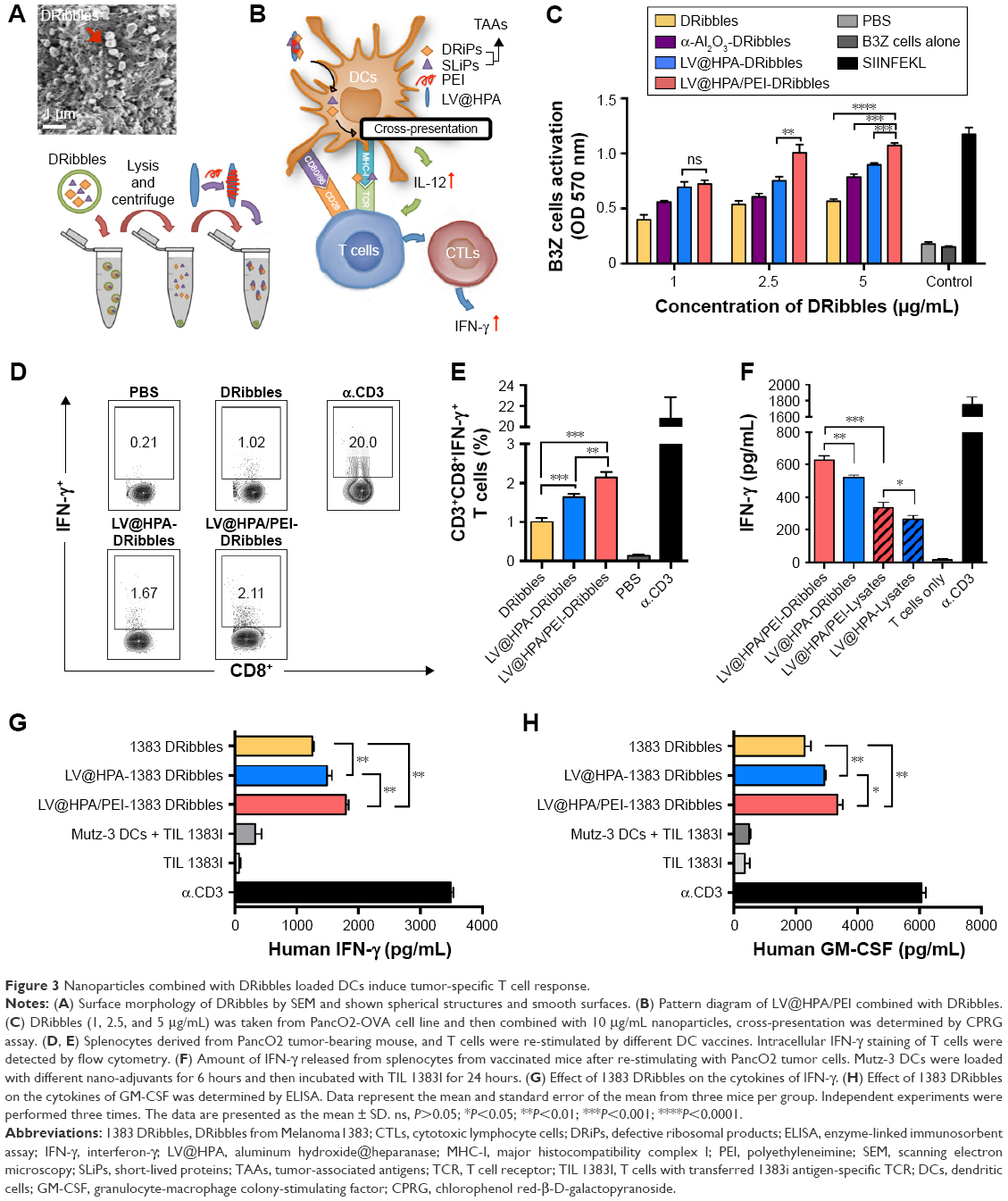 | Figure 3 Nanoparticles combined with DRibbles loaded DCs induce tumor-specific T cell response. |
LV@HPA/PEI-DRibbles had a potent therapeutic efficacy on the established PancO2 tumor model
Following the injection of four different vaccines, tumor volumes were measured every 3 days for a total of 54 days (Figure 4A). No obvious body weight loss or noticeable abnormality could be detected in groups receiving vaccines, indicating no onset of acute toxicity of the LV@HPA/PEI-DRibble-DCs and DRibble-DCs. In contrast, evident weight gain could be observed in the PBS and DC-alone treated groups, and the increase of body weight is due to rapid tumor growth (Figure 4B). The different effects of immunizations on tumor-bearing mice were observed by constructing survival curves, in which LV@HPA/PEI-DRibble-DCs could significantly improve the survival rate compared with DRibble-DCs and DCs alone (Figure 4C). The group of mice immunized with LV@HPA/PEI-DRibble-DCs showed the smallest tumor size, which could be considered as the obvious suppression of tumor growth (Figure 4D). This is shown in respective survival curves and tumor growth curves for each group. Most of the mice in the PBS group died before 33 days, and the tumor growth curve was very steep. All mice in the DC-alone group survived for more than 33 days, and two mice survived nearly 42 days. In DRibble-DCs group, all mice survived beyond 39 days. However, the growth curves of the LV@HPA/PEI-DRibble-DCs group was more gentle and smooth, and all mice survived for more than 51 days, with four mice surviving for more than 54 days. In addition, the tumors on LV@HPA/PEI-DRibble-DCs group were significantly suppressed. Compared with other vaccines, the results showed that LV@HPA/PEI-DRibble-DCs possessed excellent anti-tumor effects in vivo.
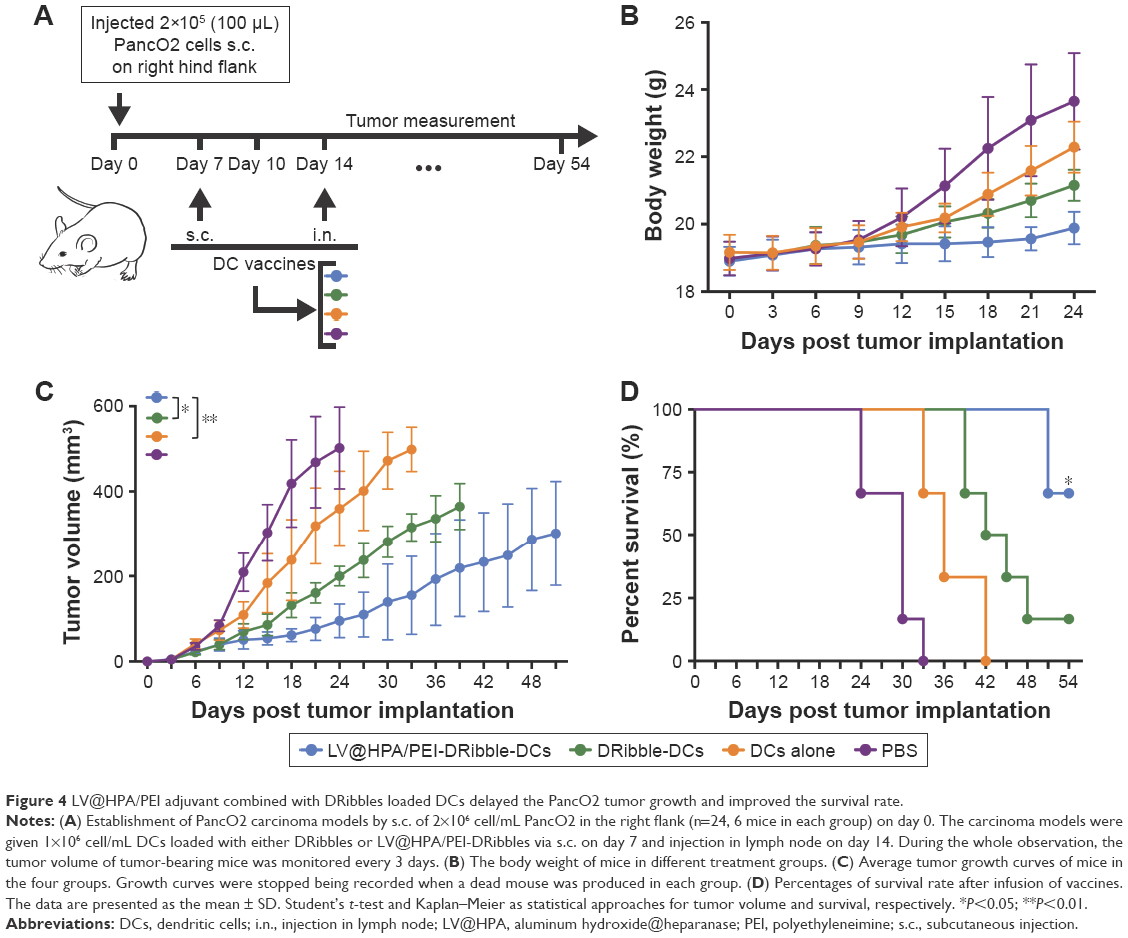 | Figure 4 LV@HPA/PEI adjuvant combined with DRibbles loaded DCs delayed the PancO2 tumor growth and improved the survival rate. |
Discussion
In order to suppress tumors, professional APCs need effective adjuvants to present TAA to active CTL responses by antigen cross-presentation. DCs have a key character to produce antitumor T cell responses due to their ability to take in antigens, process them into peptides with MHC I, and then present them to CD8+ T cells through a process known as cross-presentation.33 Rehydragel® (LV@HPA), an aluminum hydroxide nanoparticle with remarkable adsorption properties, has a strong adsorption capacity to carry soluble proteins. However, there are few systematic studies that evaluated the validity of LV@HPA as immune adjuvants. Recently, more attention has been given to PEI, a surface modifier, which is a powerful positive charge modified nano-material that can modify charged surface of nanoparticles.34 In order to investigate the performance of surface modification of LV@HPA with PEI, LV@HPA/PEI was explored as antigen vector to promote the function of DCs in our study. Through the zeta-potential detection, it was found that LV@HPA could be charged from negative to positive surface charge after being modified with PEI. Furthermore, the adsorbing performance of LV@HPA/PEI was improved compared to that of LV@HPA alone. Cellular internalization of cationic nanoparticle-PEI may trigger cytotoxicity with early necrotic cell damage and later apoptotic cell death.35 However, by modification with PEI, cytotoxicity of LV@HPA/PEI is almost negligible below 10 μg/mL. High-quality nano-adjuvants have been shown to be useful not only for their excellent biocompatibility but also for more antigens into APCs presented with lower concentration. Our results showed that LV@HPA/PEI could be internalized more effectively compared to LV@HPA in DCs, which were demonstrated by TEM detection. Compared with negative charged nanoparticles, the positive charged nanoparticles of LV@HPA/PEI were more likely to be internalized by DCs. Thus, it is quite possible that the positive-charged nanoparticles might be wrapped into endosomes and negative-charged nanoparticles into autolysosomes.31 From the flow cytometry and Western blot detection, our results showed that LV@HPA/PEI could significantly improve the transportation of OVA protein into DCs compared with either LV@HPA or OVA protein alone. This possibly means more OVA protein will be present on the DC surface as the peptide–MHC complex molecules are transported by LV@HPA/PEI to induce tumor-specific T cells activation.36
After confirming the efficient protein transport ability of LV@HPA/PEI, we further explored whether it could help to promote DC maturation and enhance their antigen cross-presenting ability. First, the secretion of one of the most potent Th1-cytokines, IL-12, was detected. IL-12 has been used to improve the effect of DC-based anti-cancer immunotherapy.37 We found that LV@HPA/PEI could promote more secretion of IL-12 by DCs than LV@HPA in a dose dependent manner. The co-stimulatory molecules of CD80 and CD86 on DC surfaces are important molecules in the identification and binding DCs and T cells for T cell activation.38 LV@HPA/PEI-OVA induced the upregulation of CD80 and CD86 expression on DCs, suggesting that the LV@HPA/PEI could effectively promote the maturation of DCs in vitro.
To determine whether OVA protein could be processed and presented to MHC restricted–specific CD8+ T cells by DCs, BMDCs were treated with LV@HPA/PEI-OVA and then co-cultured with CFSE-labeled OT-1 T cells, respectively. The results clearly showed that LV@HPA/PEI-OVA could effectively load on DCs and obviously active the proliferation of OT-1 T cells, which means LV@HPA/PEI could help antigens produce tumor-associated immune response. Based on our previous reports, antigens could be brought into the cytoplasm by positive-charged magnetic nanoparticles and degraded into peptides by the proteasome, then transported into the endoplasmic reticulum by the transporter TAP1 protein during antigens cross-presentation in DCs.39 The TAP1-knockout DCs with complete elimination of TAP1 was introduced in our study; the results indicated that the cross-presentation could be activated by the nano-adjuvants; this pathway was TAP1 dependent in DCs.
OVA protein is a soluble model antigen, whereas numbers of unconfirmed TAAs contained in DRibbles will be recognized and presented by DCs to inhibit tumor growth.40 With the assistance of suitable nano-adjuvants in a previous study, we showed that α-Al2O3 (60 nm) was a high-quality aluminum adjuvant that could bind to tumor autophagy antigens (DRibbles) to induce tumor-specific T cells activation, expansion, and tumor regression in mice bearing lung tumors.25 The DRibbles, including a number of TAAs, can load on DCs and inhibit the growth of tumors. Therefore, we would like to know whether LV@HPA/PEI has the ability to combine DRibbles and enhance DC cross-presentation. Then, we inhibited proteasome and lysosome of PancO2-OVA cells, a mouse pancreatic tumor cell line, to induce the DRibbles, as in our previous method.41,42 After DRibbles was combined with LV@HPA/PEI and co-cultured with DCs, the CRRG assay was performed and the result showed LV@HPA/PEI-DRibbles was the best adjuvant to active DCs and to induce T cell activation, compared with either LV@HPA-DRibbles or α-Al2O3-DRibbles. In order to explore the anti-tumor immune response of LV@HPA/PEI-DRibbles, splenocytes were separated from PancO2 tumor-bearing mice and co-cultured with DCs loaded with LV@HPA/PEI-DRibbles after expanding in vitro. The results showed that the proportions of activated T cells, CD3+CD8+IFN-γ+ T cells, increased obviously, meaning that our DC vaccines can stimulate a highly anti-tumor immune response in vitro. To determine whether LV@HPA/PEI-DRibbles could induce a more effective tumor-associated immune response than LV@HPA/PEI-Lysates, C57/BL6 mice were subcutaneously vaccinated with LV@HPA/PEI-DRibbles, LV@HPA-DRibbles, LV@HPA/PEI-Lysates, and LV@HPA-Lysates. After re-stimulation with PancO2 tumor cells, splenocytes from mice vaccinated with LV@HPA/PEI-DRibbles produced a higher level of IFN-γ than other groups. The result of IFN-γ indicated DRibbles could effectively activate immunoreaction than Lysates, indicating that LV@HPA/PEI could enhance the effect of DRibbles efficiently and effectively. As well as in murine T cell activation, the application of LV@HPA/PEI-DRibbles was also studied in human DCs. In the human T cells activation assay, the APCs were human acute myeloid leukemia cell line (Mutz-3)-derived DCs; the response cell was a human T cell line that was established from tumor-infiltrating lymphocyte (TIL 1383I) and could recognize an MHC class I (HLA-A2)-restricted epitope of the melanoma antigen tyrosinase. Compared with other control groups, we found that LV@HPA/PEI-1383 DRibbles load to Mutz-3 DCs played an important role in secretion of IFN-γ and GM-CSF by TIL 1383I, and both elevated cytokines indicated tumor-specific T cells were activated. These results proved that LV@HPA/PEI-DRibbles have a similar effect both on human and murine DCs and T cell activation.
Furthermore, PancO2 tumor cells were used to establish the tumor animal model for further study. It was found that LV@HPA/PEI-DRibble-DCs could inhibit tumor growth and prolong survival in tumor-bearing mice by injecting different activated DC vaccines. There were significant changes in four groups of tumor growth curve and the survival rate of mice, and mice in the group of LV@HPA/PEI-DRibble-DCs had smaller tumor size and slower growth compared with DRibble-DC groups. These results demonstrated that the new vaccine LV@HPA/PEI-DRibble-DCs, having an outstanding anti-tumor effect and excellent curative effect, provides a theoretical basis for clinical application in the future.
Conclusion
In summary, we identified excellent biological characteristics and potential adjuvant function of LV@HPA/PEI for DC antigen cross-presentation in anti-tumor immunotherapy. To combine with the tumor autophagic antigen, DRibbles, the novel cancer adjuvant (LV@HPA/PEI-DRibbles) has excellent abilities to mature DCs. Furthermore, the DC vaccine (LV@HPA/PEI-DRibble-DCs) exhibits excellent efficacy in the activation of tumor-associated T cells, inhibition of tumor volume and prolongation of survival rate in tumor-bearing mice, and may be a candidate for an effective antitumor vaccine in the future.
Acknowledgments
The authors are grateful for grants from the National Natural Science Foundation of China (81371680 and 81571800). We acknowledge the kind support of Jaina Patel for technical assistance and Guohua Xia and Yu Zhuang for language editing.
Disclosure
The authors report no conflicts of interest in this work.
References
Wei XX, Perry J, Chang E, et al. Clinical variables associated with overall survival in metastatic castration-resistant prostate cancer patients treated with sipuleucel-t immunotherapy. Clin Genitourin Cancer. Epub 2017 Dec 27. | ||
Verboogen DR, Dingjan I, Revelo NH, Visser LJ, ter Beest M, van den Bogaart G. The dendritic cell side of the immunological synapse. Biomol Concepts. 2016;7(1):17–28. | ||
Han JA, Kang YJ, Shin C, et al. Ferritin protein cage nanoparticles as versatile antigen delivery nanoplatforms for dendritic cell (DC)-based vaccine development. Nanomedicine. 2014;10(3):561–569. | ||
Mou Y, Hou Y, Chen B, et al. In vivo migration of dendritic cells labeled with synthetic superparamagnetic iron oxide. Int J Nanomedicine. 2011;6:2633–2640. | ||
Cintolo JA, Datta J, Mathew SJ, Czerniecki BJ. Dendritic cell-based vaccines: barriers and opportunities. Future Oncol. 2012;8(10):1273–1299. | ||
Mou Y, Chen B, Zhang Y, et al. Influence of synthetic superparamagnetic iron oxide on dendritic cells. Int J Nanomedicine. 2011;6:1779–1786. | ||
Chen HM, Wang PH, Chen SS, et al. Shikonin induces immunogenic cell death in tumor cells and enhances dendritic cell-based cancer vaccine. Cancer Immunol Immunother. 2012;61(11):1989–2002. | ||
Paulis LE, Mandal S, Kreutz M, Figdor CG. Dendritic cell-based nanovaccines for cancer immunotherapy. Curr Opin Immunol. 2013;25(3):389–395. | ||
Mody N, Dubey S, Sharma R, Agrawal U, Vyas SP. Dendritic cell-based vaccine research against cancer. Expert Rev Clin Immunol. 2015;11(2):213–232. | ||
Romero Mendez IZ, Shi Y, HogenEsch H, Hem SL. Potentiation of the immune response to non-adsorbed antigens by aluminum-containing adjuvants. Vaccine. 2007;25(5):825–833. | ||
Mannhalter JW, Neychev HO, Zlabinger GJ, Ahmad R, Eibl MM. Modulation of the human immune response by the non-toxic and non-pyrogenic adjuvant aluminium hydroxide: effect on antigen uptake and antigen presentation. Clin Exp Immunol. 1985;61(1):143–151. | ||
O’Hagan DT, MacKichan ML, Singh M. Recent developments in adjuvants for vaccines against infectious diseases. Biomol Eng. 2001;18(3):69–85. | ||
Lu F, Boutselis I, Borch RF, Hogenesch H. Control of antigen-binding to aluminum adjuvants and the immune response with a novel phosphonate linker. Vaccine. 2013;31(40):4362–4367. | ||
Rhie GE, Park YM, Han JS, Yu JY, Seong WK, Oh HB. Efficacy of non-toxic deletion mutants of protective antigen from Bacillus anthracis. FEMS Immunol Med Microbiol. 2005;45(2):341–347. | ||
Usinger WR. A comparison of antibody responses to veterinary vaccine antigens potentiated by different adjuvants. Vaccine. 1997;15(17–18):1902–1907. | ||
Li X, Aldayel AM, Cui Z. Aluminum hydroxide nanoparticles show a stronger vaccine adjuvant activity than traditional aluminum hydroxide microparticles. J Control Release. 2014;173:148–157. | ||
Cui X, Belo S, Kruger D, et al. Aluminium hydroxide stabilised MnFe2O4 and Fe3O4 nanoparticles as dual-modality contrasts agent for MRI and PET imaging. Biomaterials. 2014;35(22):5840–5846. | ||
He P, Zou Y, Hu Z. Advances in aluminum hydroxide-based adjuvant research and its mechanism. Hum Vaccin Immunother. 2015;11(2):477–488. | ||
Becker LC, Boyer I, Bergfeld WF, et al. Safety assessment of alumina and aluminum hydroxide as used in cosmetics. Int J Toxicol. 2016;35(3 Suppl):16s–33s. | ||
Thomas TJ, Tajmir-Riahi HA, Thomas T. Polyamine-DNA interactions and development of gene delivery vehicles. Amino Acids. 2016;48(10):2423–2431. | ||
Anselmo AC, Mitragotri S. A review of clinical translation of inorganic nanoparticles. AAPS J. 2015;17(5):1041–1054. | ||
Zhao J, Pan N, Huang F, et al. Vx3-functionalized alumina nanoparticles assisted enrichment of ubiquitinated proteins from cancer cells for enhanced cancer immunotherapy. Bioconjug Chem. 2018;29(3):786–794. | ||
Li YH, Wang LX, Pang PY, et al. Tumor-derived autophagosome vaccine: mechanism of cross-presentation and therapeutic efficacy. Clin Cancer Res. 2011;17(22):7047–7057. | ||
Su H, Luo Q, Xie H, et al. Therapeutic antitumor efficacy of tumor-derived autophagosome (DRibble) vaccine on head and neck cancer. Int J Nanomed. 2015;10:1921–1930. | ||
Li HY, Li YH, Jiao J, Hu HM. Alpha-alumina nanoparticles induce efficient autophagy-dependent cross-presentation and potent antitumour response. Nat Nanotechnol. 2011;6(10):645–650. | ||
Fuertes Marraco SA, Grosjean F, Duval A, et al. Novel murine dendritic cell lines: a powerful auxiliary tool for dendritic cell research. Front Immunol. 2012;3:331. | ||
van de Ven R, Reurs AW, Wijnands PG, et al. Exposure of CD34+ precursors to cytostatic anthraquinone-derivatives induces rapid dendritic cell differentiation: implications for cancer immunotherapy. Cancer Immunol Immunother. 2012;61(2):181–191. | ||
Wang S, Tian Y, Tian W, et al. Selectively sensitizing malignant cells to photothermal therapy using a CD44-targeting heat shock protein 72 depletion nanosystem. ACS Nano. 2016;10(9):8578–8590. | ||
Mayorek N, Naftali-Shani N, Grunewald M. Diclofenac inhibits tumor growth in a murine model of pancreatic cancer by modulation of VEGF levels and arginase activity. PLoS One. 2010;5(9):e12715. | ||
Roszkowski JJ, Yu DC, Rubinstein MP, McKee MD, Cole DJ, Nishimura MI. CD8-independent tumor cell recognition is a property of the T cell receptor and not the T cell. J Immunol. 2003;170(5):2582–2589. | ||
Mou Y, Xing Y, Ren H, et al. The effect of superparamagnetic iron oxide nanoparticle surface charge on antigen cross-presentation. Nanoscale Res Lett. 2017;12(1):52. | ||
Ren H, Zhao S, Li W, et al. Therapeutic antitumor efficacy of B cells loaded with tumor-derived autophagasomes vaccine (DRibbles). J Immunother. 2014;37(8):383–393. | ||
Win SJ, Ward VK, Dunbar PR, Young SL, Baird MA. Cross-presentation of epitopes on virus-like particles via the MHC I receptor recycling pathway. Immunol Cell Biol. 2011;89(6):681–688. | ||
Li X, Chen Y, Wang M, Ma Y, Xia W, Gu H. A mesoporous silica nanoparticle – PEI – fusogenic peptide system for siRNA delivery in cancer therapy. Biomaterials. 2013;34(4):1391–1401. | ||
Wang GD, Tan YZ, Wang HJ, Zhou P. Autophagy promotes degradation of polyethyleneimine-alginate nanoparticles in endothelial progenitor cells. Int J Nanomedicine. 2017;12:6661–6675. | ||
Mellman I, Steinman RM. Dendritic cells: specialized and regulated antigen processing machines. Cell. 2001;106(3):255–258. | ||
Vogt A, Sievers E, Lukacs-Kornek V, et al. Improving immunotherapy of hepatocellular carcinoma (HCC) using dendritic cells (DC) engineered to express IL-12 in vivo. Liver Int. 2014;34(3):447–461. | ||
Jiang H, Zhang Y, Yin X, et al. Construction and evaluation of rats’ tolerogenic dendritic cells (DC) induced by NF-κB decoy method. Afr Health Sci. 2014;14(3):626–633. | ||
Amigorena S, Savina A. Intracellular mechanisms of antigen cross presentation in dendritic cells. Curr Opin Immunol. 2010;22(1):109–117. | ||
Ye W, Xing Y, Paustian C, et al. Cross-presentation of viral antigens in dribbles leads to efficient activation of virus-specific human memory T cells. J Transl Med. 2014;12:100. | ||
Su S, Zhou H, Xue M, et al. Anti-tumor efficacy of a hepatocellular carcinoma vaccine based on dendritic cells combined with tumor-derived autophagosomes in murine models. Asian Pac J Cancer Prev. 2013;14(5):3109–3116. | ||
Angel LP, Divino CM, Brower ST, Chen SH. A novel immunocytolytic factor secreted by pancreatic adenocarcinoma. J Surg Res. 2000;91(2):154–158. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.